Abstract
The GTPase RhoA and the transcriptional factor c–Myc are closely intertwined in cancer cells. Although this cross–talk results in potent synergistic effects that favor the transformed phenotype of cancer cells, recent results from our laboratory indicate that c–Myc also participates in a negative feed–back loop that blocks specific RhoA signaling branches connected to the induction of stress fibers, focal adhesions and actomyosin contractility. Using microarray analysis, we have unveiled a RhoA/c–Myc–dependent gene signature in charge of this negative cross–talk. This signature is composed of up–regulated and repressed transcripts encoding cytoskeletal modulators located downstream of both RhoA and Rock. Our results also indicate that this negative feed–back loop modifies the invasion and adhesion properties of RhoA–transformed cells, suggesting that it may be important to ensure fluid cytoskeletal dynamics of cancer cells. Preliminary data indicate that c–Myc may also use a different transcriptional program to interfere with the RhoA/Rock–dependent cytoskeletal branch in non–transformed cells.
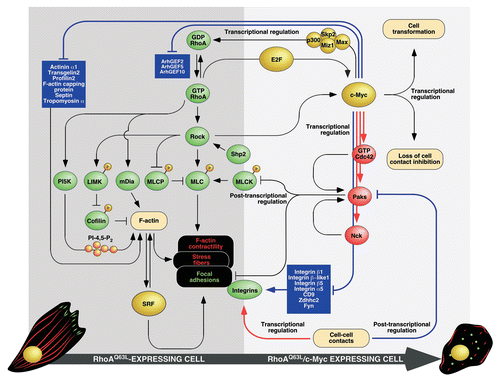
Accumulating evidence indicates that RhoA contributes to the malignant properties of cancer cells.Citation1,Citation2 However, the hyperactivation of RhoA-dependent routes has its pros and cons in this pathological setting. Thus, it obviously favors proliferative and survival responses that increase the fitness of cancer cells. Though, such positive action is counterbalanced by the negative effects that hyperactivated RhoA may have in cell migration. Those include, depending on the cell context, the induction of too rigid stress fibers and focal adhesions that inhibit polarity and migration or, alternatively, the stimulation of abnormally high levels of actomyosin contractility that could lead to cell detachment and anoikis. To deal with this problem, cancer cells have implemented a number of negative regulatory steps to tune down the activity of the RhoA pathway (). Those include, among others, the release of RhoA from the plasma membrane via phosphorylation by protein kinase A (PKA) and protein kinase G (PKG)Citation3,Citation4 (), the proteosomal degradation of RhoA induced by E3 ubiquitin ligases (Smurf1, Cullin)Citation5,Citation6 (), the interference with the RhoA activation cycle via inhibition of RhoA guanosine nucleotide exchange factors (GEFs) and stimulation of GTPase activating proteins (GAPs),Citation7–Citation9 the obstruction of effector binding by the previous interaction of RhoA with cell cycle inhibitors (p27Kip1),Citation10 or the inhibition of Rock activity by the direct interaction of this serine/threonine kinase with GTPases (RhoE/Rnd3, Gem, R-Rad)Citation11–Citation13 and cell cycle inhibitors (p21Waf1)Citation14 (). The biological output from the RhoA pathway can be also influenced indirectly by transcriptional-dependent mechanisms. For example, the tumor suppressor p53 can promote the distal inhibition of RhoA-dependent routes by stimulating transcription from Rnd3 and Cdkn1a genesCitation15,Citation16 (). Despite the fact that most of those inhibitory loops are well known at the structural/signaling level, we still lack substantial information about the actual role they play in vivo. Due to this, we do not know still whether these inhibitory steps can be generalized to most cell types or, instead, whether they are only operative in specific cell types or tumors.
Given their inhibitory nature, it is likely that the above regulatory steps represent mechanisms to repress all RhoA-(in the case of PKA, PKG, Smurf1, Cullin and p27Kip1) and Rock-dependent (in the case of RhoE/Rnd3, R-Rad, Gem, p21Waf1) signals. This may be a good option in the case of normal cells for the promotion of, for example, cell motility and proliferative responses at the same time. However, the biological complexity of cancer cells may require more sophisticated regulatory mechanisms that could induce the disengagement of specific signaling branches of the RhoA pathway. In the scientific article highlighted here,Citation17 we demonstrated that the transcriptional factor c-Myc could be a crucial element to accomplish such a schizophrenic role, since it can favor the promotion of RhoA transforming potential and, at the same time, interfere with the cytoskeletal changes induced by this GTPase ().
Previous reports have shown that the biological activities of c-Myc and RhoA are closely interrelated. Thus, recent work from Ling's lab has shown that c-Myc promotes RhoA gene transcription in cancer cells via the formation of transcriptionally active complexes composed of the E3 ubiquitin ligase Skp2 and the transcriptional co-activators p300 and Miz1,Citation18 ( and ). Furthermore, we published three years ago that a constitutively active version of RhoA (Q63L mutant) induces high c-Myc expression levels in fibroblasts by stimulating Rock- and E2F-dependent routesCitation19 ( and ). This interconnection is important in functional terms, because it has been demonstrated that the overexpression of c-Myc confers RhoA-like properties to cancer cellsCitation18 and, reciprocally, that a significant percentage of the RhoAQ63L-induced transcriptome is c-Myc-dependent.Citation19 Moreover, we could demonstrate in that earlier study that the expression of c-Myc was a condition sine qua non for the transforming activity of RhoAQ63L in fibroblasts.Citation19 At first sight, the above results appeared to represent the typical open-and-shut case: given that RhoA and c-Myc are both important for cell transformation, it was fully expected that the latter could be essential for the oncogenic activity of RhoA in vivo. Against this simplistic view, the analysis of cell lines generated in our initial work indicated that the case was more complex than anticipated, for we observed that the transformed fibroblasts co-expressing RhoAQ63L and c-Myc displayed a significant lower number of stress fibers and focal adhesions than those expressing RhoAQ63L alone (). Consistent with a negative role of c-Myc on the RhoA/Rock-dependent cytoskeleton, we also observed that the repression of the endogenous c-Myc protein led to the generation of more robust stress fibers and higher numbers of focal adhesions in RhoAQ63L-transformed cells. Using an Affymetrix microarray approach, we could identify a gene signature specifically present in RhoAQ63L/c-Myc-expressing fibroblasts that can explain the effects of c-Myc on the RhoA/Rock-dependent cytoskeleton. The close examination of these genes indicated that it does not include transcripts encoding previously reported RhoA and Rock inhibitors and, instead, is characterized by the deregulated expression of 535 transcripts encoding proteins involved in quite disparate biological functions. Interestingly, we observed the presence in that signature of an important subset of genes encoding structural and regulatory proteins of the cytoskeleton that, prima facie, could be considered as good targets to explain this cytoskeletal phenotype. To study this possibility, we decided to focus our attention on upregulated transcripts encoding molecules known to counteract RhoA routes (Cdc42, Pak1) as well as on repressed mRNAs encoding a significant large number of integrin subunits and integrin-associated proteins (). We selected those molecules because previous results had shown that Cdc42 and Pak1 influence negatively the RhoA-dependent cytoskeleton via the inhibition of myosin light chain kinase and the acceleration of focal adhesion turn-over ratesCitation20–Citation25 (). Likewise, we assumed that the inhibition of integrin subunits could contribute to the downmodulation of the RhoA/Rock-dependent cytoskeleton by reducing the numbers and/or size of focal adhesions and, consequently, weakening the connectivity of those cellular structures to the extracellular matrix. Consistent with our hypothesis, we could recapitulate the cytoskeletal phenotype of RhoAQ63L/c-Myc-expressing cells by overexpressing Cdc42 and Pak1 in RhoAQ63L-transformed cells. This effect was not observed with Cdc42 mutants incapable of activating Pak1. A similar reduction in stress fibers and focal adhesions was also observed when we knocked down specific integrin subunits in cells expressing RhoAQ63L. Further supporting the relationship of these proteins with the effects of c-Myc on the F-actin cytoskeleton, we could rescue “normal” levels of stress fibers and focal adhesions in RhoAQ63L/c-Myc-expressing fibroblasts by inhibiting the endogenous levels of Pak1.
I believe that it is important to underscore three functional features of the RhoA/c-Myc cross-talk discussed in this Extra View. The first one is its synergistic nature, since it requires the coalescence of signals from both RhoAQ63L and c-Myc to be turned on. Accordingly, the genes responsible for counteracting the RhoA-dependent cytoskeleton are only found deregulated in transformed cells coexpressing RhoAQ63L and c-Myc but not in those expressing each of those proteins alone. These observations indicate that this biological program is probably restricted to normal or disease-derived cells containing high levels of RhoA and c-Myc. A second property that the readers must keep in mind is that c-Myc influences negatively the RhoA/Rock-dependent cytoskeleton of RhoAQ63L-transformed cells but, at the same time, has quite positive effects in other RhoA-dependent routes essential for the maintenance of the transformed phenotypeCitation19 (). This high level of signaling specificity is even observed within the Rock pathway, because the overexpression of c-Myc reduces the phosphorylation levels of well-known Rock targets that regulate actomyosin contractility (myosin light chain and myosin light change phosphatase) but, instead, it does not block the Rock-dependent transcriptome present in RhoAQ63L-transformed cells.Citation17 This high level of signaling selectivity is probably achieved by the fact that Pak1 and integrins, unlike the case of other inhibitors of the RhoA/Rock routeCitation11–Citation14 (), work downstream of Rock proteins in this new route (). This ensures the preservation of active pools of Rock not directly connected to the F-actin cytoskeleton. A third feature of this gene signature is that it can be easily reversed by stimuli elicited by the establishment of intercellular contacts or the acquisition of high cell densities ( and ). Although this pathway has not been fully dissected in our work, the available data are consistent with the idea that the establishment of cell-to-cell contacts engages both transcriptional and a post-transcriptional mechanisms that counterbalance the action of c-Myc (). The transcriptional mechanisms restore normal integrin subunit mRNA expression levels whereas the post-transcriptional routes impact on the stability of Pak1 protein (). Regardless of the mechanisms involved, it is worth noting that the biological program activated by cell-to-cell contacts is not related to the stimulation of contact inhibition pathways, because RhoAQ63L/c-Myc-expressing cells are highly transformed and have lost completely that type of regulation.Citation19
Although the data presented in this Extra View article formally demonstrated the role of Pak1 and integrin subunits in the c-Myc-dependent biological program, the readers should keep in mind that many more proteins could be implicated in this process. Thus, we have identified transcripts encoding additional integrin subunits (integrin-β1l, integrin-α5) and cytoskeletal regulators (actinin α1, transgelin2, profilin2 or F-actin capping protein) in our microarray experiments whose implication in this negative feedback loop has not been dissected yet (). Moreover, we have discarded in our functional analyses a large number of deregulated genes present in the microarray data due to the lack of any obvious functional link to the F-actin cytoskeleton. However, we cannot rule out that proteins encoded by some of those transcripts could still participate in this new biological program by as yet unknown mechanisms. An obvious strategy to identify all the culprits of this RhoAQ63L/c-Myc-dependent program is to analyze the effect of each microarray-identified gene in the cytoskeleton of RhoAQ63L- and RhoA/c-Myc-transformed cells using gain- and loss-of-function studies similar to those used in the Extra View article discussed here. Finally, we have not considered in our studies a subset of genes encoding GEFs for RhoA (ArhGEF2, AhrGEF3, ArhGEF10) that were seen clearly repressed upon c-Myc overexpression (). This was because the alteration in the expression levels of these proteins should not affect the biological activity of the oncogenic, GTPase deficient RhoAQ63L protein expressed in the transformed fibroblasts used in this study. However, the identification of those proteins is potentially interesting if we consider that the hyperactivation of the RhoA pathway in cancer cells derives from the alteration in the normal regulatory cycle of wild type RhoA rather than from the development of oncogenic mutations in its locus.Citation2 Due to this, these proteins may have an effective role in the modulation of the overall RhoA signaling output in cell settings different to those used in our work. Other candidates that could be important in a similar biological context are the polarity protein Par6 and the Rac1 exchange factor Ect2, since previous results have shown that they contribute to the stimulation of the catalytic activities of p190RhoGAP and Pak1,Citation26–Citation29 that, in turn, could contribute to the down-modulation of the cytoskeleton by promoting the inactivation of endogenous RhoA (in the case of p190RhoGAP) and faster focal adhesion turn-over rates (in the case of Pak1). In Drosophila, the Ect2 homology Pebble also promotes Rac1 activation during mesodermal migration,Citation30,Citation31 offering an extra mechanism to counterbalance RhoA-dependent signals in this functional context.
Although the effect of c-Myc overexpression in RhoAQ63L-transformed cells is more conspicuous, it is worth noting that we have found that the overexpression of this transcriptional factor also affects the stress fiber status of normal fibroblasts. This indicates that the negative action of c-Myc on the cytoskeleton could be a generalized event independently of the transformed status of cells. However, the action of c-Myc in normal fibroblast must involve a different transcriptional program because, as discussed above, the gene signature described in the article object of this Extra View results exclusively from synergistic interactions between strong RhoA and c-Myc signals. The transcriptional program involved in the c-Myc-dependent downmodulation of the F-actin cytoskeleton remains to be determined, but it could be identified in the near future by comparing the transcriptomes of c-Myc-expressing fibroblasts and parental cells. Given the availability of inducible c-Myc knock-in mice, an interesting experiment in the future will be to verify whether the inactivation of c-Myc in vivo contributes to alterations in the F-actin cytoskeleton similar to those described here. This may be actually the case, because we have observed that the previously described rat c-Myc−/− HO15.19 cells display more robust and abundant stress fibers and focal adhesions than their wild type TGR-1 counterparts (Sauzeau V, Berenjeno IM, Citterio C and Bustelo XR, unpublished data). Further work in this area will allow us to obtain a better view of the influence of c-Myc on the cytoskeletal dynamics of normal and cancer cells and to identify all the molecules/pathways involved in this negative feedback mechanism.
Figures and Tables
Figure 1 Schematic representation of RhoA signaling elements connected to the regulation of the F-actin cytoskeleton and known inhibitory steps operating in this route. Signaling proteins are shown in green. Transcriptional factors are depicted in yellow. Molecules that inhibit either RhoA or Rock are shown in blue. Activation and inactivation steps are shown as arrows and blunted lanes, respectively. Stress fibers, focal adhesions and nuclei are depicted in red, green and yellow, respectively. Further details can be found in the main text. LIMK, LIM-domain kinase; mDia, mouse Diaphanous homologue; MLC, myosin light chain; MLCK, MLC kinase; MLCP, MLC phosphatase; P, phosphorylation site; PI5K, phosphatidylinositol 5-kinase; SRF, serum responsive factor. Other abbreviations have been described in the main text.
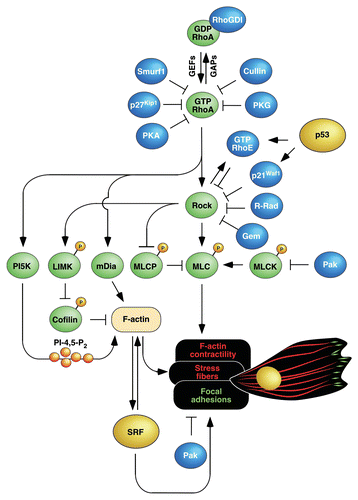
Figure 2 Overexpression of c-Myc leads to positive synergistic interactions with RhoA that favor cell transformation and, at the same time, the generation of a transcriptomal-based genetic program that blocks the RhoA/Rock-dependent F-actin cytoskeleton. This latter program is counterbalanced by signals derived from cell-to-cell contacts. Proteins and cytoskeletal structures follow the same color code used in . This scheme has been designed taking into consideration the data published in the article object of this Extra View and from previous publications in this area.Citation18,Citation19,Citation32
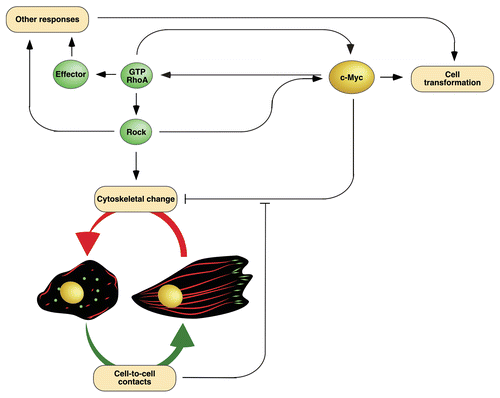
Figure 3 Schematic representation of the cytoskeletal-related RhoA pathways (dark gray area, left) and the transcriptional programs discovered in RhoA/c-Myc-transformed cells that are described in this Extra View (light gray area, right). Signaling proteins operating in the RhoA route connected to F-actin cytoskeletal regulation are shown in green. Transcriptional factors and associated proteins are depicted in yellow. Known connections among signal transduction elements are indicated with black thin lanes. Connections revealed in the work summarized in this Extra View are depicted with thick blue (when indicating gene repression events) or red (when indicating gene upregulation events). Activation and inactivation steps are shown as arrowheads and blunted lanes, respectively. Upregulated protein products are shown in red. Downmodulated protein products have been enclosed in dark blue boxes. Further details can be found in the main text. For abbreviations, see legend to .
Acknowledgements
X.R.B. work is supported by grants from the NIH (5R01-CA73735-13), the Spanish Ministry of Science and Innovation (SAF2009-07172 and RD06/0020/0001), the Castilla y León Autonomous Government (GR97) and the 7th Framework European Union Program (FP7-HEALTH-2007-A-201862). X.R.B.'s lab has been recognized as a Consolidated Cancer Research Group by the Asociación Española contra el Cáncer.
The content of this work is solely the responsibility of the author and does not necessarily represent the official views of the National Cancer Institute, the National Institutes of Health or the rest of funding agencies that supported this work.
Extra View to: Sauzeau V, Berenjeno IM, Citterio C, Bustelo XR. A transcriptional cross-talk between RhoA and c-Myc inhibits the RhoA/Rock-dependent cytoskeleton. Oncogene 2010; 29:3781 - 3792; PMID: 20453885; http://dx.doi.org/10.1038/onc.2010.134
References
- Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer 2002; 2:133 - 142
- Gomez del Pulgar T, Benitah SA, Valeron PF, Espina C, Lacal JC. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays 2005; 27:602 - 613
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 1996; 15:510 - 519
- Sauzeau V, Sevilla MA, Montero MJ, Bustelo XR. The Rho/Rac exchange factor Vav2 controls nitric oxide-dependent responses in mouse vascular smooth muscle cells. J Clin Invest 2010; 120:315 - 330
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003; 302:1775 - 1779
- Chen Y, Yang Z, Meng M, Zhao Y, Dong N, Yan H, et al. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell 2009; 35:841 - 855
- Alberts AS, Qin H, Carr HS, Frost JA. PAK1 negatively regulates the activity of the Rho exchange factor NET1. J Biol Chem 2005; 280:12152 - 12161
- Callow MG, Zozulya S, Gishizky ML, Jallal B, Smeal T. PAK4 mediates morphological changes through the regulation of GEF-H1. J Cell Sci 2005; 118:1861 - 1872
- Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol 2000; 10:719 - 722
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862 - 876
- Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol 2003; 23:4219 - 4229
- Hatzoglou A, Ader I, Splingard A, Flanders J, Saade E, Leroy I, et al. Gem associates with Ezrin and acts via the Rho-GAP protein Gmip to downregulate the Rho pathway. Mol Biol Cell 2007; 18:1242 - 1252
- Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B, et al. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J Cell Biol 2002; 157:291 - 302
- Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem 2004; 279:1885 - 1891
- Ongusaha PP, Kim HG, Boswell SA, Ridley AJ, Der CJ, Dotto GP, et al. RhoE is a pro-survival p53 target gene that inhibits ROCK I-mediated apoptosis in response to genotoxic stress. Curr Biol 2006; 16:2466 - 2472
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817 - 825
- Sauzeau V, Berenjeno IM, Citterio C, Bustelo XR. A transcriptional cross-talk between RhoA and c-Myc inhibits the RhoA/Rock-dependent cytoskeleton. Oncogene 2010; 29:3781 - 3792
- Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol 2010; 12:457 - 467
- Berenjeno IM, Nunez F, Bustelo XR. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene 2007; 26:4295 - 4305
- Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999; 283:2083 - 2085
- Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, De Lanerolle P, et al. Inhibition of contraction and myosin light chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J Physiol 2003; 549:489 - 500
- Frost JA, Khokhlatchev A, Stippec S, White MA, Cobb MH. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J Biol Chem 1998; 273:28191 - 28198
- Coniglio SJ, Zavarella S, Symons MH. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol Cell Biol 2008; 28:4162 - 4172
- Chan PM, Lim L, Manser E. PAK is regulated by PI3K, PIX, CDC42 and PP2Calpha and mediates focal adhesion turnover in the hyperosmotic stress-induced p38 pathway. J Biol Chem 2008; 283:24949 - 24961
- Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, et al. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol 1997; 17:1129 - 1143
- Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene 2009; 28:3597 - 3607
- Zhang H, Macara IG. The PAR-6 polarity protein regulates dendritic spine morphogenesis through p190RhoGAP and the Rho GTPase. Dev Cell 2008; 14:216 - 226
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem 2005; 280:31109 - 31115
- Liu XF, Ishida H, Raziuddin R, Miki T. Nucleotide exchange factor ECT2 interacts with the polarity protein complex Par6/Par3/protein kinase Czeta (PKCzeta) and regulates PKCzeta activity. Mol Cell Biol 2004; 24:6665 - 6675
- van Impel A, Schumacher S, Draga M, Herz HM, Grosshans J, Muller HA. Regulation of the Rac GTPase pathway by the multifunctional Rho GEF Pebble is essential for mesoderm migration in the Drosophila gastrula. Development 2009; 136:813 - 822
- Schumacher S, Gryzik T, Tannebaum S, Muller HA. The RhoGEF Pebble is required for cell shape changes during cell migration triggered by the Drosophila FGF receptor Heartless. Development 2004; 131:2631 - 2640
- Berenjeno IM, Bustelo XR. Identification of the Rock-dependent transcriptome in rodent fibroblasts. Clin Transl Oncol 2008; 10:726 - 738