Abstract
Mutations in IQSEC2, a guanine nucleotide exchange factor for the ADP-ribosylation factor (Arf) family of small GTPases have recently been shown to cause non-syndromic X-linked intellectual disability (ID), characterised by substantial limitations in intellectual functioning and adaptive behaviour. This discovery was revealed by a combination of large-scale resequencing of the X chromosome, and key functional assays that revealed a reduction, but not elimination, of IQSEC2 GEF activity for mutations affecting conserved amino acids in the IQ-like and Sec7 domains. Compromised GTP binding activity of IQSEC2 leading to reduced activation of selected Arf substrates (Arf1, Arf6) is expected to impact on cytoskeletal organization, dendritic spine morphology and synaptic organisation. This study highlights the need for further investigation of the IQSEC gene family and Arf GTPases in neuronal morphology and synaptic function, and suggests that the genes encoding the ArfGEFs IQSEC1 and IQSEC3 should be considered as candidates for screening in autosomal ID.
Introduction
Intelligence describes the ability of the brain to comprehend, infer, make decisions, reason, use language, learn and plan. These cognitive abilities require the correct assembly of complex neuronal structures during development and a continued ability to adapt in response to internal and external cues. Disruption of any of these elements by environmental and/or genetic factors can lead to deficits in cognitive function. For example, intellectual disability (ID) is definedCitation1 as significantly impaired cognitive functioning coupled with a deficit in adaptive behaviour, with onset before the age of 18. ID is characterised by significantly below-average scores in test of mental ability or intelligence and limitations in the ability to function in areas of daily life, such as communication, self-care, getting along in social situations or school activities. ID is frequent in the population, with as many as 1 in every 50 individualsCitation2 worldwide directly affected. Whilst the causes of ID are highly heterogeneous, genetic studies of X chromosome-linked intellectual disability (XLID) are relatively advanced. Subdividing XLID into ‘non-syndromic’ (where ID is the only consistent clinical abnormality) and ‘syndromic’ forms (where other, consistent defects may manifest) and the collection of large cohorts of families by international consortia (e.g., the IGOLD project; http://goldstudy.cimr.cam.ac.uk/) has facilitated the discoveryCitation3 of associated genes. However, although more than 90 XLID genes have been reported, the underlying defect remains unresolved in over 50% of families with non-syndromic XLID. Until recently, identification of the causative mutations in XLID has been achieved primarily by positional and functional candidate gene approaches and targeted re-sequencing of specific regions of the X chromosome. The application of the systematic re-sequencing of the entire X chromosome exomeCitation4 has removed bias associated with candidate gene selection and revealed previously unidentified XLID-associated genes such as SYP, ZNF711 or CASK. Despite the success of this approach, several challenges remain to be overcome, including incomplete coverage due to PCR failure, lack of analysis of gene elements outside coding regions (e.g., promoters/enhancers, non-coding RNAs, etc.,) or copy number variations and reliance on automated sequence analysis. Even when changes were detected, distinguishing pathogenic mutations from rare synonymous or non-synonymous variants is not easy in such large data sets, particularly when non-pathogenic truncating variants are presentCitation4,Citation5 in 1% of X-chromosome genes. In a small-scale follow-up studyCitation6 on one gene, IQSEC2—a GEF for Arf family GTPases ()—we have highlighted how comprehensive data analysis, genetic and functional assays can be used to link DNA sequence variation to ID pathology.
Mutations in IQSEC2 Cause XLID
Three non-synonymous SNPs in IQSEC2 were identified by Tarpey et al. in separate, multigenerational families with non-syndromic ID (). In MRX1, the first family where non-syndromic ID was mappedCitation7 to the X chromosome, a c.2587C>T change was found in exon 8 leading to a p.R863W substitution. By contrast, in the MRX18 family, a c.2402A>C change in exon 6 leading to a p.Q801P substitution was noticed. A third family ascertained in the USA (US166) had a c.2273G>A change in exon 5 leading to a p.R758Q substitution. Although these variants were not detected in the rest of the resequenced cohortCitation4 or numerous control DNAs, IQSEC2 was not immediately assigned as a new XLID gene.Citation4 Part of the reason for this was that these variants were amongst over 550 unique non-recurrent variants identified in 208 XLID families. Due to the size of this data set, tracking of all variants in all relevant families could not be performed in a timely manner. In addition, these IQSEC2 variants were not amongst the most highly ranked in an assessmentCitation4 of likely pathogenicity based on conservation score. However, our follow-up studyCitation6 demonstrated that these changes and an additional c.1075C>T change in exon 4 leading to a p.R359C substitution (found in an Australian family, AU128) clearly segregated with the ID phenotype in the respective families. IQSEC2 is also known as BRAG1 or IQ-ArfGEF () and occurs in two isoformsCitation6,Citation8 generated by alternative splicing. IQSEC2 is enriched in the PSD at excitatory synapsesCitation8,Citation9 and associates with PSD95, a PDZ domain-containing synaptic protein involved in NMDAR clustering (). Curiously, IQSEC2 transcripts are localised in both hippocampal neuronal cell bodies and dendritic processes,Citation9 suggesting that local translation of IQSEC2 mRNA in an activity-dependent manner could contribute to synaptic plasticity. Other binding partnersCitation10,Citation11 include a 53 kDa insulin receptor tyrosine kinase substrate IRSp53 and RIBEYE, a unique scaffolding protein found at retinal ribbon synapses. The IQSEC2 protein contains an IQ-like domain, a Sec7 domain, a PH domain and a C-terminal PDZ domain binding motif (). The p.R359C substitution is found in the IQ-like domain, which in other GEFs has been shownCitation12 to bind calmodulin and accelerate the exchange activity. Interestingly, the IQSEC2 IQ-like motif lacks the G and second basic residue of the complete motif () suggesting that the binding of calmodulin to IQSEC2 is calcium dependent. Since the p.R359C substitution disrupts the conserved basic residue in the IQ-like domain consensus motif (FILV)Qxxx(RK)xxxxxx, this change is predicted to result in disrupted calcium-dependent calmodulin binding, leading to an altered exchange of GDP for GTP on Arf6. Consistent with this theory, the other three sequence variants result in the substitutions p.R758Q, p.Q801P, p.R863W in the Sec7 domain (), which catalyses GDP-GTP exchange on Arf GTPasesCitation8,Citation9 such as Arf1 and Arf6.
Functional Analysis of IQSEC2 Mutations
Since all four amino acid substitutions were located within key functional domains of IQSEC2 and conserved in the related IQSEC1 and IQSEC3 proteins ( and C) we conducted further bioinformatic analysis using online analysis tools such as SIFT (http://sift.jcvi.org/) and Polyphen (http://genetics.bwh.harvard.edu/pph/) which indicated that these changes were likely to be damaging. This suggested that a loss of GEF activity was the most likely underlying molecular mechanism for IQSEC2 XLID. To obtain definitive proof of IQSEC2 dysfunction, we assessed the effect of the three different Sec7 domain mutations on the ability of IQSEC2 to catalyze GDP-GTP exchange on Arf6 in vitro, using recombinant GST-tagged Sec7 domain fusion proteins. GTP binding mediated by the Sec7 domain mutants p.R758Q, p.Q801P and p.R863W was similar to that of a control dominant-negative mutation (p.E849A), demonstrating significantly diminished GTP binding to Arf6 compared to wild-type IQSEC2 (). Subsequently, we tested the GEF activity of full-length IQSEC2 in a cellular model, using a pull-down assay with the adaptor protein Golgi-localized, γ ear-containing Arf-binding protein 3 (GGA3). GGAs specifically interact with active, GTP-bound Arf, but not inactive Arfs. HA-tagged Arf6 and FLAG-tagged wild-type or mutant versions of IQSEC2 or empty vector were co-expressed in HEK293 cells and cell lysates were subjected to a pull-down assay with GST-GGA3 immobilized on glutathione-agarose. Co-transfection of Arf6 with wild-type IQSEC2 resulted in an approximately 12-fold increase in GTP-bound Arf6, but only a 6- to 8-fold increase when co-expressed with IQSEC2 harbouring mutations in the Sec7 domain (). These subtle changes in function may provide a plausible reason why IQSEC2 variants showed only moderate conservation scoresCitation4 and highlight the importance of functional testing of missense variants. Taken together, these results strongly suggested all four missense changes in IQSEC2 were both functionally relevant and likely to be the disease-causing mutations in these families. We would also note that some carrier females from the four families present with a mild learning disability (although this could not be unequivocally assigned to their IQSEC2 mutation carrier status) and that a complete loss of IQSEC2 function might be expected to yield a more severe neuro-developmental phenotype.
Future Prospects
Having established that mutations in IQSEC2 are causative for XLID, a major challenge is to discover the changes in neuronal function triggered by defective IQSEC2. These could include changes in cell morphology, neuronal migration or synapse formation, function and plasticity. Clearly, the regulation of actin dynamics, membrane trafficking and synaptic function play a pivotal role in this process, since mutations in the several GEFs have been implicated in non-syndromic XLID (). These include ARHGEF6 encoding α-PIX/Cool-2,Citation13 ARHGEF9 encoding collybistinCitation14–Citation16 and the faciogenital dysplasia (Aarskog-Scott syndrome) gene,Citation17–Citation20 FGD1. In addition, mutations in GTPase-activating proteins (GAPs) and effector proteins for other small GTPases (Rho and Rab families), such as oligophrenin-1 and PAK3, also contribute to ID. Deficits in the function of GEFs, GAPs and effectors affecting actin dynamics have provided a mechanistic link to microstructural deficits (e.g., abnormalities of dendritic spines) associated with intellectual disability.Citation21,Citation22 Certainly, pathways affecting synaptic function and the remodelling of neuronal networks have been suggestedCitation23 as an underlying mechanism for a broad spectrum of neurocognitive and neuro-psychiatric phenotypes.
IQSEC2 is currently undergoing additional screening for genetic variants in families with XLID that were not part of recent studies (e.g., the EURO MRX Consortium; http://www.euromrx.com/). Given their similar domain structure, neuronal expression and localisation at synapses (), other IQSEC gene family members—IQSEC1 and IQSEC3—should also be considered as candidates for genetic studies of autosomal ID. In this regard, it is noteworthy that a maternally inherited 1.65 Mb deletion on 12p13.33 encompassing 10 genes (including IQSEC3) was reportedCitation24 to cause moderate intellectual disability and behavioural problems in a 15-year-old boy. However, whether dominant de novo mutations or autosomal recessive forms of ID (ARID) result from mutations in IQSEC1 or IQSEC3 remains to be determined. Large scale re-sequencing is clearly impacting on studies of XLID,Citation4 providing a wealth of data for detailed follow-up studies. The discovery of new disease genes in these disorders also opens new avenues for the development of rational pharmacotherapies, which together with novel and most accurate diagnostic testsCitation25 will provide a brighter outlook for affected individuals and their families.
Abbreviations
| Arf | = | ADP-ribosylation factor |
| GEF | = | guanine nucleotide exchange factor |
| NMDA | = | N-methyl-Daspartate |
| XLID | = | X-linked intellectual disability |
Figures and Tables
Figure 1 Role of Arf6 in neuronal development and synaptic plasticity. Small GTPases of the ADP ribosylation factor (Arf) family play a crucial role in the regulation of vesicular transport, organelle structure, lipid modification, membrane trafficking and actin dynamics.Citation26 In mammals there are six ubiquitously expressed genes/paralogs, divided into three classes based on sequence similarity (Class I: Arf1–3, Class II: Arf4–5, Class III: Arf6). Class I and II Arfs are localised to the Golgi and endosomal compartments. By contrast, Arf6 is localised to cell periphery in association with the plasma membrane and a subset of endosomes.Citation27 The exchange of GDP for GTP on Arfs is catalysed by guanine nucleotide exchange factors (GEFs). There are at least fifteen ArfGEFs in the human genome,Citation28 each characterised by a catalytic Sec7 domain with sequence identity to the yeast ArfGEF sec7p. Active Arf6 (Arf6-GTP) activates several downstream effectors such as phospholipase D (PLD), phosphatidylinositol-4-phosphate 5-kinases (PIP5K) and Rac1, leading to changes in membrane trafficking and actin dynamics. PA, phosphatidic acid; PIP2, phosphatidylinositol bisphosphate. Modified from reference Citation29.
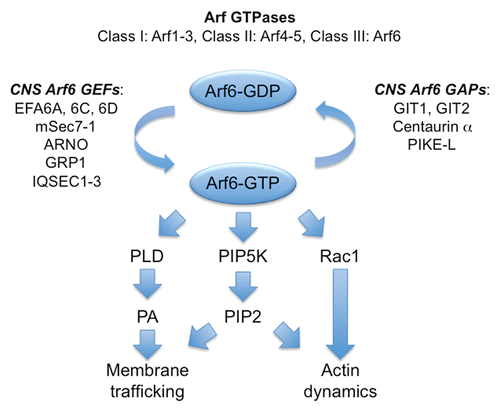
Figure 2 IQSEC2 mutations in XLID. (A) Schematic of the human IQSEC2 protein with a regulatory IQ-like motif, a catalytic Sec7 domain, a pleckstrin homology (PH) domain and a PDZ binding motif. The relative locations of mutations found in each family are shown. In the MRX 1 family (MIM 309530), a c.2587C>T change was identified in exon 8 leading to a p.R863W substitution. In the MRX 18 family, a c.2402A>C change in exon 6 was detected, leading to a p.Q801P substitution. A third, unpublished family from the USA (US166) had a c.2273G>A change in exon 5 leading to a p.R758Q substitution. In a fourth, Australian family (AU128) a c.1075C>T change in exon 4 was noted, leading to a p.R359C substitution in the IQ-like domain of IQSEC2. (B and C) Sequence alignments of IQSEC1, IQSEC2 and IQSEC3 showing the location of mutations (bold red type above alignments) in the IQ-like and Sec7 domains. Note that IQ-like motifs lack the G and second basic residue found in canonical IQ motifs. Characters within parentheses can substitute for each other. The R359C mutation disrupts the basic (R) residue. Note that IQSEC2 Sec7 domain mutations do not affect predicted GTP binding residues (bold blue type below alignments) in the structure of the related Sec7 domain in the GEF ARNO. Grey shading indicates amino acids that are identical in all four sequences.
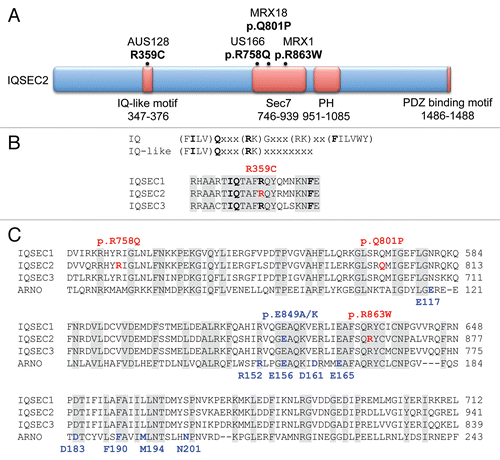
Figure 3 Missense mutations in IQSEC2 that cause non-syndromic XLID reduce guanine nucleotide exchange activity. (A) Radiometric analysis of the effects of mutations in the Sec7 domain of IQSEC2 on guanine nucleotide exchange activity. Recombinant wild-type (dark blue) or mutated versions of the Sec7 domain of IQSEC2 were incubated with recombinant Arf6 and GTPγ35S to catalyze binding of GTPγ35S to Arf6. Arf6-GTPγ35S was isolated on nitrocellulose membrane and the amount of bound radiolabel was quantified in a scintillation counter. Each XLID mutation in the Sec7 domain (light blue) significantly reduced GTP binding to Arf6. *p < 0.0001 vs. wild-type. Note that mutations to E849 (sky blue) are artificial dominant-negative mutants that strongly reduce GEF activity of the Sec7 domain. (B) Guanine nucleotide exchange activity in vivo analyzed using a pull-down assay. Arf6 and either wild-type (dark blue) or mutated (light blue) full-length IQSEC2 were co-expressed in HEK293 cells. The cells were lysed and Arf6-GTP was isolated on beads coated with the adaptor protein Golgi-localized, γ ear-containing Arf binding protein 3 (GGA), which binds to GTP-bound but not GDP-bound Arfs. Arf6-GTP was stripped from the beads and quantified by western blotting. Each mutation reduced activation of Arf6 in this cellular assay.
Table 1 IQSEC family of proteins
Table 2 Guanine nucleotide exchange factors implicated in X-linked intellectual disability
Acknowledgements
The authors acknowledge the support of the Australian NHMRC (J.G. and C.S.), the SMILE Foundation (J.G.), the Women's and Children's Hospital Foundation (C.S.), D. Harwood (J.G.) and the Medical Research Council (R.J.H.).
Extra View to: Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 2010; 42:486 - 488; PMID: 20473311; http://dx.doi.org/10.1038/ng.588
References
- Luckasson R. Mental retardation: definition, classification and systems of support 2002; 10th Edn Washington, DC American Association on Mental Retardation
- Ropers HH. Genetics of intellectual disability. Curr Opin Genet Dev 2008; 18:241 - 250
- Gecz J, Shoubridge C, Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet 2009; 25:308 - 316
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009; 41:535 - 543
- Raymond FL, Whibley A, Stratton MR, Gecz J. Lessons learnt from large-scale exon re-sequencing of the X chromosome. Hum Mol Genet 2009; 18:60 - 64
- Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 2010; 42:486 - 488
- Suthers GK, Turner G, Mulley JC. A non-syndromal form of X-linked mental retardation (XLMR) is linked to DXS14. Am J Med Genet 1988; 30:485 - 491
- Murphy JA, Jensen ON, Walikonis RS. BRAG1, a Sec7 domain-containing protein, is a component of the postsynaptic density of excitatory synapses. Brain Res 2006; 1120:35 - 45
- Sakagami H, Sanda M, Fukaya M, Miyazaki T, Sukegawa J, Yanagisawa T, et al. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci Res 2008; 60:199 - 212
- Sanda M, Kamata A, Katsumata O, Fukunaga K, Watanabe M, Kondo H, Sakagami H. The postsynaptic density protein, IQ-ArfGEF/BRAG1, can interact with IRSp53 through its proline-rich sequence. Brain Res 2009; 1251:7 - 15
- Katsumata O, Ohara N, Tamaki H, Niimura T, Naganuma H, Watanabe M, Sakagami H. IQ-ArfGEF/BRAG1 is associated with synaptic ribbons in the mouse retina. Eur J Neurosci 2009; 30:1509 - 1516
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 1995; 376:524 - 527
- Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet 2000; 26:247 - 250
- Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci 2004; 24:5816 - 5826
- Marco EJ, Abidi FE, Bristow J, Dean WB, Cotter P, Jeremy RJ, et al. ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J Med Genet 2008; 45:100 - 105
- Kalscheuer VM, Musante L, Fang C, Hoffmann K, Fuchs C, Carta E, et al. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression and mental retardation. Hum Mutat 2009; 30:61 - 68
- Pasteris NG, Cadle A, Logie LJ, Porteous ME, Schwartz CE, Stevenson RE, et al. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: a putative Rho/Rac guanine nucleotide exchange factor. Cell 1994; 79:669 - 678
- Lebel RR, May M, Pouls S, Lubs HA, Stevenson RE, Schwartz CE. Non-syndromic X-linked mental retardation associated with a missense mutation (P312L) in the FGD1 gene. Clin Genet 2002; 61:139 - 145
- Orrico A, Galli L, Buoni S, Hayek G, Luchetti A, Lorenzini S, et al. Attention-deficit/hyperactivity disorder (ADHD) and variable clinical expression of Aarskog-Scott syndrome due to a novel FGD1 gene mutation (R408Q). Am J Med Genet A 2005; 135:99 - 102
- Orrico A, Galli L, Faivre L, Clayton-Smith J, Azzarello-Burri SM, Hertz JM, et al. Aarskog-Scott syndrome: clinical update and report of nine novel mutations of the FGD1 gene. Am J Med Genet A 2010; 152:313 - 318
- Vaillend C, Poirier R, Laroche S. Genes, plasticity and mental retardation. Behav Brain Res 2008; 192:88 - 105
- Ramakers GJA. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 2002; 25:191 - 199
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 2008; 455:912 - 918
- Baker E, Hinton L, Callen DF, Haan EA, Dobbie A, Sutherland GR. A familial cryptic subtelomeric deletion 12p with variable phenotypic effect. Clin Genet 2002; 61:198 - 201
- Turner G, Boyle J, Partington MW, Kerr B, Raymond FL, Gécz J. Restoring reproductive confidence in families with X-linked mental retardation by finding the causal mutation. Clin Genet 2008; 73:188 - 190
- Myers KR, Casanova JE. Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol 2008; 18:184 - 192
- D'Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 2006; 7:347 - 358
- Gillingham AK, Munro S. The small G proteins of the Arf family and their regulators. Annu Rev Cell Dev Biol 2007; 23:579 - 611
- Jaworski J. ARF6 in the nervous system. Eur J Cell Biol 2007; 86:513 - 524
- Nagase T, Ishikawa K, Suyama M, Kikuno R, Miyajima N, Tanaka A, et al. Prediction of the coding sequences of unidentified human genes. XI. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 1998; 5:277 - 286
- Someya A, Sata M, Takeda K, Pacheco-Rodriguez G, Ferrans VJ, Moss J, Vaughan M. ARF-GEP(100), a guanine nucleotide-exchange protein for ADP-ribosylation factor 6. Proc Natl Acad Sci USA 2001; 98:2413 - 2418
- Hiroi T, Someya A, Thompson W, Moss J, Vaughan M. GEP100/BRAG2: activator of ADP-ribosylation factor 6 for regulation of cell adhesion and actin cytoskeleton via E-cadherin and alpha-catenin. Proc Natl Acad Sci USA 2006; 103:10672 - 10677
- Dosemeci A, Makusky AJ, Jankowska-Stephens E, Yang X, Slotta DJ, Markey SP. Composition of the synaptic PSD-95 complex. Mol Cell Proteomics 2007; 6:1749 - 1760
- Scholz R, Berberich S, Rathgeber L, Kolleker A, Köhr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron 2010; 66:768 - 780
- Inaba Y, Tian QB, Okano A, Zhang JP, Sakagami H, Miyazawa S, et al. Brain-specific potential guanine nucleotide exchange factor for Arf, synArfGEF (Po), is localized to postsynaptic density. J Neurochem 2004; 89:1347 - 1357
- Hattori Y, Ohta S, Hamada K, Yamada-Okabe H, Kanemura Y, Matsuzaki Y, et al. Identification of a neuron-specific human gene, KIAA1110, that is a guanine nucleotide exchange factor for ARF1. Biochem Biophys Res Commun 2007; 364:737 - 742