Abstract
Mutations of the inositol-5-phosphatase OCRL1 cause Lowe syndrome. Lowe syndrome is an inherited disease characterized by renal dysfunction and impaired development of the eye and the nervous system. OCRL1 is a Rab effector protein that can bind to a large number of different Rab proteins. We have recently determined the X-ray structure of the Rab-binding domain of OCRL1 in complex with Rab8. Furthermore, we have characterized point mutations that abolish binding to Rab proteins and cause Lowe syndrome. Here we shortly review our recent biophysical and structural work and discuss possible functional implications of our finding that Rab8 binds with the highest affinity to OCRL1 among the Rab proteins tested. This could direct further work on OCRL1 leading to a better understanding of the complex disease mechanism of Lowe syndrome.
Keywords: :
Mutations in the gene for the protein OCRL1 are responsible for oculo-cerebro-renal syndrome of Lowe (Lowe syndrome).Citation1 Lowe syndrome is an X-linked disease affecting the development of the eye (congenital cataracts) and the nervous system (neonatal hypotonia, mental retardation). Lowe syndrome is also characterized by dysfunction of renal proximal tubule cells resulting in low molecular weight proteinuria and acidosis.Citation2 The precise molecular mechanism explaining how mutations in OCRL1 lead to the described phenotype affecting the different organs is still unclear. OCRL1 is a phosphatidylinositol-5-phosphatase with preferred substrate specificity for phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate. OCRL1 is involved in regulation of vesicular trafficking and cell migration.Citation3,Citation4 It has been demonstrated that OCRL1 regulates transport from early endosomes to the trans-Golgi network and that it is involved in receptor recycling.Citation5,Citation6 Furthermore, it was shown that OCRL1 transiently localizes to late clathrin coated pits.Citation7 OCRL1 contains two clathrin- and one AP-2 binding site important for clathrin coated pit association.Citation8-Citation10 OCRL1 has a highly modular structure and consists of an N-terminal pleckstrin homology (PH) domain harboring one clathrin binding site followed by an inositol-5-phosphatase domain. In addition, OCRL1 contains an ASPM-SPD2-Hydin (ASH) domain and a catalytically inactive RhoGAP homology domain, which contains the second clathrin binding site.Citation7 Several interacting proteins have been identified beside clathrin. The endocytic proteins APPL1 and SES1/2 interact with the RhoGAP homology domain.Citation5,Citation7,Citation11 OCRL1 also interacts with a number of different Rab proteins, which are, like phosphatidylinositols, major regulators of intracellular membrane transport. Precisely, OCRL1 interacts with Rab1, Rab3, Rab5, Rab6, Rab8 as well as Rab14 and Rab35.Citation12,Citation13 Interaction with such a broad array of Rab proteins involving completely different subfamilies is very unusual among Rab binding proteins. Given the fact that several Lowe syndrome causing mutations are located within the Rab binding region of OCRL1, precise understanding of the OCRL1/Rab binding properties might give important insights into the molecular disease mechanism.
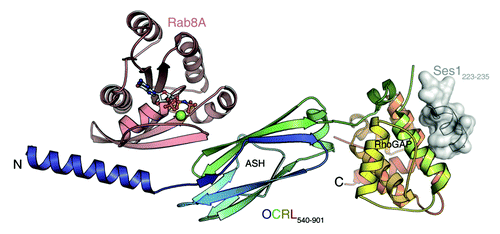
In an attempt to better understand the Rab binding properties of OCRL1, we determined the binding affinities of several OCRL1/Rab protein pairs in our recent publication.Citation14 Furthermore, we solved the X-ray structure of the OCRL1 Rab binding domain in complex with Rab8a. Previously, the NMR-structures of the N-terminal PH domain as well as the crystal structure of the ASH domain together with the RhoGAP-like domain were solved. In our study we have presented the first structure of OCRL1 in complex with one of its binding partners. Using a number of different N- and C-terminal truncations we first determined the minimal Rab binding domain of OCRL1 to be amino acids 540–678. This region comprises the complete ASH domain as well as an α-helix from the 5-phosphatase domain. The X-ray structure of the Rab-binding fragment 540–678 in complex with Rab8a (amino acids 6–176) revealed a novel Rab effector binding mode. Whereas other known Rab effector proteins bind their respective Rab protein via α-helical structural elements, OCRL1 interacts with Rab8a via one α-helix but additionally via the β-sheet structures of the ASH domain. Two binding sites mediate interaction with Rab8a. The first site involves the switch I region of Rab8a and the α-helix from the linker region between the inositol-5-phosphatase domain and the ASH domain of OCRL1. The molecular interactions constituting binding site I are mostly of a polar nature. The second site (binding site II) involves the switch I and switch II region of Rab8a and the ASH domain of OCRL1. The ASH domain is an all β-sheet structure previously suggested to play an important role for Rab binding.Citation13 In binding site II, polar as well as hydrophobic interactions are involved. In particular, F668 located in the ASH domain of OCRL1 makes hydrophobic contacts with I41, G42 in switch I and F70 in switch II of Rab8a. Interestingly, the hydrophobic triad (F45, W62, Y77), which has been suggested to be a main determinant for Rab/effector binding specificity, seems to play a minor role in binding of Rab8 to OCRL1.Citation15 This is in line with the observed broad Rab binding specificity of OCRL1 across Rab subfamilies.
Previous studies showed that the ASH domain is also important for OCRL1 binding to the F&H motif of APPL1 and Ses1/2.Citation7,Citation11 We studied whether binding of APPL1 or Ses1 peptides to OCRL1 influences the binding affinity to Rab8a. However, we could not detect any modulatory effect on the stability of the OCRL1/Rab8a complex via F&H peptides, suggesting no overlap or crosstalk between the two binding sites. Recently, the crystal structure of the ASH-RhoGAP-like domain of OCRL1 in complex with the F&H peptide of Ses1 was published.Citation16 Ses1 binds to a conserved groove on the posterior surface of the RhoGAP-like domain. Thus, the binding site for Ses1 is clearly distinct from the binding site for Rab proteins, confirming the independence of the two sites (). The requirement of the ASH domain for binding of OCRL1 to APPL1/Ses1/2 seems to be indirect, by stabilizing the RhoGAP-like domain fold.
Figure 1. OCRL1 (c-terminal portion) in complex with Rab8a and the F&H peptide of Ses1. Rab proteins and F&H motif proteins bind to two distinct binding sites. Rab8a is binding to the ASH domain and to a c-terminal α-helix of the inositol-5-phosphatase domain. Ses1 binds to the posterior surface of the RhoGAP-like domain.
The residue F668, which is involved in binding site II of the OCRL1/Rab8 complex, drew our special attention because mutation of F668 to valine is known to cause Lowe syndrome.Citation11 Thus, we investigated in detail the Rab binding characteristics of this point mutation and the behavior of the F668V mutant OCRL1 protein in cells. Mutation of F668 to valine led to a 5.8 fold decrease of the Rab8 binding affinity. This reduction was even more pronounced on replacing F668 by alanine. The decrease in binding affinity to Rab8a was confirmed by pull-down assays using recombinant OCRL1 harboring the F668V mutation and endogenous Rab8. Moreover, we also tested the subcellular localization of OCRL1 F668V in Hela cells and observed extensive delocalization of mutant OCRL1 to the cytosol with clearly reduced vesicular appearance. This is in line with the idea that Rab proteins are the main membrane-recruiting element for OCRL1.Citation13 F668V is so far the only point mutation selectively affecting interaction of OCRL1 with Rab proteins. Given the fact that this mutation causes Lowe syndrome, this is evidence for the crucial role of OCRL1/Rab interactions in vivo.
As mentioned above, in our recent study we have determined the binding affinities of several OCRL1/Rab binding pairs (OCRL1 with Rab1b, Rab5a, Rab6a, Rab8a). A surprising outcome of this investigation was the fact that the preferred in vitro binding partner of OCRL1 among the tested Rab proteins is Rab8. So far, OCRL1 has mainly been detected on the Golgi apparatus and on early endosomes, pointing to interaction with Rab1, Rab6 or Rab5. Here we would like to discuss briefly the possible implications of the interaction of OCRL1 with Rab8 and the relevance to Lowe syndrome. Rab8 is involved in several membrane dependent processes, including exocytosis, membrane recycling and ciliogenesis ().Citation17 All three processes could be of relevance to the molecular mechanism of Lowe syndrome. A major phenotype of Lowe syndrome is proximal tubule dysfunction with amino aciduria, phosphaturia and proteinuria.Citation18 This phenotype could be mainly explained by improper function of the receptor Megalin. Megalin is a large cell surface receptor important for re-absorption localized at the apical domain of polarized proximal tubule cells. In line with a possible role of Megalin in Lowe syndrome is the finding of decreased levels of the extracellular domain of Megalin in the urine of Lowe syndrome patients, which could reflect lower steady-state levels of Megalin cell surface expression.Citation19 Lower cell surface expression could be the result of impaired receptor recycling or impairment of the biosynthetic transport pathway or a combination of both. Rab8 has been demonstrated to be important for cargo delivery to the apical domain in polarized epithelial cells in vivo.Citation20 Knockout of Rab8 in mice leads to improper localization of apical proteins and defects in the absorption of various nutrients in the small intestine, reminiscent of the renal reabsorption defect observed in Lowe syndrome. Moreover, several studies demonstrate a role for Rab8 in the biosynthetic pathway regulating transport of newly synthesized protein to the plasma membrane.Citation21,Citation22 It has been suggested that newly synthesized proteins are transported in a Rab8 regulated manner to recycling endosomes followed by transport to the plasma membrane.Citation23 Thus, it will be important to analyze the potential role for OCRL1 in the biosynthetic delivery of Megalin to the plasma membrane. In fact we observed strong co-localization of OCRL1 to Rab8 positive tubules. Rab8 positive tubules have been suggested to play a role in membrane delivery to the plasma membrane.Citation24
Figure 2. Overview of subcellular distribution of Rab8 and OCRL1 . Rab8 is localized to membrane ruffles and regulates transformation of macropinosomes into tubules. Rab8 also regulates transport to the recycling endosome within the biosynthetic pathway and is involved in the biogenesis of cilia. Potential involvement of OCRL1 is discussed in the main text. n = nucleus; TGN = Trans-Golgi-Network.
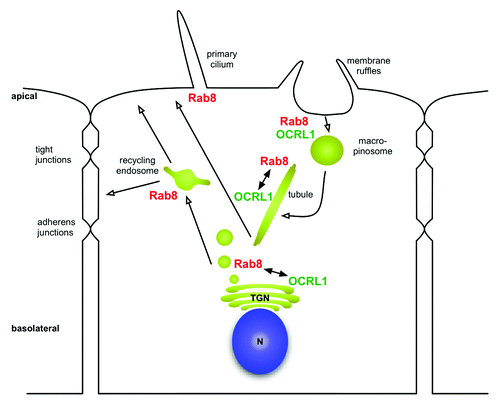
Rab8 also regulates recycling of plasma membrane receptors, but a major role for the OCRL1/Rab8 module for recycling of Megalin in proximal tubule cells is unlikely in the light of a recent report.Citation6 In this study, a defect in Megalin recycling in proximal tubule cells derived from Lowe syndrome patients was demonstrated. This recycling defect appears to take place at early endosomes caused by increased phosphatidylinositol-4,5-bisphosphate levels and aberrant actin polymerization. Given the absence of Rab8 on early endosomes, recycling of Megalin is probably mediated via the OCRL1/Rab5 module. However, Rab8 has been reported to localize to macropinosomes and to regulate membrane recycling to the plasma membrane, which is especially important during cell migration.Citation24 Interestingly, OCRL1 has been reported to localize to macropinosomes as well.Citation4 Moreover, fibroblasts derived from Lowe syndrome patients show a defect in fluid phase endocytosis and in cell migration.Citation4 The mechanism of OCRL1 recruitment to macropinosomes is elusive, and Rab8 could be a promising candidate, but it is possible that several Rab proteins synergize in recruiting OCRL1. It is tempting to speculate that OCRL1 could also be involved in membrane recycling from macropinosomes.
The neuronal phenotype of Lowe syndrome is hallmarked by central hypotonia and mental retardation, which probably reflects developmental defects of the nervous system. However, the role of OCRL1 in the nervous system is completely unclear. Depletion of Rab8 from hippocampal neurons inhibits neurite outgrowth.Citation25 In addition, Rab8 is required for the delivery of AMPA-receptors to the cell surface of dendrites and regulates AMPA-receptor recycling.Citation26 Thus, the OCRL1/Rab8 module could be involved in two fundamental processes (neurite outgrowth and synaptic transmission) shaping the nervous system.
Finally, there is considerable interest in Rab8 because of its involvement in ciliogenesis. Primary cilia are microtubule-based membrane projections located at the cell surface of many cells. Defects in primary cilia formation is implicated in a number of genetic disorders like Bardet-Biedl syndrome or polycystic kidney disease. The guanine nucleotide exchange factor (GEF) for Rab8 Rabin8 interacts with BBS1, which is a component of the BBsome, a multiprotein complex important for cargo delivery to the cilium.Citation27 Interestingly, in a study reporting the existence of the ASH domain the presence of this domain in proteins associated with cilia, flagella and centrosomes was noted. However, a role of OCRL1 in ciliogenesis has not been reported so far.Citation28
Our recent study of the Rab binding properties of the Lowe syndrome protein, OCRL1, gives a structural explanation for some Lowe syndrome causing mutations. Moreover, we have confirmed OCRL1 to bind to a large array of Rab proteins, with OCRL1/Rab8 among the tested OCRL1/Rab pairs displaying the highest affinity. Although several OCRL1/Rab interactions play an important role in OCRL1 function, focusing on known functions of Rab8 could guide part of the future work on OCRL1 and lead to a better mechanistic understanding of Lowe syndrome.
Acknowledgments
The work was supported by a Grant from the Deutsche Forschungsgemeinschaft SFB642, projects A4 and A17 to RSG and KSE, respectively.
References
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, et al. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 1992; 358:239 - 42; http://dx.doi.org/10.1038/358239a0; PMID: 1321346
- Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Nephrol 2009; 5:529 - 38; http://dx.doi.org/10.1038/nrneph.2009.124; PMID: 19701229
- Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell 2005; 16:3467 - 79; http://dx.doi.org/10.1091/mbc.E05-02-0120; PMID: 15917292
- Coon BG, Mukherjee D, Hanna CB, Riese DJ 2nd, Lowe M, Aguilar RC. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Hum Mol Genet 2009; 18:4478 - 91; http://dx.doi.org/10.1093/hmg/ddp407; PMID: 19700499
- Noakes CJ, Lee G, Lowe M. The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol Biol Cell 2011; 22:606 - 23; http://dx.doi.org/10.1091/mbc.E10-08-0730; PMID: 21233288
- Vicinanza M, Di Campli A, Polishchuk E, Santoro M, Di Tullio G, Godi A, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P₂-dependent regulation of endosomal actin. EMBO J 2011; 30:4970 - 85; http://dx.doi.org/10.1038/emboj.2011.354; PMID: 21971085
- Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, et al. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell 2007; 13:377 - 90; http://dx.doi.org/10.1016/j.devcel.2007.08.004; PMID: 17765681
- Mao Y, Balkin DM, Zoncu R, Erdmann KS, Tomasini L, Hu F, et al. A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J 2009; 28:1831 - 42; http://dx.doi.org/10.1038/emboj.2009.155; PMID: 19536138
- Choudhury R, Noakes CJ, McKenzie E, Kox C, Lowe M. Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J Biol Chem 2009; 284:9965 - 73; http://dx.doi.org/10.1074/jbc.M807442200; PMID: 19211563
- Ungewickell A, Ward ME, Ungewickell E, Majerus PW. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A 2004; 101:13501 - 6; http://dx.doi.org/10.1073/pnas.0405664101; PMID: 15353600
- Swan LE, Tomasini L, Pirruccello M, Lunardi J, De Camilli P. Two closely related endocytic proteins that share a common OCRL-binding motif with APPL1. Proc Natl Acad Sci U S A 2010; 107:3511 - 6; http://dx.doi.org/10.1073/pnas.0914658107; PMID: 20133602
- Fukuda M, Kanno E, Ishibashi K, Itoh T. Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics 2008; 7:1031 - 42; http://dx.doi.org/10.1074/mcp.M700569-MCP200; PMID: 18256213
- Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J 2006; 25:3750 - 61; http://dx.doi.org/10.1038/sj.emboj.7601274; PMID: 16902405
- Hou X, Hagemann N, Schoebel S, Blankenfeldt W, Goody RS, Erdmann KS, et al. Structural basis for Lowe syndrome caused by mutations in the Rab binding domain of OCRL1. EMBO J 2011; 30:1659 - 70; http://dx.doi.org/10.1002/humu.9239; PMID: 15108291
- Merithew E, Hatherly S, Dumas JJ, Lawe DC, Heller-Harrison R, Lambright DG. Structural plasticity of an invariant hydrophobic triad in the switch regions of Rab GTPases is a determinant of effector recognition. J Biol Chem 2001; 276:13982 - 8; PMID: 11278565
- Pirruccello M, Swan LE, Folta-Stogniew E, De Camilli P. Recognition of the F&H motif by the Lowe syndrome protein OCRL. Nat Struct Mol Biol 2011; 18:789 - 95; http://dx.doi.org/10.1038/nsmb.2071; PMID: 21666675
- Peränen J. Rab8 GTPase as a regulator of cell shape. Cytoskeleton (Hoboken) 2011; 68:527 - 39; http://dx.doi.org/10.1002/cm.20529; PMID: 21850707
- Bockenhauer D, Bokenkamp A, van’t Hoff W, Levtchenko E, Kist-van Holthe JE, Tasic V, et al. Renal phenotype in Lowe Syndrome: a selective proximal tubular dysfunction. Clin J Am Soc Nephrol 2008; 3:1430 - 6; http://dx.doi.org/10.2215/CJN.00520108; PMID: 18480301
- Norden AG, Lapsley M, Igarashi T, Kelleher CL, Lee PJ, Matsuyama T, et al. Urinary megalin deficiency implicates abnormal tubular endocytic function in Fanconi syndrome. J Am Soc Nephrol 2002; 13:125 - 33; PMID: 11752029
- Sato T, Mushiake S, Kato Y, Sato K, Sato M, Takeda N, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 2007; 448:366 - 9; http://dx.doi.org/10.1038/nature05929; PMID: 17597763
- Ang AL, Fölsch H, Koivisto UM, Pypaert M, Mellman I. The Rab8 GTPase selectively regulates AP-1B-dependent basolateral transport in polarized Madin-Darby canine kidney cells. J Cell Biol 2003; 163:339 - 50; http://dx.doi.org/10.1083/jcb.200307046; PMID: 14581456
- Huber LA, Pimplikar S, Parton RG, Virta H, Zerial M, Simons K. Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J Cell Biol 1993; 123:35 - 45; http://dx.doi.org/10.1083/jcb.123.1.35; PMID: 8408203
- Henry L, Sheff DR. Rab8 regulates basolateral secretory, but not recycling, traffic at the recycling endosome. Mol Biol Cell 2008; 19:2059 - 68; http://dx.doi.org/10.1091/mbc.E07-09-0902; PMID: 18287531
- Hattula K, Furuhjelm J, Tikkanen J, Tanhuanpää K, Laakkonen P, Peränen J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J Cell Sci 2006; 119:4866 - 77; http://dx.doi.org/10.1242/jcs.03275; PMID: 17105768
- Huber LA, Dupree P, Dotti CG. A deficiency of the small GTPase rab8 inhibits membrane traffic in developing neurons. Mol Cell Biol 1995; 15:918 - 24; PMID: 7823956
- Gerges NZ, Backos DS, Esteban JA. Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J Biol Chem 2004; 279:43870 - 8; http://dx.doi.org/10.1074/jbc.M404982200; PMID: 15297461
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007; 129:1201 - 13; http://dx.doi.org/10.1016/j.cell.2007.03.053; PMID: 17574030
- Ponting CP. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics 2006; 22:1031 - 5; http://dx.doi.org/10.1093/bioinformatics/btl022; PMID: 16443634