Abstract
Rho-GTPases have been found to be crucial for cytoskeleton remodelling and cell polarity, as well as key players in directed cell migration in various tissues and organs, therefore becoming good candidates for involvement in neuronal migration disorders. We recently found that genetic deletion of the small GTPase RhoA in the developing mouse cerebral cortex results in three distinct cortical malformations: a defect in the proliferation of progenitor cells during development that leads to a bigger cerebral cortex in the adult mouse, a change in the morphology of radial glial cells that results in the formation of a subcortical band heterotopia (SBH, also called Double Cortex) and an increase in the speed of migrating newborn neurons. The latter, together with the aberrant radial glial shape, is likely to be the cause of cobblestone lissencephaly, where neurons protrude beyond layer I at the pial surface of the brain.
Introduction
Neuronal migration in the cerebral cortex of mice and humans occurs primarily during development. During the first stage, the preplate stage, the first early generated neurons migrate from the ventricular zone, where they have been produced by asymmetric cell division from neuroepithelial/radial glial cells. These neurons are called the Cajal-Retzius cells.Citation1-Citation3 As neurogenesis progresses, groups of postmitotic neurons exit the proliferative ventricular and subventricular zones and migrate radially in an inside-out manner toward the pial surface to later form the organized cortical layers.Citation4 The interneurons migrate from the proliferative zone of the ventral telencephalon in a tangentially oriented manner toward the cerebral cortex and intermingle with the excitatory neurons generated directly in the cortex.Citation5
Excitatory and inhibitory neurons which are initially found in the dorsal and ventral telencephalon, respectively, use different modes of migration. Both radially migrating projection neurons (the excitatory neurons) and tangentially migrating interneurons (the inhibitory neurons) are guided mainly by the processes of other cells. In the case of radial migration, dorsal radial glial cells often provide the scaffold for these neurons to migrate, while tangentially migrating interneurons do not use the radial processes for their invasion into the cerebral cortex, but instead use neuronal processes. However, once arrived in the cerebral cortex, the interneurons may also be guided to the final location by switching to a radial guided migration.Citation6 The radially migrating neurons use two different modes of migration: early in development, when the cortical plate is still very thin, they extend a leading process toward the basement membrane and the soma then follows the process (somal translocation); at later stages, they start to use radial glial processes as a guide to reach the distant cortical plate (glial-guided locomotion).Citation6 Different defects in the neuronal migration steps result in distinct cortical malformations, namely the neuronal migration disorders. Genetic analyses of human brain malformations have highlighted a number of cytoskeletal-associated proteins underlying these functions.Citation4,Citation7
For instance, when neurons fail to accomplish the first step of neuronal migration and do not manage to start migrating along the radial glial processes, they accumulate in the proliferative zone, next to the ventricles. This ectopic accumulation of cells results in a human malformation called periventricular heterotopia (PH) and is caused by mutations of FilaminA and Arfgef2.Citation4 Alternatively, neurons can indeed begin migrating, but eventually arrest before reaching the cortical plate and distribute in a disorganized manner in the developing cerebral cortex. This is often the case of mutations in microtubule associated proteins, like Double-Cortin (Dcx) and Lis1 and results in lissencephaly or subcortical band heterotopia (SBH or Double Cortex) in humans.Citation4,Citation7 In some cases neurons fail to correctly laminate, like in the case of the reeler mouse, showing an inverted or scattered organization of the neuronal layers.Citation4 Lasty, neurons can miss the correct position to terminate migration, resulting in the invasion of those neurons into layer I and this is often correlated with the formation of neuronal ectopias at the basement membrane and associated with lissencephaly type II.Citation4
Neurogenesis and cell migration are controlled by extracellular and intracellular signals, with these signals eventually converging on the cytoskeleton. Certainly, many brain developmental disorders are associated with mutations in genes that encode cytoskeletal proteins and their modulators.Citation8,Citation9 However, mutations of the genes so far identified in human patients fail to mimic the phenotypes in mouse models.
Cdc42, Rac1 and RhoA, the most studied members of the Rho GTPase family, are expressed in the developing brain and most specifically in the ventricular/subventricular zone, where progenitor cells are located.Citation10 Until now we know very little about the functions of Rho GTPases in radial glial cells. Cdc42 plays an important role in cell fate specification and it is required for maintenance of the adherens junctions at the ventricular surface.Citation11 Rac1 is also required for maintaining cell proliferation, since when mutated, differentiation takes over, leading to a smaller brain size.Citation12,Citation13 Moreover, abnormal signaling through Rho family GTPases is an important cause of mental retardation.Citation14 More recently, deletion of RhoA in the spinal cord and midbrain has revealed insights into its role at early stages of CNS development, highlighting not only common functions in the maintenance of adherens junction coupling, but also its opposite role in regulating cell proliferation in the spinal cord vs. midbrain.Citation15,Citation16 Rho-GTPases are therefore suitable candidates for playing a role in neuronal migration disorders and cortical malformation.
RhoA Deletion Results in Three Distinct Cortical Malformations
The increase in the cortical size—defect in cell proliferation
The first cortical malformation observed in the RhoA mutant cortex is a defect in cell proliferation. The size of the adult RhoA mutant cortex is enlarged, suggesting changes in proliferation probably occurring during development. Indeed, the number of mitotic cells is transiently increased in the RhoA knockout cerebral cortex (cKO) and their localization is changed: scattered cells are distributed in the whole developing cerebral cortex in contrast to their alignment at the ventricular surface in the cerebral cortex of control animals. In accordance with the abnormal location of progenitors, some neurons are located in ectopic positions at the apical surface already at early stages.
Surprisingly, a few days later, all progenitor cells clustered into a large band situated in the middle of the cerebral cortex, while the neurons split into two layers, framing the proliferative zone. In this reorganization, the progenitors did not change cell identity and kept their features of radial glial cells or basal progenitors, which is in contrast to the Cdc42 cKO cortex where mutant progenitors, while changing the location of division, also shift their fate toward the more differentiated basal progenitors.Citation11 RhoA deletion in radial glial cells therefore altered the normal cycling proprieties of progenitors, leading to a transient expansion in the progenitors of the developing cerebral cortex and resulting in an enlargement of the adult cortexCitation17 (see also schematic drawing in ). Interestingly, the number of proliferating cells is later reduced until it again reaches the normal levels, suggesting a mechanism of compensation.
Figure 3. The cortical malformations of the RhoA cKO.
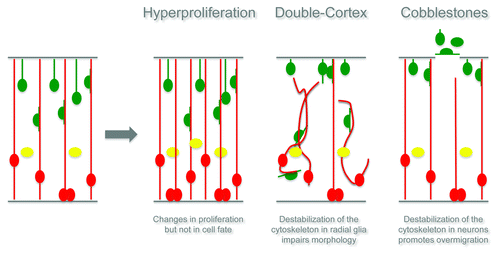
Interestingly, RhoA regulates cell proliferation in a region-specific manner. For instance it was recently demonstrated that conditional deletion of RhoA in the midbrain and forebrain results in accelerated proliferation of neural progenitors, reduction of cell cycle exit with consequent expansion of neural progenitor cells and exencephaly-like protrusions.Citation16 On the contrary, conditional deletion of RhoA in the spinal cord neuroepithelium leads to decreased neuroepithelial cell proliferation and premature cell cycle exit.Citation15 Rho-GTPases are essential regulators of both the actin and the microtubule cytoskeleton; therefore these region-specific phenotypes certainly open new questions: how is the cytoskeleton able to regulate cell proliferation and/or differentiation in the developing cerebral cortex? One possible explanation is that the accumulation of actin monomers observed in the RhoA cKO forces the transcriptional co-activator MAL into the cytoplasm, resulting in an inhibition of the serum response factor (SRF) activation, known to be responsible for transcription of genes that are required for differentiation.Citation18 However, additional pathways may also be involved and regulated, for instance by the changes in the microtubule stability.
The Double Cortex—defect in radial glial morphology
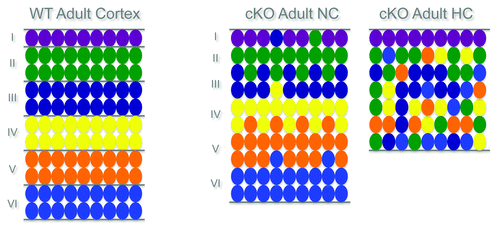
The second cortical malformation observed in the RhoA cKO mutants is a neuronal migration defect, namely a huge heterotopia composed of neurons underneath an apparently normal cortex. Two types of migrational disorders include this type of heterotopia: the periventricular heterotopia (PH) and the subcortical band heterotopia (SBH or Double Cortex). Interestingly, in the case of the RhoA cKO, ectopic neurons are embedded within the white matter and not directly opposed to the ventricle (like in the case of PH), defining this malformation as a form of SBH. The laminar identity of the neurons in the upper normotopic cortex (NC) is maintained, but all of the different neurons are also clearly present in the lower disorganized heterotopic cortex (SBH). However, most neurons in the SBH have primarily an upper layer identity, suggesting a strong contribution of late generated neuronsCitation17 (see also schematic drawing in ).
Figure 1. Layer organization of the cerebral cortex in WT and RhoA cKO.
Moreover, the radial glial processes in the cKO cortices are arranged in a strongly disorganized manner and only few processes reach the ventricular zone and the basement membrane.
We therefore hypothesize two different mechanisms that could explain the formation of the Double Cortex. One possible explanation is that abnormalities in the radial glial cells, which are used by the projection neurons as main guides for migration, contribute to the failure of many neurons to reach their final positionCitation17 (see also schematic drawing in ). Alternatively, RhoA may be crucial for one type of neuronal migration, either somal translocation or glial-guided locomotion and therefore, only one population of neurons would be affected and contribute to the formation of the heterotopiaCitation17 (see also schematic drawing in ).
Figure 2. Hypothesis of the function of RhoA in migrating neurons and radial glial cells.
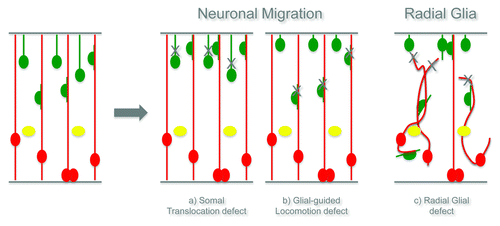
Live imaging and transplantation experiments strongly support the hypothesis that RhoA deletion does not directly affect neuronal migration, but rather the radial glial scaffold that neurons use as a guidance to migrate and find their final destinationCitation17 (see also schematic drawing in ).
These data clearly suggest a new concept, namely that the formation of a Double Cortex is not only due (if at all) to intrinsic defects in the migration capacity of neurons, but primarily to a radial glial abnormality. So far, it is still not possible to conclude that this is the only way of generating a SBH, because many different heterotopias have been observed in human, of different size and located in different cortical areas. But, from these data we can certainly recognize the prominent role of radial glia cells in migrational disorders and, in particular, in the formation of the Double Cortex. Analysis of other mouse models of SBH is therefore crucial to determine if the radial glial cells are prevalently responsible for the formation of the Double Cortex.
Additionally, this model can be adopted to understand how neurons located in the NC or in the SBH are functionally connected and communicate with other brain areas and vice versa. A first analysis of GFP-labeled neurons in the upper NC and in the lower SBH reveals a normal pyramidal neuron morphology with projections, suggesting that neurons in the NC and SBH develop a grossly normal dendrite-axon polarity.Citation17 Further analysis of the neuronal morphology and the reciprocal connectivity between the NC and SBH, as well as electrophysiological properties of single neurons in different layers of the NC and SBH, could give additional insight and reveal the reciprocal communication between the upper and lower cortices.
Patients affected with SBH often develop epileptic seizures.Citation19 Interestingly, tangential migration is reduced in the RhoA cKO cortices, resulting in a significantly lower number of interneurons in the upper NC. This phenotype is certainly relevant because incorrect balance of excitatory and inhibitory neurons is often one of the cause for increased susceptibility to epileptic seizures.Citation19 This raises a new question: how WT tangentially migrating interneurons generated in the ventral telencephalon of the RhoA cKO mouse integrate into the upper NC and lower SBH. For instance, we can imagine that interneurons early generated in the ventral telencephalon can still find the correct road to the mutant cortex, but later on, when the mutant cortex is completely disorganized, they probably have the choice of migrating to the upper NC or to the SBH, eventually preferring to populate the second one.
The RhoA signaling pathway ultimately stabilizes the cytoskeleton by promoting actin polymerization and microtubule assembly. Lack of RhoA could therefore influence the balance between stable vs. dynamic microtubules as well as the actin polymerization and hence affect the radial morphology. Indeed these alterations in the radial glia scaffold are caused by destabilization of both the actin and the microtubule cytoskeleton, demonstrating a crucial role of RhoA in maintaining the integrity of the radial glial morphology.Citation17
Interestingly, the stabilization of the actin cytoskeleton has recently been shown to be an essential regulator for radial vs. tangential migration of cortical neurons. Indeed, downregulation of Lamellipodin, an actin-remodeling protein, caused changes in the ratio of polymerized to unpolymerized actin in the pyramidal neurons that then adopt a tangential, rather than radial, migration mode.Citation20
Is then the stability of the microtubules and actin the key to understanding PH and SBH? Interestingly, actin regulating genes seem to play a major role in the formation of PH in human patients,Citation21 while mutations in genes regulating microtubules are often associated with the SBH formation.Citation4 Therefore, the microtubule destabilization observed in the RhoA mutant radial glial cells has perhaps a more relevant role than the actin depolymerization. Additionally, the microtubule destabilization in the RhoA cKO was observed mainly in the radial glial cells and not (or much less) in neurons, highlighting again the prominent role of microtubule stability and radial cells in SBH formation. Interestingly, the loss of the actin nucleator mDia, downstream of RhoA, results in disruption of only apical actin fibers with consequent formation of PH and not SBH.Citation22
Taken together, these data suggest that the microtubule destabilization in radial glia is the main cause of SBH formation while fine regulation of actin fibers is the key for understanding the formation of PH. Alternatively, the border between these two disorders is very thin and therefore, temporal and spatial cellular conditions are crucial for the establishment of the development of different types of heterotopias.
The cobblestones—defect in radial glia and/or neuronal migration
The third and last cortical malformation observed in the RhoA cKO is also a neuronal migration defect and is the formation of neuronal ectopias at the basal side of the developing cerebral cortex, namely the cobblestones, with patches of basement membrane missing already from embryonic stages. This phenotype was also observed previously in several mouse models.Citation23-Citation27 Interestingly, a few of these mice have mutations in genes directly or indirectly linked to RhoA, for instance G protein-coupled receptor (GPR) 56,Citation25,Citation26 Gα12/13Citation27, α6 integrinCitation28 and focal adhesion kinase (FAK).Citation23 Different mouse models showing cobblestones share at least two of the three common features of this malformation: neurons do not stop migrating, the radial glial endfeet are not morphologically normal and the basement membrane is disrupted in small patches. Interestingly, it is not clear what the leading event is. For example, deletion of FAK with the Emx1Cre driver (for all cells in the dorsal telencephalon) results in the formation of cobblestones, while using a specific neuronal driver (NexCre) does not affect cortical development.Citation23 Additionally, specific deletion of FAK in the meningeal fibroblasts only results in the formation of a neuronal ectopia.Citation23 On the contrary, in the Gα12/13 mutant, the NexCre mediated recombination of neurons was sufficient to promote the formation of neuronal ectopia,Citation27 suggesting that all three events can initiate the formation of cobblestones.
In order to figure out if this phenotype was also the result of a non-cell-autonomous defect, downregulation of RhoA was performed in single cells by Cre electroporation. Already three days later many more neurons were found in the cortical plate, suggesting a faster neuronal migration, and the first cobblestones were observed five days after electroporation. On the contrary, overexpression of a fast cycling form of RhoA clearly slows neurons down and results in changes in the morphology of the migrating cells. This result clearly demonstrates that RhoA in the developing cerebral cortex seems to act as a brake for migrating neurons. This faster migration observed, presumably together with the aberration in some radial glial endfeet, is likely to be responsible for the formation of basal cobblestones in a cell-autonomous way. It remains unclear if it is the faster neuronal migration or the aberration of the radial glial endfeet that primarily drives the formation of the cobblestone, as they could simply both be required for the formation of the ectopia. Certainly, in the case of the RhoA cKO the disruption of the basement membrane is a consequence of one or the other, because the basement membrane is not depleted of RhoA in our cKO mutantsCitation17 (see also schematic drawing in ).
Taken together these observations highlight the emerging role of cytoskeleton stability in cerebral cortex development, suggesting that fine-tuning of the actin and microtubule dynamics is key to understanding several cortical malformations, including neuronal migration disorders.
Acknowledgments
I would like to thank Magdalena Götz, Pia Johansson and Emily Violette Baumgart for helpful comments and input to the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 1999; 19:7881 - 8; PMID: 10479690
- Meyer G, Wahle P. The paleocortical ventricle is the origin of reelin-expressing neurons in the marginal zone of the foetal human neocortex. Eur J Neurosci 1999; 11:3937 - 44; http://dx.doi.org/10.1046/j.1460-9568.1999.00818.x; PMID: 10583482
- Monuki ES, Porter FD, Walsh CA. Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-Lhx2 pathway. Neuron 2001; 32:591 - 604; http://dx.doi.org/10.1016/S0896-6273(01)00504-9; PMID: 11719201
- Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol 2004; 20:593 - 618; http://dx.doi.org/10.1146/annurev.cellbio.20.082503.103047; PMID: 15473853
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 1997; 278:474 - 6; http://dx.doi.org/10.1126/science.278.5337.474; PMID: 9334308
- Marín O, Valiente M, Ge X, Tsai LH. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol 2010; 2:a001834; http://dx.doi.org/10.1101/cshperspect.a001834; PMID: 20182622
- Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci 2001; 24:1041 - 70; http://dx.doi.org/10.1146/annurev.neuro.24.1.1041; PMID: 11520927
- Leventer RJ, Guerrini R, Dobyns WB. Malformations of cortical development and epilepsy. Dialogues Clin Neurosci 2008; 10:47 - 62; PMID: 18472484
- Reiner O, Sapir T. Polarity regulation in migrating neurons in the cortex. Mol Neurobiol 2009; 40:1 - 14; http://dx.doi.org/10.1007/s12035-009-8065-0; PMID: 19330467
- Pinto L, Mader MT, Irmler M, Gentilini M, Santoni F, Drechsel D, et al. Prospective isolation of functionally distinct radial glial subtypes--lineage and transcriptome analysis. Mol Cell Neurosci 2008; 38:15 - 42; http://dx.doi.org/10.1016/j.mcn.2008.01.012; PMID: 18372191
- Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch-Bräuninger M, et al. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci 2006; 9:1099 - 107; http://dx.doi.org/10.1038/nn1744; PMID: 16892058
- Chen L, Melendez J, Campbell K, Kuan CY, Zheng Y. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev Biol 2009; 325:162 - 70; http://dx.doi.org/10.1016/j.ydbio.2008.10.023; PMID: 19007770
- Leone DP, Srinivasan K, Brakebusch C, McConnell SK. The rho GTPase Rac1 is required for proliferation and survival of progenitors in the developing forebrain. Dev Neurobiol 2010; 70:659 - 78; PMID: 20506362
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 2002; 25:191 - 9; http://dx.doi.org/10.1016/S0166-2236(00)02118-4; PMID: 11998687
- Herzog D, Loetscher P, van Hengel J, Knüsel S, Brakebusch C, Taylor V, et al. The small GTPase RhoA is required to maintain spinal cord neuroepithelium organization and the neural stem cell pool. J Neurosci 2011; 31:5120 - 30; http://dx.doi.org/10.1523/JNEUROSCI.4807-10.2011; PMID: 21451048
- Katayama K, Melendez J, Baumann JM, Leslie JR, Chauhan BK, Nemkul N, et al. Loss of RhoA in neural progenitor cells causes the disruption of adherens junctions and hyperproliferation. Proc Natl Acad Sci U S A 2011; 108:7607 - 12; http://dx.doi.org/10.1073/pnas.1101347108; PMID: 21502507
- Cappello S, Böhringer CR, Bergami M, Conzelmann KK, Ghanem A, Tomassy GS, et al. A radial glia-specific role of RhoA in double cortex formation. Neuron 2012; 73:911 - 24; http://dx.doi.org/10.1016/j.neuron.2011.12.030; PMID: 22405202
- Luxenburg C, Pasolli HA, Williams SE, Fuchs E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat Cell Biol 2011; 13:203 - 14; http://dx.doi.org/10.1038/ncb2163; PMID: 21336301
- McManus MF, Golden JA. Neuronal migration in developmental disorders. J Child Neurol 2005; 20:280 - 6; http://dx.doi.org/10.1177/08830738050200040301; PMID: 15921227
- Pinheiro EM, Xie Z, Norovich AL, Vidaki M, Tsai LH, Gertler FB. Lpd depletion reveals that SRF specifies radial versus tangential migration of pyramidal neurons. Nat Cell Biol 2011; 13:989 - 95; http://dx.doi.org/10.1038/ncb2292; PMID: 21785421
- Zhang J, Neal J, Lian G, Shi B, Ferland RJ, Sheen V. Brefeldin A-inhibited guanine exchange factor 2 regulates Filamin A phosphorylation and neuronal migration. J Neurosci 2012; 32:12619 - 29; http://dx.doi.org/10.1523/JNEUROSCI.1063-12.2012; PMID: 22956851
- Thumkeo D, Shinohara R, Watanabe K, Takebayashi H, Toyoda Y, Tohyama K, et al. Deficiency of mDia, an actin nucleator, disrupts integrity of neuroepithelium and causes periventricular dysplasia. PLoS One 2011; 6:e25465; http://dx.doi.org/10.1523/JNEUROSCI.4773-10.2011; PMID: 21389223
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, et al. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 2003; 40:501 - 14; http://dx.doi.org/10.1016/S0896-6273(03)00666-4; PMID: 14642275
- Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 1999; 147:1109 - 22; http://dx.doi.org/10.1083/jcb.147.5.1109; PMID: 10579729
- Iguchi T, Sakata K, Yoshizaki K, Tago K, Mizuno N, Itoh H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J Biol Chem 2008; 283:14469 - 78; http://dx.doi.org/10.1074/jbc.M708919200; PMID: 18378689
- Li S, Jin Z, Koirala S, Bu L, Xu L, Hynes RO, et al. GPR56 regulates pial basement membrane integrity and cortical lamination. J Neurosci 2008; 28:5817 - 26; http://dx.doi.org/10.1523/JNEUROSCI.0853-08.2008; PMID: 18509043
- Moers A, Nürnberg A, Goebbels S, Wettschureck N, Offermanns S. Galpha12/Galpha13 deficiency causes localized overmigration of neurons in the developing cerebral and cerebellar cortices. Mol Cell Biol 2008; 28:1480 - 8; http://dx.doi.org/10.1128/MCB.00651-07; PMID: 18086886
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmüller A. Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol 1998; 8:983 - 6; http://dx.doi.org/10.1016/S0960-9822(98)70402-6; PMID: 9742403