Abstract
Rho GTPases regulate numerous cellular processes including apoptosis. Chp/RhoV is an atypical Rho GTPase which functions are poorly understood. Here we investigated the role of Chp in regulation of cell viability using PC12 cells with inducible expression of Chp as a model. We found that expression of Chp results in apoptosis in PC12 cells. Chp-induced apoptosis was accompanied by activation of JNK signaling and both death receptor-mediated and mitochondrial apoptotic pathways as justified by caspase-8 and caspase-9 activation, respectively. Moreover, inhibition of JNK by SP600125 rescued PC12 cells from Chp-triggered cell death and attenuated activation of caspases-9 and -3/7 suggesting that activation of JNK mediates pro-apoptotic effect of Chp. Expression of Chp resulted in increased phosphorylation of c-Jun in PC12 cells, and Chp expression in HEK293 cells up-regulated AP-1-dependent transcription in a JNK-dependent manner. Together results of our study reveal the role of Chp GTPase as a putative regulator of JNK-dependent apoptotic death in PC12 cells, similarly to previously described pro-apoptotic activity of the related Cdc42 and Rac1 GTPases.
Introduction
Small GTPases act as “molecular switches” cycling between inactive GDP-bound state and active GTP-bound state in which they interact with effector proteins.Citation1 Rho GTPases are known to regulate a variety of cellular processes including cytoskeleton dynamics, cell adhesion, gene expression and apoptotic cell death.Citation2 It has been shown that constitutively active forms of Cdc42 and Rac1 induce apoptosis of primary sympatic neurons via activation of JNK and AP-1 transcription factor and are required for apoptosis of neurons after NGF withdrawal.Citation3 Cdc42 is also capable of inducing apoptosis in other cell types such as lymphocytes,Citation4 and is required for resveratrol-induced apoptosis of HL-60 promyelocytes.Citation5
Chp/RhoV is an atypical Rho GTPase which functions are poorly understood. To date, several effectors of Chp have been identified including p21-activated kinases (Pak1, Pak2 and Pak4), N-WASP and MLK3.Citation6–Citation8 It has been reported that ectopic expression of Chp leads to lamellipodia formation,Citation6,Citation7 focal adhesion assemblyCitation7 and oncogenic transformation of mouse NIH3T3 fibroblasts.Citation9,Citation10
JNK-dependent apoptosis is important for the proper development of the nervous system in many organisms including mammals.Citation11 Chp was shown to stimulate JNK in HEK293 cells upon transient expression.Citation6 Chp is expressed early in embryonic development of chick,Citation12 frogCitation13 and zebrafish,Citation14 being one of the earliest expressed neural crest markers in X. laevis embryos and playing an important role in differentiation of neural crest cells.Citation13 In mammals RHOV transcript encoding Chp is found both in fetal and adult brainCitation6,Citation15 but functions of Chp in the nervous system of mammals remain to be elucidated. PC12 rat pheochromocytoma cell line is often used as a model of neuronal differentiation, and activation of JNK pathway is crucial for apoptosis in these cells.Citation16 In the present work we used PC12 cells with inducible expression of Chp as a model to identify cellular processes regulated by Chp. Here we showed that Chp induces apoptotic cell death of undifferentiated PC12 cells. Our data suggest that activation of JNK pathway in PC12 cells by Chp is one of the molecular mechanisms underlying Chp-induced cell death.
Results and Discussion
Expression of Chp reduces viability of PC12TetOn cells.
To identify cellular processes affected by Chp GTPase we established PC12TetOn cell lines with tetracycline-regulated expression of the N-terminally FLAG-tagged wild type Chp (wt5 cells) and its constitutively active mutant ChpG40V (gv2, gv3, gv5 and gv7 cells) (). No detectable transgene expression was observed in all clones in the absence of doxycycline (DOX) ().
Rho GTPases Cdc42 and Rac1 were shown to induce apoptotic death in different types of cells including neurons.Citation2–Citation5 Chp is a Cdc42-related GTPase and it shares with Cdc42 several effectors (such as MLK3 and Pak2) with pro-apoptotic activity.Citation6,Citation7 This prompted us to question if Chp GTPase is also involved in the regulation of apoptosis. First we assayed if Chp expression affects viability of PC12 cells. We found that expression of Chp or its G40V mutant led to a statistically significant decrease in viability of PC12TetOn clones grown on collagen IV (p < 0.005 for all clones) compared to cells without transgene expression (). The most pronounced decrease in viability was observed for clones expressing ChpG40V (by 41%, 66%, 76% and 55% in gv2, gv3, gv5 and gv7 cells, respectively) compared to 38% for clone wt5 expressing wild type Chp GTPase. Visual examination of wells revealed that in a course of the experiment, the number of cells became lower in the presence of DOX compared to non-induced cells (). Importantly, viability of PC12TetOn cells expressing unrelated protein (namely protein kinase MAK-V/HUNK) was not compromised upon induction of transgene expression. On the contrary, MAK-V expression improved cell viability.Citation17 Finally, no decrease in viability has been observed for parental PC12TetOn cells upon DOX treatment (data not shown). Together these observations rule out the possibility that the decrease in cell viability is non-specifically caused by addition of DOX. As the effect of Chp on cell viability was observed in five independent clones, it is also unlikely that the reduced viability of PC12TetOn cells upon Chp expression is a sporadically acquired clonal feature.
Chp is cytotoxic to PC12TetOn cells.
The observed decrease in viability of DOX-treated cells could be both due to increase in cell death and/or reduction in proliferation activity. Importantly, after 3 days of incubation in the presence of DOX, the number of cells in wells was dramatically reduced as revealed by microscopic examination (). This suggests that expression of Chp promotes death of PC12TetOn cells. To test this hypothesis we measured lactate dehydrogenase (LDH) release from cells into culture medium, an indicator of cell death. We found that induction of Chp expression resulted in statistically significant (p < 0.005) increase in LDH release in all the clones assayed compared to untreated cells (by 18%, 32%, 11%, 61% and 50% for wt5, gv2, gv3, gv5 and gv7 cells, respectively) (). These data indicate that Chp induces death of PC12TetOn cells. The most pronounced cytotoxic effect was observed for gv5 and gv7 clones, which is in line with the strongest reduction in cell viability observed for these clones upon induction of ChpG40V expression. gv3 cells did not grow well on collagen IV even in the absence of DOX (data not shown), therefore the relative LDH release was quite moderate. These data confirm that expression of Chp is toxic for PC12 cells and suggest that the compromised viability of PC12TetOn clones is likely due to increased cell death rather than results from suppressed proliferation.
Chp induces apoptosis in PC12 cells.
Cdc42 and Rac1 were reported to induce apoptosis in PC12 cells.Citation16 Thus we assumed that expression of Chp also triggers apoptosis in PC12 cells. Caspase activation is a hallmark of apoptotic cell death.Citation18 Therefore we first assessed the activation of executioner caspases-3/7 upon the induction of Chp expression. We found that induction of Chp or its G40V mutant expression in wt5, gv5 and gv7 clones resulted in ∼1.4-fold, ∼3.5-fold and ∼1.6-fold increase in caspase-3/7 activity, respectively (). Maximal activation of caspase-3/7 was observed in gv5 clone which is in line with the most pronounced growth suppression and LDH release observed for this clone (, and ). Thus induction of apoptosis likely underlies compromised cell viability caused by Chp expression in PC12 cells. Importantly, caspase-3/7 activation has not been altered in parental PC12TetOn cells upon DOX treatment further confirming specificity of the observed effects of Chp expression (). In addition, treatment of wt5 cells with DOX resulted in increased Annexin V-FITC reactivity (), a hallmark of apoptotic cell death. Together these data indicate that Chp induces apoptosis in PC12 cells.
Chp activates both death-receptor and mitochondrial apoptotic pathways in pc12 cells.
There are two main apoptotic pathways in cells: death-receptor mediated pathway and intrinsic mitochondrial pathway. In a death receptor pathway, activation of executioner caspases-3/7 is mediated by initiator caspase-8 while in a mitochondrial pathway caspase-9 is the principal activator of executioner caspases.Citation18 To reveal the contribution of these pathways in Chp-induced apoptotic death of PC12 cells, we assayed the effect of Chp expression on activity of caspases-8 and -9. We found that both initiator caspases are activated by Chp ( and C). Although there was a trend for activation of caspase-8 in wt5 clone, the difference was not statistically significant. Activities of both caspase-8 and -9 were not elevated in parental PC12TetOn cells upon DOX treatment, again confirming specificity of effects of Chp expression ( and C). Thus Chp promotes activation of both death receptor and mitochondrial apoptotic pathways in PC12 cells.
Chp activates JNK in PC12 cells.
Activation of JNK signaling cascade plays a central role in apoptosis in different cell types.Citation19 In particular, JNK is activated downstream of Cdc42 and Rac1 to induce apoptosis of PC12 cells.Citation16 It has been shown that upon transient expression in HEK293 cells, Chp activates JNK but not ERK or p38MAPK kinases.Citation6 Therefore, we assessed if Chp expression activates JNK in wt5 and gv7 cells. JNK activation was monitored by western blot analysis with anti-phospho-specific antibodies. Indeed, we found that expression of both wild type Chp and its G40V mutant resulted in about 2 ± 0.22 and 3.79 ± 1.24 fold increase in JNK activity, respectively ( and B). In gv7 clone, increase in JNK phosphorylation was accompanied by ∼2.7-fold increase in phosphorylation of c-Jun at Ser73, a target site for JNK, suggesting that activation of JNK downstream of Chp results in activation of AP-1 transcriptional factor. In wt5 clone increase in c-Jun phosphorylation was not obvious. JNK was also activated in PC12gv5 cells (data not shown). In addition, we analyzed phosphorylation status of p38MAPK in wt5 and gv7 cells after expression of Chp, since activation of p38MAPK is able to induce apoptosis in PC12 cells.Citation20,Citation21 In agreement with the previously published data in reference Citation6, we failed to detected activation of p38MAPK by Chp (). Together these findings indicate that Chp-induced cell death is accompanied by activation of pro-apoptotic JNK pathway in PC12 cells.
Cytotoxic effect of Chp is reversed by JNK inhibitor, SP600125.
To confirm that the cytotoxic effect of Chp expression in PC12TetOn cells depends on JNK activity we measured LDH release from gv5 cells cultured in the presence of different concentrations of a JNK inhibitor, SP600125. We used gv5 cells as we observed the highest cytotoxic effect of Chp in this clone. We found that treatment with SP600125 in a dose-dependent manner rescued gv5 cells from Chp-induced cytotoxicity and reduced LDH release. Treatment of DOX-induced cells with 5 µM SP600125 reduced LDH release ∼1.8-fold (p < 0.0001) compared to untreated with SP600125 cells (). Moreover, treatment with SP600125 rescued gv5 cells from Chp-induced death in a dose-dependent manner (). Viability of cells treated with 5 µM SP600125 and with DOX did not differ significantly from that of untreated cells (p = 0.363), and was ∼2.1-fold higher than the viability of cells treated with DOX alone (p = 0.0001) (). Together these findings show that Chp-induced cell death of PC12TetOn cells, at least in part, requires JNK activation.
SP600125 treatment attenuates activation of caspases by Chp in PC12 cells.
In order to further prove that Chp-induced apoptosis of PC12 cells is JNK-dependent, we investigated effect of SP600125 treatment on Chp-induced activation of caspases. Along with induction of Chp expression cells were exposed to 10 µM of SP600125 or left untreated with SP600125. Forty eight hrs later activities of caspases-3/7, 8 and 9 were analyzed. We found that caspase-8 activity was not affected by SP600125 (), indicating that Chp activates caspase-8 via JNK-independent mechanism. On the contrary, Chp-induced activity of caspase-9 in wt5 and gv5 cells was reduced by SP600125 treatment by about 28% and 30%, respectively (p < 0.05) (). Based on this finding we concluded that caspase-9 is activated by Chp via JNK-dependent mechanism. Accordingly, activity of caspase-3/7 was also reduced in SP600125-treated wt5 and gv5 cells by 23% and 37%, respectively (p < 0.05) (). Together these data confirm that Chp-induced apoptosis of PC12 cells involves activation of JNK. In addition, it seems likely that Chp activates two parallel apoptotic pathways: JNK-dependent pathway that activates mitochondrial apoptotic cascade involving caspase-9, and JNK-independent death receptor-mediated pathway involving caspase-8.
Chp activates JNK and AP-1-dependent transcription in HEK293 cells.
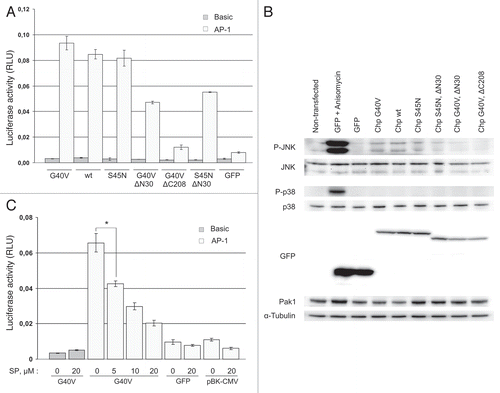
AP-1 transcription factor is a downstream target of JNK, and activation of AP-1-dependent transcription is required for JNK-induced apoptosis of neuronal cells.Citation22 Although it is expected that AP-1 transcription factor is activated by JNK downstream of Chp, it has not been demonstrated so far. We found that expression of Chp results in c-Jun phosphorylation at JNK target site in gv7 cells () suggesting upregulation of AP-1 dependent transcription. Indeed, transient expression of ChpG40V, Chp or ChpS45N fused to green fluorescent protein (GFP) induced about 10-fold activation (p < 0.0001) of AP-1-dependent transcription in HEK293 cells compared to cells expressing GFP alone (). Western blot analysis showed that transient expression of GFP-ChpG40V, GFP-Chp and GFP-Chp S45N induced activation of JNK (). Similarly to data obtained on PC12 cells, expression of GFP-Chp had no effect on p38MAPK phosphorylation at Thr180/Tyr182 that was observed only in cells treated with anisomycin (). As expected, anisomycin also induced robust JNK phosphorylation in HEK293 cells. Taken together, these data indicate that Chp indeed activates JNK pathway and AP-1 dependent transcription in HEK293.
Inhibition of JNK blocks AP-1 activation by Chp.
To confirm that activation of AP-1 reporter by Chp occurs via JNK, we questioned if treatment with JNK inhibitor SP600125 would block AP-1-dependent reporter gene activation by GFP-Chp in HEK293 cells. HEK293 cells were cultured in the presence of SP600125 at different concentrations for 48 hrs before measuring luciferase activity. We found that treatment with SP600125 inhibited Chp-induced AP-1-dependent reporter gene activity in a dose-dependent manner (). Inhibition of JNK with 5, 10 and 20 µM SP600125 led to about 1.5-, 2.2- and 3.2-fold reduction in the level of AP-1-dependent transcription, respectively (p < 0.005). Although treatment with 20 µM SP600125 also resulted in about 2-fold decrease in AP-1-dependent reporter gene activity in GFP-transfected cells, it likely resulted from the suppression of basal level of AP-1 activity rather than was the effect of GFP expression since the suppression also occurred in cells transfected with pBK-CMV plasmid (). Based on these observations we conclude that Chp activates JNK pathway to induce c-Jun phosphorylation and increase AP-1 transcriptional activity.
Membrane targeting and N-terminal domain of Chp are required for its ability to activate JNK pathway.
To get insight into molecular mechanisms of JNK pathway activation by Chp we analyzed the effect of Chp N-terminal (1–28 aa; ) and C-terminal domain (208–236 aa; ) deletions on its ability to activate AP-1 reporter and JNK. We found that the effect of GFP-ChpG40V on AP-1-dependent transcription and JNK activation was abrogated upon deletion of the C-terminal domain ( and B). This finding reinforces the importance of membrane association for the function of Chp GTPase which is defined by its C-terminal domainCitation9 and further confirms the specificity of the effect of Chp on AP-1-dependent transcription.
Surprisingly, presumably inactive GDP-bound ChpS45N mutant also induced AP-1-dependent transcription (about 10-fold over GFP alone, p < 0.0001) and JNK activation ( and B). ChpS45N does not bind to effector proteins such as Pak2,Citation6 or Pak1 (data not shown) and, in agreement with previous report in reference Citation8, we found that expression of ChpG40V or wild type Chp but not ChpS45N reduced the total level of endogenous Pak1 thus confirming that GFP-ChpS45N is indeed inactive ( and Pak1 part). Moreover, unlike wild type Chp or G40V mutant, GFP-ChpS54N was unable to induce actin cytoskeleton rearrangements (our unpublished observation). S45N mutation is expected to lock Chp in GDP-bound state but is not likely to affect the proline-rich domain of Chp or its membrane localization. Recently it has been reported that Chp binds to POSH,Citation8 an SH3-domain containing scaffold protein that is essential for the activation of JNK pathway during apoptosis in PC12 cells.Citation16 One possibility is that membrane-bound ChpS45N in its GDP-bound state recruits endogenous POSH to membrane compartment to mediate partial activation of JNK pathway in HEK293 cells. Indeed, deletion of N-terminal domain of ChpG40V that is likely to mediate interaction with POSH, decreased the effect of ChpG40V on the AP-1-dependent transcription by about 50% (p = 0.0001) and reduced JNK activation ( and B). Similarly, deletion of N-terminal domain of GFP-ChpS45N has led to 1.5-fold decrease in AP-1-dependent reporter gene activation (p = 0.0018) compared to full-length GFP-ChpS45N ().
Materials and Methods
Plasmids.
pTRE-Tight (631059), pHygro (Cat. number is unavailable), pEGFP-C2 (6083-1) and pEYFP-N1 (6083-1) plasmids were obtained from Clontech. Luciferase reporter plasmids pGL3-Basic (E1751) and pRL-CMV (E2261) were from Promega. pBK-CMV plasmid is from Agilent Technologies (212209). pfLuc based AP-1 reporter plasmidCitation25 was a gift from Dr. Eugene Tulchinsky (University of Leicester, Leicester, UK). pCan plasmids encoding myc-tagged wild type Chp, its constitutively active GTPase-deficient mutant (ChpG40V) and its GDP-bound dominant-negative mutant (ChpS45N),Citation6 were provided by Dr. Arie Abo (Nuvelo, Inc.). To produce pTRE-Tight plasmids for expression of N-terminally FLAG-tagged Chp, oligonucleotides 5′-AAT TCT ACC ATG GAC TAC AAA GAC GAT GAC GAC AAG GGT AC-3′ and 5′-CCT TGT CGT CAT CGT CTT TGT AGT CCA TGG TAG-3′ encoding FLAG epitope were annealed and cloned into EcoRI/KpnI sites of pTRE-Tight to produce pTRE-Tight-FLAG plasmid. Then cDNA fragments encoding Chp or its G40V mutant were subcloned as EcoRI (blunted with Klenow fragment)/XhoI fragments from pCan-myc plasmids into SmaI/SalI sites of the pTRE-Tight-FLAG. pEGFP-Chp, pEGFP-ChpG40V and pEGFP-ChpS45N plasmids for expression of N-terminally EGFP-tagged Chp or its G40V and S45N mutants were generated by subcloning the respective cDNAs as EcoRI/XhoI fragments from pCan-myc vectors into EcoRI/SalI sites of pEGFP-C2 vector. pEGFP-ChpG40VΔN30 and pEGFP-ChpG40VΔC208 for expression of EGFP C-terminally fused to ChpG40V mutant lacking amino acids 1–30 and 208–236, respectively, were constructed by PCR and subsequent cloning of amplified cDNAs into EcoRI/SalI sites of pEGFP-C2. pEGFP-ChpS45NΔN30 plasmid for expression of EGFP C-terminally fused to ChpS45N mutant lacking amino acids 1–30 was constructed by PCR and subsequent cloning of amplified cDNA as EcoRI/BglII fragment into EcoRI/BamHI sites of pEGFP-C2. Nucleotide sequences of all vectors were confirmed by double strand DNA sequencing either at “Genome” Center (Engelhardt Institute of Molecular Biology, Russian Academy of Sciences) or at DNA sequencing facility (Fox Chase Cancer Center).
Cell culture, reagents and transfection.
PC12TetOn cells (Clontech, 631137) were cultured on Dulbecco's modified Eagle's medium without sodium pyruvate (Gibco, 11960-044) supplemented with 10% heat-inactivated horse serum (Gibco, 26050-070), 5% tetracycline-screened fetal bovine serum (HyClone, SH30070.03T) and 100 µg/ml of G418 sulfate (Calbiochem, 345812). To produce clones with the inducible expression of N-terminally FLAG-tagged Chp or its G40V mutant PC12TetOn cells were co-transfected with the respective pTRE-Tight-FLAG-Chp and pHygro plasmids. Clones were selected in medium containing 200 µg/ml of hygromycin B (Calbiochem, 400052) and further maintained in growth medium supplemented with 100 µg/ml of hygromycin B. To induce transgene expression cells were treated with 1 µg/ml of DOX (Clontech, 631311) for at least 2 days. HEK293 cells (European Collection of Cell Cultures, 85120602) were cultured on Dulbecco's modified Eagle's medium (HyClone, SH30285.01) supplemented with 10% fetal bovine serum (HyClone, SV30160.03). All cell lines were grown at +37°C in humidified atmosphere containing 5% CO2. Collagen IV from human placenta (Sigma, C5533-5MG) was used to coat cell culture dishes as recommended by manufacturer. Cells were transfected using Unifectinࡊ-56 transfection reagent (Unifect Group, RBL R48-452). Anisomycin was purchased from Calbiochem (176880). SP600125 was purchased from Santa Cruz Biotechnology (sc-200635).
Cell viability measurement.
PC12TetOn cells (wt5, gv2, gv3, gv5 and gv7 clones) were seeded at a density of 1 × 104 per well of 96-well plate (Costar, 3599) coated with collagen IV. Next day, expression of Chp was induced by addition of DOX and cells were grown for 7 days with the medium replaced every 2 days. Cell viability was determined on day 7 using CellTiter96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, G5421). Images of live cells in wells were acquired using CloneSelect Imager (Genetix). Brightness and contrast of acquired images were adjusted with Photoshop 7.0 software (Adobe Systems).
LDH measurement.
Cells were seeded at a density of 1 × 104 per well of 96-well plate (Costar, 3599) coated with collagen IV. Next day, expression of Chp was induced by addition of DOX and cells were grown for 72 hrs. Then, LDH activity released to cell culture medium was measured with CytoTox96® Non-Radioactive Cytotoxicity Assay (Promega, G1780) using 50 µl of cell culture medium. In addition, cell viability in the same experimental wells was determined with CellTiter96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, G5421) as described above.
Caspase activation assays.
PC12TetOn cells (parental cell line and clones wt5, gv5 and gv7) were seeded at a density of 1 × 104 per well of white 96-well plate with transparent bottom (Costar, 3610) coated with collagen IV. Next day expression of Chp was induced by DOX and cells were grown for 48 hrs. When appropriate, cells were grown in presence of SP600125 (10 µM) for 48 hrs. At the end of incubation caspase activities were measured with Caspase-Glo® 3/7 Assay (Promega, G8091), Caspase-Glo® 8 Assay (Promega, G8201) and Caspase-Glo® 9 Assay (Promega, G8211). Cell viability in duplicate wells was assayed with CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7570) by measuring intracellular ATP content. Caspase activities were normalized to the quantity of viable cells.
Flow cytometry.
wt5 cells were seeded at a density of 1 × 106 per 60 mm dish (Costar, 430166) coated with collagen IV. Next day, expression of Chp was induced by adding DOX and cells were grown for 72 hrs. For flow cytometry cells were trypsinized, washed with phosphate-buffered saline (PBS, Sigma, P5368-10PAK) pooled with culture medium and PBS washes to assure that dead and detached cells are not lost, and washed one more time with PBS. Cells were stained with Annexin V-FITC using Annexin V-FITC Apoptosis Detection Kit (Sigma, APO-AF) and immediately analyzed on Cytomics FC500 MPL flow cytometer (Beckman Coulter).
AP-1 reporter activation assay.
HEK293 were seeded at a density of 1 × 105 cells per well of 24-well plate (Costar, 3524). Next day cells were transfected with the appropriate GFP-Chp expression plasmids together with firefly (pGL3-Basic or pfLuc-AP-1) and Renilla (pRL-CMV) luciferase reporter plasmids. Cells were grown for 48 hrs and luciferase activities were measured with Dual Luciferase Reporter Assay System (Promega, E1910) using GloMax 20/20 luminometer (Promega) with integration time set to 10 sec. The firefly luciferase activity was normalized to the Renilla luciferase activity. Cells were cultured in the presence of 5, 10 and 20 µM of SP600125 for 48 hrs before proceeding with luciferase activity measurements, where indicated.
Western blot analysis and antibodies.
gv7 and wt5 cells were seeded at a density of 0.5 × 105 per 35 mm dish (Costar, 430165) coated with collagen IV. Next day, expression of Chp was induced by adding DOX and cells were grown for 48 hrs. Cells were lysed in 200 µl of 1x SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane Hybond-P (GE Healthcare, RPN303F). Blots were blocked with tris-buffered saline (Sigma, T6664-10PAK) supplemented with 0.1% Tween-20 (BioRad, 161-0781) (TBS-T) and 5% non-fat dry milk for 1 h at room temperature and incubated with primary antibodies overnight at +4°C. Blots were washed three times with TBS-T and incubated with secondary horse radish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies as appropriate (GE Healthcare, NA931V or NA934V, respectively), which were detected using ECL Plus western Blotting Detection System (GE Healthcare, RPN2133). Chemiluminescence was imaged with ChemiDoc XRS gel documentation system (BioRad) and quantified with QuantityOne 4.6.2 software (BioRad). Rabbit polyclonal anti-FLAG (F7425-.2MG) and mouse monoclonal anti-α-tubulin, clone DM1α (T9026-.2ML), antibodies were from Sigma. Mouse monoclonal anti-EGFP antibodies, clone 3A9 (PSM001), were from Proteinsynthesis. Rabbit polyclonal anti-phospho-Ser73-c-Jun (9164S), anti-Pak1 (2602), anti-p38MAPK (9212) and rabbit monoclonal anti-SAPK/JNK (9258), anti-phospho-Thr183/Tyr185-SAPK/JNK (4668), anti-c-Jun (9165) and anti-phospho-Thr180/Tyr182 p38MAPK (4511) antibodies were from Cell Signaling.
Quantification of phoshoprotein levels in PC12 cell lysates.
To quantify the activation state of JNK the cell lysates were first equalized by α-tubulin levels. Then two western blottings were run in parallel. One blot was probed with anti-total protein antibodies and another blot was probed with anti-phosphoprotein antibodies. After washing each blot was re-probed with anti-α-tubulin antibodies. Chemiluminescence was detected and quantified as described above. The levels of total and phosphorylated proteins were first normalized to α-tubulin level on each blot, and then the phoshoprotein level was normalized to the total level of the protein.
Statistics.
Cell viability, LDH release, caspase activation and AP-1 reporter activation were measured in at least three independent experiments. Data of representative experiments are shown as mean value of three independent measurements ± SD indicated by error bars. An unpaired t-test was used to calculate two-tailed p-value with GraphPad software (www.graphpad.com).
Conclusions
In this work, we focused on the atypical Rho GTPase Chp which functions are poorly characterized. We used PC12TetOn cells with inducible expression of FLAG-tagged Chp to uncover biological processes affected by Chp. We found that expression of Chp leads to a profound decrease in viability of PC12TetOn cells. Expression of Chp had cytotoxic effect in PC12 cells marked by increased LDH release, activation of caspases-8, -9 and -3/7 and increased Annexin V reactivity. Together these observations clearly indicate that expression of Chp induces apoptotic cell death of undifferentiated PC12 cells. In both PC12 and HEK293 cells, Chp activated JNK but not p38MAPK. Inhibition of JNK with the specific inhibitor, SP600125, rescued PC12 cells from Chp-induced death and attenuated activation of caspase-9 and executioner caspases-3/7 but had no effect on caspase-8 activation. Together these data confirm that Chp-induced apoptosis of PC12 cells, at least in part, occurs via activation of JNK. In addition, our findings suggest that Chp activates two parallel apoptotic pathways: JNK-dependent pathway leading to mitochondrial caspase-9 apoptotic cascade activation, and JNK-independent death receptor-mediated pathway involving caspase-8. In gv7 cells, Chp expression induced phosphorylation of c-Jun on Ser73, a JNK target site, implying that AP-1 transcriptional factor could be activated by JNK downstream of Chp. Indeed, we analyzed AP-1-dependent transcriptional activity in HEK293 cells and found that AP-1 is activated in a Chp-dependent manner via JNK. We assume that AP-1 is activated in a similar manner downstream of Chp in PC12 cells to mediate apoptotic cell death, although it remains to be proved experimentally. Although our data clearly indicate that JNK is one of the mediators of Chp-induced apoptosis of PC12 cells, the observed SP600125 JNK inhibitor-dependent suppression of caspase activation was not complete. In addition, treatment with SP600125 failed to suppress Chp-induced caspase-8 activation. Together this indicates that JNK is not the sole executioner of apoptotic effect of Chp, and other molecular mechanisms of Chp induced apoptosis remain to be identified.
The data obtained suggest that Chp-induced apoptosis requires Chp to be in active, GTP-bound state as expression of ChpG40V mutant apparently resulted in more pronounced effects compared to the expression of the wild type protein. Surprisingly, inactive GFP-ChpS45N mutant activated JNK and AP-1-dependent transcription in HEK293 cells to the same extent as wild type Chp or its G40V mutant. Although nucleotide binding properties of Chp and regulation of its activity and GTPase cycle are currently unknown, previous report demonstrated that ChpS45N is inactive, at least in terms of binding to its effectors such as Pak1.Citation8 We also observed that GFP-ChpS45N and GFP-ChpS45NΔN30 did not reduced Pak1 level in HEK293 lysates indicating that these two forms of Chp are inactive. How presumably inactive Chp can activate JNK pathway? One of the possible explanations is that membrane-targeted Chp upon overexpression can activate JNK pathway to some extent via nucleotide-independent binding to unknown factors. One might speculate that C-terminal sequences of Chp might be involved in JNK activation since ChpG40VΔC208 is barely active. It is feasible that deletion of C-terminal domain not only mislocalizes Chp to cytosol rendering its inactive but also disrupts binding to unknown factors. For example, the related GTPase Wrch-1 was shown to be phosphorylated at Y254 residue in a Src-dependent manner to regulate its localization and ability to interact with effectors.Citation23 It remains to be elucidated if Chp is regulated through its C-terminal domain in a similar manner. Taken together our findings reveal that both N-terminal and C-terminal domains of Chp are important for its ability to activate JNK pathway, while nucleotide-bound state seems to be dispensable.
Although JNK is likely to mediate the apoptotic effect of Chp, molecular mechanisms of JNK activation downstream of Chp are yet to be uncovered. MLK3 could be one of the possible links between Chp and JNK. Chp was shown to interact with MLK3 in yeast two-hybrid systemCitation7 but it remains unknown if these proteins interact in mammalian cells and what is the functional consequence of their interaction. MLK3 plays an important role in PC12 apoptotic death acting upstream of JNK.Citation24 Importantly, if Chp contributes to MLK3 activation, then activation of JNK by MLK3 downstream of Chp might be one of the mechanisms of Chp-dependent apoptosis in PC12 cells although this hypothesis remains to be tested experimentally. Another possible molecular mechanism of JNK activation by Chp could be the recruitment of POSH scaffold to plasma membrane in a manner similar to that described for Rac1 GTPase.Citation16
Together, our data highlight Chp as a putative regulator of JNK-dependent apoptosis in PC12 cells. Further studies are required to establish the physiological role of Chp-induced apoptosis in mammalian cells, for example, during the embryonic development of the nervous system. Since the effects of JNK are cell type specific, the activation of JNK pathway by Chp in other cell types might result in different biological outcomes which are yet to be uncovered.
Abbreviations
| DOX | = | doxycycline |
| SD | = | standard deviation |
| AU | = | arbitrary units |
| LDH | = | lactate dehydrogenase |
| GFP | = | green fluorescent protein |
Figures and Tables
Figure 1 Expression of Chp reduces viability of PC12TetOn cells. (A) Schematic structure of Chp GTPase. N-terminal proline-rich domain (amino acids 1–28), effector domain (53–77), C-terminal domain (208–236) and conserved amino acids G40 and S45 are shown by boxes. N-terminal domain binds to the SH3 domain of adaptor protein Grb2.Citation9 Effector domain is supposed to mediate binding to effector proteins. C-terminal domain is subjected to palmitoylation and regulates subcellular targeting of Chp.Citation9,Citation10 Substitution of Gly40 for Val (G40V) renders Chp constitutively active (GTP-bound). Substitution of Ser45 for Asp (S45N) renders Chp inactive (GDP-bound). (B) PC12TetOn clones with inducible expression of FLAG-Chp (wt5) and FLAG-ChpG40V (gv2, gv3, gv5 and gv7). Cells were grown on plastic for 48 hrs in the presence (+) or absence (−) of DOX. Cell lysates were analyzed by western blotting with indicated antibodies. (C) Quantification of cell viability in PC12TetOn clones grown for 7 days on collagen IV in 96-well plate in the presence (+) or absence (−) of DOX. Data are presented in arbitrary units (AU) of cellular dehydrogenase activity as mean values of three independent measurements ± SD indicated by error bars. Viability of non-treated cells (−) is set as 1.0. The data are representative of at least three independent experiments for each clone. An unpaired t-test was used to calculate two-tailed p-value. Expression of both wild type (wt5 clone) and active form (gv2, gv3, gv5 and gv7 clones) of Chp resulted in statistically significant decrease in viability of PC12TetOn cells. *p < 0.005. (D) Representative images of the same well of PC12gv5 cells grown as in (C) at the day of DOX addition (Day 0) and after 3 days (Day 3) of incubation in DOX-containing medium or medium without DOX are shown. On the day 3, the number of cells incubated with DOX-containing medium is clearly lower than the number of cells cultured in medium without DOX. Scale bar-100 µm.
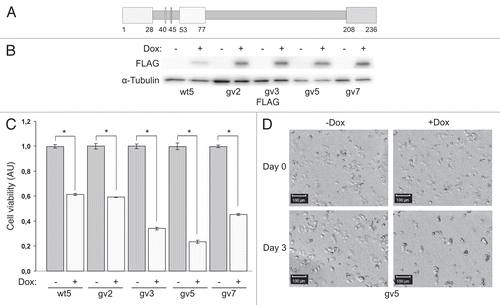
Figure 2 Expression of Chp results in PC12TetOn cell death. (A) Quantification of LDH release from PC12TetOn clones grown for 3 days on collagen IV in 96-well plate in the presence (+) or absence (−) of DOX. Data are presented in arbitrary units (AU) of LDH activity as mean values of three independent measurements ± SD indicated by error bars. LDH release from non-treated cells (−) is set as 1.0. The data are representative of at least two independent experiments for each clone. An unpaired t-test was used to calculate two-tailed p-value. Expression of both wild type (wt5 clone) and active form (gv2, gv3, gv5 and gv7 clones) of Chp resulted in statistically significant increase in cytotoxicity of PC12TetOn clones. *p < 0.005. (B) LDH release from gv5 cells cultured as in (A) but in the presence of different concentration of SP600125 (SP; 0, 0.5, 1, 2 and 5 µM). Data are presented in arbitrary units (AU) of LDH activity as mean values of three independent measurements ± SD indicated by error bars and are representative of two independent experiments. Treatment with 5 µM SP600125 has led to a statistically significant decrease in LDH release from gv5 cells compared to untreated cells. *p < 0.0001. (C) Quantification of gv5 cell viability. Cell viability was determined in the same experimental wells that were used for LDH activity measurement in (B). Data are presented in arbitrary units (AU) of cellular dehydrogenase activity as mean values of three independent measurements ± SD indicated by error bars. Treatment with 5 µM SP600125 (SP) along with DOX restored viability of gv5 cells. *p = 0.0001; n.s.—the difference is not statistically significant.
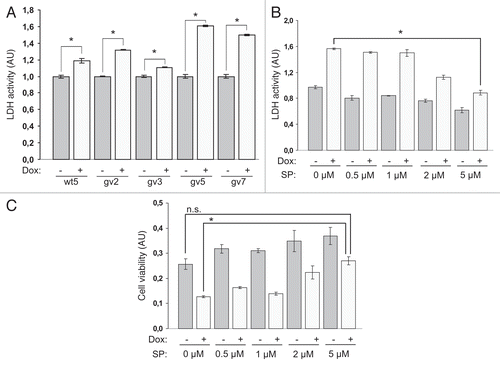
Figure 3 Chp induces apoptosis in PC12TetOn cells. Caspase-3/7 (A), caspase-8 (B) and caspase-9 (C) activities in parental PC12TetOn and wt5, gv5 and gv7 cells grown on collagen IV in 96-well plate for 48 hrs in the presence (+) or absence (−) of DOX. Data are presented in arbitrary units (AU) of caspase activity after normalization to cell viability as mean values of three independent measurements ± SD indicated by error bars. The data are representative of two independent experiments. An unpaired t-test was used to calculate two-tailed p-value. *p < 0.005; **p < 0.05; n.s.—the difference is not statistically significant. (D) Flow cytometry analysis of wt5 cells stained with Annexin V-FITC. Cells were grown on collagen IV for 72 hrs in the presence (+) or absence (−) of DOX, then stained with Annexin V-FITC and analyzed by flow cytometry. Chp-expressing cells (+DOX) show enhanced Annexin V-FITC reactivity indicating of onset of apoptosis.
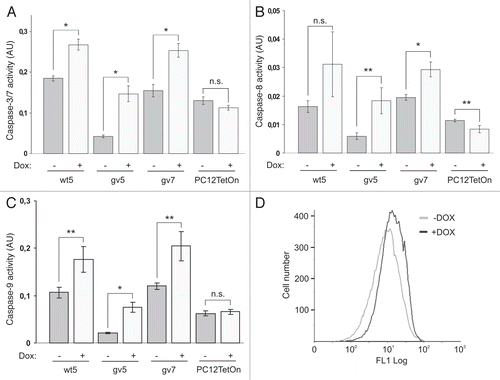
Figure 4 Expression of Chp activates JNK in PC12TetOn cells. (A) wt5 and gv7 cells were grown on collagen IV in 35 mm dishes for 48 hrs in the presence (+) or absence (−) of DOX. Lysates were analyzed by western blotting to detect total levels of JNK, c-Jun, p38MAPK and their phosphorylated forms. Total protein level was monitored by staining with anti-α-tubulin antibodies. Representative results of one out of three independent experiments are shown. (B) Quantification of JNK activation. Data (fold changes in JNK activity) are shown relative to nontreated (−) samples as mean values of three independent measurements ±SD indicated by error bars. Average data for both isoforms of JNK are shown. *p < 0.05.
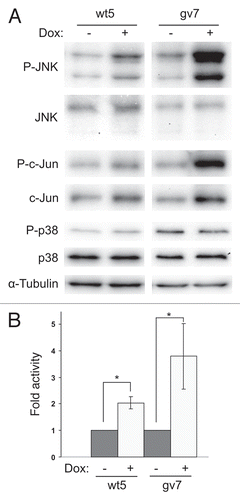
Figure 5 SP600125 treatment attenuates caspase activation by Chp in PC12 cells. Caspase-3/7 (A), caspase-8 (B) and caspase-9 (C) activities in parental PC12TetOn and wt5 and gv5 cells grown on collagen IV in 96-well plate for 48 hrs in the presence of DOX and in the presence (+) or absence (−) of 10 µM of SP600125 (SP). Data are processed and presented as in . *p < 0.05; n.s.—the difference is not statistically significant.
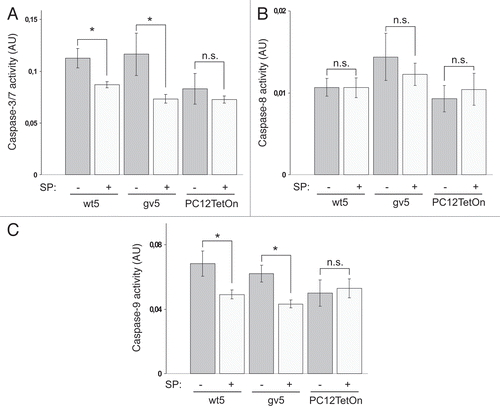
Figure 6 Chp activates AP-1-dependent transcription in HE K293 cells. (A) HE K293 cells were cultured in 24-well plate and transfected with plasmids for expression of GFP or indicated GFP-Chp mutants together with firefly luciferase reporter plasmids (‘Basic’-pGL3-Basic, ‘AP-1’-pfLuc-AP-1) and Renilla luciferase reporter plasmid pRL-CMV. Forty eight hrs after transfection cells were lysed and luminescence was measured with Dual Luciferase Reporter Assay System (Promega). Activity of firefly luciferase was normalized to Renilla luciferase activity. Data are presented in relative luminescence units (RLU) as mean values of three independent measurements ± SD indicated by error bars. The data are representative of three independent experiments. An unpaired t-test was used to calculate two-tailed p-value. (B) HE K293 cells were transiently transfected with plasmids for expression of indicated proteins or left non-transfected (lane 1) and grown for 48 hrs. Prior to lysis positive control cells were treated with 10 µg/ml of anisomycin for 30 min to induce activation of JNK (lane 2). Cell lysates were analyzed by western blotting with indicated antibodies. (C) HE K293 cells were cultured and transfected as in (A). In addition, cells were transfected with reporter plasmids and pBK-CMV. After transfection cells were incubated for 48 hrs in growth medium containing SP600125 (SP) at indicated concentration (0, 5, 10 and 20 µM) prior to luciferase activity measurements. Data are processed and presented as in (A). *p = 0.0018.
Acknowledgements
We are grateful to Dr. Arie Abo for providing the myc-Chp cDNAs and to Dr. Eugene Tulchinsky for providing AP-1 reporter plasmid. We thank Dr. Anna Chernyshova for help with FACS analysis. The work was supported by a grant from Russian Foundation for Basic Research (10-04-01772), (http://www.rfbr.ru), by the Molecular and Cell Biology Program of the Russian Academy of Sciences and by a grant from the Ministry of Education and Science of the Russian Federation (02.522.12.2008). Jonathan Chernoff was supported by a grant from the NIH (R01CA117884).
References
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 2000; 348:241 - 255
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420:629 - 635
- Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci USA 1998; 95:3984 - 3989
- Chuang TH, Hahn KM, Lee JD, Danley DE, Bokoch GM. The small GTPase Cdc42 initiates an apoptotic signaling pathway in Jurkat T lymphocytes. Mol Biol Cell 1997; 8:1687 - 1698
- Su JL, Lin MT, Hong CC, Chang CC, Shiah SG, Wu CW, et al. Resveratrol induces FasL-related apoptosis through Cdc42 activation of ASK1/JNK-dependent signaling pathway in human leukemia HL-60 cells. Carcinogenesis 2005; 26:1 - 10
- Aronheim A, Broder YC, Cohen A, Fritsch A, Belisle B, Abo A. Chp, a homologue of the GTPase Cdc42Hs, activates the JNK pathway and is implicated in reorganizing the actin cytoskeleton. Curr Biol 1998; 8:1125 - 1128
- Aspenstrom P, Fransson A, Saras J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J 2004; 377:327 - 337
- Weisz Hubsman M, Volinsky N, Manser E, Yablonski D, Aronheim A. Autophosphorylation-dependent degradation of Pak1, triggered by the Rho-family GTPase, Chp. Biochem J 2007; 404:487 - 497
- Chenette EJ, Abo A, Der CJ. Critical and distinct roles of amino- and carboxyl-terminal sequences in regulation of the biological activity of the Chp atypical Rho GTPase. J Biol Chem 2005; 280:13784 - 13792
- Chenette EJ, Mitin NY, Der CJ. Multiple sequence elements facilitate Chp Rho GTPase subcellular location, membrane association and transforming activity. Mol Biol Cell 2006; 17:3108 - 3121
- Yeo W, Gautier J. Early neural cell death: dying to become neurons. Dev Biol 2004; 274:233 - 244
- Notarnicola C, Le Guen L, Fort P, Faure S, de Santa Barbara P. Dynamic expression patterns of RhoV/Chp and RhoU/Wrch during chicken embryonic development. Dev Dyn 2008; 237:1165 - 1171
- Guemar L, de Santa Barbara P, Vignal E, Maurel B, Fort P, Faure S. The small GTPase RhoV is an essential regulator of neural crest induction in Xenopus. Dev Biol 2007; 310:113 - 128
- Tay HG, Ng YW, Manser E. A vertebrate-specific Chp-PAK-PIX pathway maintains E-cadherin at adherens junctions during zebrafish epiboly. PLoS One 5:10125
- Katoh M. Molecular cloning and characterization of WRCH2 on human chromosome 15q15. Int J Oncol 2002; 20:977 - 982
- Xu Z, Kukekov NV, Greene LA. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 2003; 22:252 - 261
- Korobko IV, Kalinichenko SV, Korobko EV, Ninkina NN, Kiselev SL, Buchman VL. Pro-survival activity of the MAK-V protein kinase in PC12 cells. Cell Cycle 2010; 9:4248 - 4249
- Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene 2003; 22:8543 - 8567
- Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene 2008; 27:6245 - 6251
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270:1326 - 1331
- Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem 1997; 272:20490 - 20494
- Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol 2000; 60:1015 - 1021
- Alan JK, Berzat AC, Dewar BJ, Graves LM, Cox AD. Regulation of the Rho family small GTPase Wrch-1/RhoU by C-terminal tyrosine phosphorylation requires Src. Mol Cell Biol 2010; 30:4324 - 4338
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol 2001; 21:4713 - 4724
- Cohn MA, Hjelmso I, Wu LC, Guldberg P, Lukanidin EM, Tulchinsky EM. Characterization of Sp1, AP-1, CBF and KRC binding sites and minisatellite DNA as functional elements of the metastasis-associated mts1/S100A4 gene intronic enhancer. Nucleic Acids Res 2001; 29:3335 - 3346