Abstract
The RNA polymerase III pre-initiation complex (PIC) assembled on yeast tRNA genes naturally causes replication fork pausing that contributes to genome instability. Mechanistic coupling of the fork pausing activity of tRNA genes to replication has long been considered likely, but only recently demonstrated. In contrast to the expectation that this coupling might occur by a passive mechanism such as direct disruption of transcription factor-DNA complexes by a component of the replisome, it turns out that disassembly of the RNA polymerase III PIC is actively controlled by the replication stress checkpoint signal transduction pathway. This advance supports a new model in which checkpoint-dependent disassembly of the transcription machinery at tRNA genes is a vital component of an overall system of genome stability control that also targets replication and DNA repair proteins.
Introduction
The rate of tRNA gene transcription is a critical determinant of the translational capacity of a cell. Therefore, cell growth and proliferation, as well as maintenance functions in differentiated cells, are affected by the pace of tRNA synthesis. For these reasons there has been an intense effort to understand how tRNA gene transcription is regulated. Here we review advances in budding yeast which suggest that tRNA gene regulation has previously unsuspected connections to the control of DNA replication and genome stability. Readers are referred to McFarlane and WhitehallCitation1 for an excellent complementary synopsis of some of the literature on the relationships between tRNA gene transcription and replication control.
tRNA Gene Transcription: The Core Machinery
Each unique tRNA species in eukaryotes is usually encoded by multiple unlinked tRNA genes with identical coding sequences ().Citation2,Citation3 The promoters of these genes, and the core protein components of the tRNA transcriptional machinery, are similar across the kingdom ( and C).Citation4–Citation6 Three multi-subunit complexes are essential for tRNA gene transcription: RNA polymerase III, and transcription initiation factors IIIB and IIIC (RNAP III, TFIIIB and TFIIIC respectively). These proteins assemble into a pre-initiation complex (PIC) on tDNA in a sequential fashion, starting with specific binding of TFIIIC to a bipartite, intragenic sequence motif found in all tRNA genes ().Citation7 TFIIIC binding is followed by stepwise recruitment of TFIIIB and RNAP III to form the PIC (). Unlike genes transcribed by RNAP I and II, tRNA genes are not regulated by sequences that bind trans-acting factors such as enhancer, repressor or activator proteins.
In addition to positively-acting proteins, transcriptional regulation of the tRNA genes often involves a conserved repressor called Maf1.Citation8 This small acidic protein (29–45 kDa, depending on the species) is remarkable in its ability to integrate inputs from a broad spectrum of signaling pathways to repress tRNA gene transcription under conditions of cellular stress.Citation9 Dephosphorylation of Maf1 is necessary for signal-dependent repression of transcription by a mechanism that involves its recruitment to tRNA genes. Although it is known to bind to RNAP III and TFIIIB, the precise mechanism of action of Maf1 remains ill-defined and it lacks amino acid motifs that might suggest how it functions. Because of its central role in the regulation of RNAP III transcription, Maf1 is a subject of intense ongoing study in the field.Citation10
The rather simple structure of tRNA genes compared to the genes transcribed by RNAPs I and II, the limited repertoire of transcription factors required for tRNA gene expression, and the fact that tRNAs are housekeeping molecules, might suggest that the tRNA gene transcriptional cycle is not highly regulated. However, the tRNA gene transcriptional machinery is subject to a broad spectrum of regulatory inputs that help the cell to match the activity of the RNAP III transcriptional machinery to the demand for tRNAs to participate in translation.Citation11–Citation13 In all organisms, such inputs include signals from a variety of stress pathways. In vertebrates, the tRNA gene transcriptional machinery also receives repressive signals from cell cycle regulators during the M and G1 phases of the division cycle.Citation14,Citation15
“Product-Independent” Functions of tRNA Genes and Promoter Elements
The expression of tRNA molecules that are functional in translation is a matter of life or death for nucleated cells. Therefore, it is not surprising that genetic perturbation of the tRNA genes and interference with the function of the RNAP III transcriptional machinery usually compromise fitness. What is surprising is that tRNA genes and the RNAP III transcriptional machinery can also affect cell physiology by mechanisms that do not depend on the influence of tRNA output on translation.
One classical “product-independent” function of tRNA genes in budding yeast is the modulation of heterochromatin spreading. Specifically, heterochromatin spreading from the HMR locus is normally limited by the immediately adjacent (AGU)C threonine tRNA gene.Citation16 This activity of tT(AGU)C is unlikely to be dependent on tRNA synthesis since an element that binds only TFIIIC also has boundary function.Citation17 tRNA genes and the tRNA gene transcriptional machinery also have product-independent functions that depend on tRNA gene transcription. RNAP II promoters for example are repressed by neighboring tRNA genes, and that repression depends on tRNA synthesis but not tRNA function.Citation18
The knowledge that tRNA gene transcription can be regulated and that tRNA gene transcription has product-independent effects on cellular physiology, leads to a new biological question: is the transcription process at tRNA genes ever regulated to modulate its product-independent outcomes? Below we explore this question as it pertains to interference with DNA replication, a striking product-independent outcome of the DNA-associated events that lead to tRNA gene transcription.
Replication Interference by tRNA Genes during Normal S Phase
Normal tRNA genes can interfere with normal replication. This effect was discovered in work focused on nuclear DNA replication in budding yeast. DNA is synthesized at the replication fork by processive leading- and lagging-strand DNA polymerases (DNAPs).Citation19 These enzymes, and a plethora of accessory factors including the replisome progression complex (RPC), assemble at every fork.Citation20 Replication intermediates synthesized by replisomes can be resolved by neutral-neutral 2D gel electrophoresis and detected by Southern blotting.
Desphande and NewlonCitation19 used neutral-neutral 2D gel electrophoresis to visualize the replication intermediates of a series of plasmids containing different fragments from a region of yeast chromosome III. This analysis identified a natural locus that interferes with fork movement. The critical element of this ‘fork-pausing’ locus turned out to be a tRNA gene. Desphande and Newlon further demonstrated that fork pausing by a plasmid-borne tRNA gene requires binding of TFIIIC and recruitment of RNAP III. That is, fork pausing minimally depends on formation of the pre-initiation complex. Since TFIIIC must interact with TFIIIB in order for RNAP III to be assembled on tRNA genes,Citation21 fork pausing must also depend on TFIIIB. The work using 2D gel mapping methods also clearly revealed that there is a polarity to the effect of tRNA genes on replication of plasmids: they only cause pausing if the fork moves into the gene in the direction opposite of transcription. Collectively, work from the Newlon lab supported a model in which the RNAP III pre-initiation complex at tRNA genes, when oriented to fire RNAP III into the fork that copies them, causes fork pausing (). These results did not rule out the possibility that transcription itself is necessary for fork pausing at tRNA genes. While that might indeed be the case, the absolute amount of tRNA produced by a particular tRNA gene is not the primary determinant of the capacity of that gene to cause fork pausing. We know this because pausing,Citation19 but not tRNA production,Citation18 is dampened when a tRNA gene and its immediately flanking sequences (required for optimal expression) are reversed in orientation.
Seven years after fork pausing by tRNA genes was demonstrated on plasmids, Ivessa et al. Showed by 2D gel electrophoresis of replication intermediates that chromosomal tRNA genes also cause fork pausing by a mechanism that requires promoter binding by TFIIIC.Citation22 Furthermore, only tRNA genes oriented into the forks that replicate them were observed to cause pausing.
The Zakian group also tested if RNAP III can cause fork pausing when it transcribes intergenic DNA. The natural and “backup” terminators of a fork-pausing tRNA gene were inactivated by mutation. In these situations, RNAP III transcribes well beyond the normal site of RNA release, but the size of the pause does not change.Citation22 Therefore, RNAP III transcription of intergenic DNA does not interfere with fork progression. This finding does not exclude the possibility that RNAP III transcription of tRNA genes is necessary for fork interference, since transcription elongation through intergenic DNA does not involve some molecular interactions which occur at tRNA genes. For example, RNAP III that is transcribing intergenic DNA does not have to compete with TFIIIC for binding to template DNA.Citation23 Therefore, it remains unclear if replication interference requires only the presence of a PIC at a tRNA gene, active RNAP III transcription of the gene or both.
The story surrounding the phenomenon of replication interference by tRNA genes has taken some unexpected turns recently. Two groups have analyzed association of the replisome with DNA on a genome-wide scale using chromatin immunoprecipitation (ChIP) in combination with microarray technology (the “ChIP on chip” approach). Azvolisnksy et al. reported the results of a study of leading strand DNAP (Pol2) cross-linking to DNA in unsynchronized populations of budding yeast cells.Citation24 Peaks of Pol2 cross-linking revealed by this approach were taken to correspond to regions where replication is slow. This study yielded evidence that fork pausing occurs naturally at some tRNA genes that are transcribed in the same direction as they are replicated (). It follows that pausing at tRNA genes is not exclusively due to a buildup of superhelical tension caused by convergence of RNAPs and DNAPs.Citation19,Citation22 Surprisingly, some tRNA genes identified as strong fork-pausing elements by 2D gel electrophoresis of DNA were not sites of Pol2 accumulation. The reason for this discrepancy is unknown.
In a related study, Sekedat et al. monitored replication fork movement in cells progressing synchronously through S phase. Association of a subunit of the replisome-associated GINS complex with DNA was used as a proxy for association of the replication machinery as a whole.Citation25 This ChIP on chip study yielded evidence that all 274 tRNA genes in budding yeast are difficult to replicate. Collectively, the new results obtained using the ChIP on chip method support the model that all active tRNA genes have a propensity to cause fork pausing during normal replication in budding yeast. It seems likely that this phenomenon characterizes replication in higher organisms, since non-polar fork pausing at relocated chromosomal tRNA genes has been demonstrated in fission yeast by 2D gel electrophoresis of replication intermediates.Citation26
A number of conflicting results pertaining to fork movement through tRNA genes remain to be reconciled. Specifically, there are striking discrepancies between measurements of fork movement obtained by 2D gel electrophoresis of replication intermediates, ChIP on chip analysis of the catalytic subunit of the leading strand DNAP, and ChIP on chip analysis of the GINS complex. Pausing was observed at relatively few tRNA genes in the ChIP on chip study of Pol2 cross-linking, but at every tRNA gene in the ChIP on chip study of GINS cross-linking. And as noted above, ChIP on chip analysis of Pol2 revealed pausing at tRNA genes previously thought not to cause fork interference (a subset of co-directional tRNA genes), but failed to reveal pausing at some tRNA genes at which pausing is readily detected by 2D gel analysis of replication intermediates. Until we understand the cause of these discrepancies, it will be challenging to develop a compelling model for the mechanism by which active tRNA genes affect the replication process.
Interestingly, another repeated gene transcribed by RNAP III, the 5S rRNA gene, does not cause fork pausing in wild-type cells.Citation27,Citation28 The reason why the 5S rRNA and tRNA genes affect fork movement differently is unknown. One key difference between them is that 5S rRNA gene transcription requires a sequence-specific DNA binding protein, transcription factor IIIA, that is not used at tRNA genes.Citation21 Perhaps the PIC assembled by TFIIIA on the 5S rRNA gene is less inhibitory to fork movement than the PIC assembled on tRNA genes without involvement of TFIIIA. This possibility remains to be tested.
The Good and Bad of Fork Encounters with tRNA Gene Pre-Initiation Complexes
The pausing effect of tRNA genes on replication can be thought of as fork “interference.” Because pausing has only been measured using population-based assays, it remains unknown if fork interference by individual tRNA genes is relatively constant between cells or highly variable. For example, it is not known if a moderate pausing signal in a Pol2 ChIP experiment reflects moderate pausing in all cells (consistent with “scheduled” fork interference) or extremely high pausing in some cells, but no pausing in others (consistent with “unscheduled” pausing).
Regardless of whether fork interference at tRNA genes is scheduled or unscheduled, it is not yet clear if it can be a good thing when it does occur. Rothstein et al.Citation29 have argued that there may be selective advantages to having forks pause at tRNA genes during unchallenged proliferation (proliferation in the absence of an exogenous genotoxic insult). One advantage could be to promote gene conversion events between identical tRNA genes on different chromosomes, thereby ensuring the sequence homogeneity of members of the various tRNA gene families.Citation29 Since pausing depends on assembly of a functional RNAP III PIC, a mechanism of this type would partly be favored because it selects for recombination involving those genes that have not suffered intragenic mutations that compromise transcription (such mutations might occur in the B box, which is required for TFIIIC binding; see ).
The idea that pausing at tRNA genes promotes recombination has been tested in buddingCitation30 and fissionCitation26 yeast, using reporter systems for recombination between plasmid-borne direct repeats, and between sequences on a plasmid and a chromosome, respectively. The effect on recombination of a single tRNA gene was studied in budding yeast, and a monomeric and dimeric tRNA gene were tested in Schizosaccharomyces pombe. In both systems, pausing was monitored by 2D gel analysis of replication intermediates. No correlation between fork pausing at tRNA genes and recombination was observed. Therefore, the tRNA genes studied, on their own, are not portable recombination enhancers. Furthermore, their barrier activity in a non-native chromosomal context is not sufficient to stimulate recombination.
Because the cis-acting sequences and trans-acting factors that control transcription are highly similar between tRNA genes, it seems reasonable to presume that these findings reflect the behavior of all of them. However, variations in the sequence immediately upstream of tRNA gene coding regions can affect transcription strength in vitro,Citation31 and tRNA genes that differ only in their flanking sequences can differ substantially in transcription efficiency in vivo.Citation19,Citation32 Therefore, the propensity of additional tRNA genes to stimulate recombination may be worthy of investigation. Comparison of tRNA genes and the RTS1 recombination hotspot in S. pombe could be particularly informative. In previous work, tRNA genes did not stimulate recombination or cause the same intensity of fork interference as RTS1 in its pausing orientation.Citation26 Perhaps tRNA genes that cause as much fork interference as RTS1 (if they exist) will be found to stimulate recombination during unchallenged proliferation.
While tRNA genes on their own might not be a major source of recombinogenic lesions during unchallenged proliferation, they do contribute to the formation of such lesions. Chromosomal fragile sites display recurrent instability that is normally suppressed by checkpoint controls. In normally cycling cells, tRNA genes have been shown to contribute to recombination at fragile sites (ref. Citation33 and unpublished observations of Jones H and Weinert T; reviewed in ref. Citation1). And the breakpoints of segmental duplications which arise by a replication-dependent recombination often correspond to tRNA genes.Citation34 Collectively, these recombination events likely have a strong deleterious effect on fitness. For example, recombination at fragile sites can lead to chromosome loss, and segmental duplications can disrupt gene dosage.
It is not known how tRNA genes affect recombination, but the available evidence points to a role for replication interference by the RNAP III PIC. Breakage at a site that includes a tRNA gene is elevated by treatment with hydroxyurea (HU), which causes fork movement to slow down by depleting cells of deoxyribonucleoside triphosphates (dNTPs).Citation33 Encounters between forks and active tRNA genes are also associated with an increased frequency of chromosome breakage at tRNA genes in a replication protein mutant (RRM3-null strain discussed below).Citation22,Citation35 Collectively, these results suggest a model in which replication interference by the RNAP III PIC can underlie spontaneous chromosome breakage (reviewed in ref. Citation33 and Citation36).
Overall, it seems likely that fork interference by tRNA genes has good and bad effects on genome stability. How has fork pausing at tRNA genes been preserved during evolution? On the one hand, its positive effects on fitness may simply outweigh the negative. Alternatively, the potential of active tRNA genes to cause replication interference might be protected from negative selection pressure because pausing by tRNA genes is a regulated process.
Replisome-Associated Proteins Regulate Fork Pausing at tRNA Genes
The discovery that tRNA genes can cause fork pausing led to studies aimed at understanding how this process is modulated by protein components of the replication machinery. So far two proteins, Rrm3 and Tof1, found in the replisome have been implicated in the control of pausing at tRNA genes in budding yeast (). The Zakian lab showed that the DNA helicase Rrm3 has a specialized role in promoting fork progression through tRNA genes.Citation22,Citation36,Citation37 In cells lacking Rrm3, forks that move into the front end of tRNA gene pre-initiation complexes pause for longer than normal, and fork pausing becomes apparent even at co-directional tRNA genes at which fork pausing cannot normally be detected (by 2D gel electrophoresis) in wild-type cellsCitation22 (). The latter finding suggested to Ivessa et al. that tRNA genes are normally difficult to replicate regardless of their orientation, which we now know to be the case.Citation24 It is not yet clear how Rrm3 promotes fork movement through active tRNA genes. One possibility is that Rrm3 removes components of the tRNA gene transcription machinery from DNA.Citation37 Arguing against this possibility is the evidence that tRNA gene transcription is not induced in RRM3-null mutants (ref. Citation30 and our unpublished data).
Deepak Bastia and colleagues have shown that another fork-associated protein, the Tof1 subunit of the RPC, functions in opposition to Rrm3. That is, Tof1 helps to impose the pause that is counteracted by Rrm3.Citation38 Since the biochemical function of Tof1 is unknown, the mechanism by which it promotes pausing at tRNA genes remains a matter of speculation.Citation38,Citation39
There is no evidence that the ability of Rrm3 or Tof1 to modulate fork progression through tRNA genes is subject to regulation by signaling systems that monitor or control the replication process. Therefore, although Rrm3 and Tof1 might collaborate to set the rate of replisome progression through active tRNA genes, the cell may not be able to control this rate by signaling mechanisms that impinge on Rrm3 or Tof1.
tRNA Gene Transcription is Actively Repressed by Genotoxic Stress Signals
Might regulators of tRNA gene transcription affect fork pausing at tRNA genes? In hindsight, this possibility was suggested several years ago by the discovery that tRNA gene transcription in yeast is repressed under conditions of genotoxic stress, specifically exposure to ultraviolet light and methane methylsulfonate (MMS).Citation40 Inhibition of tRNA gene transcription by MMS involves active repression of TFIIIB, partly by a mechanism that impinges one of its components, the protein kinase CK2. Constitutive CK2 activity is required for the full activity of TFIIIB. When cells are exposed to MMS, the catalytic subunit of CK2 dissociates from TFIIIB and TFIIIB activity declines. CK2 is also associated with TFIIIB in human, and downregulation of TFIIIB accounts for MMS repression of tRNA gene transcription in human cells.Citation41,Citation42 In yeast and human, Maf1 is also necessary for tRNA gene repression by MMS.Citation9,Citation43 Collectively, the data in yeast and human suggest that there is a conserved pathway for repression of tRNA gene transcription in response to genotoxic stress.
Checkpoint signal transduction pathways trigger many critical cellular responses to the appearance of abnormal chromosome structures in the nucleus. Checkpoint pathways are built around three classes of protein: upstream protein kinases that sense chromosome abnormalities, non-enzymatic kinase adaptors and transducer kinases which drive downstream responses to chromosome stress.Citation44–Citation46 The pathways come in two basic flavors. Chromosome double strand breaks (DSBs) activate ‘DNA damage’ checkpoint signaling, which involves the sensor kinases Mec1 and Tel1, Rad9 as the adaptor, and transducer kinases Rad53 and Chk1. Interference with DNA replication activates the “replication stress” checkpoint signaling pathway by a mechanism that does not require generation of a DSB. The replication stress pathway is comprised of Mec1, the adaptor Mrc1, Rad53 and the effector protein kinase Dun1. Mec1, Rad53 and Dun1 all modulate cellular processes that promote genome stability.
When repression of tRNA gene transcription by UV light and MMS was reported in 2001,Citation40 these genotoxins were thought to activate chromosome structure checkpoint signaling mainly by causing DSBs. Furthermore, CK2 had been implicated in the adaptation response to DSBs in yeast (in which cells eventually resume cell cycle progression despite the existence of an irreparably broken chromosome).Citation47 Accordingly, it was proposed that tRNA gene repression in UV/MMS-treated cells is a response to DSBs generated outside of replication forks and, therefore, controlled by the DNA damage checkpoint pathway that can control tRNA gene transcription.Citation48 Generally speaking this seemed a very plausible idea, since DNA damage checkpoint signaling was known to control the transcriptional response of various mRNA genes to ionizing radiation.Citation49 Well-characterized checkpoint signaling molecules have now been implicated in the control of tRNA gene transcription. Surprisingly, this group of proteins does not include molecules known to function preferentially in DNA damage checkpoint signaling. Rather, it is comprised of core components of the replication stress checkpoint pathway.
Replication Stress Checkpoint Proteins Control tRNA Gene Transcription in Normally Cycling Cells
The connections between tRNA gene transcription and the replication stress signal transduction pathway were recently described by Nguyen et al.Citation50 Checkpoint genes with a role in repression of tRNA gene transcription were identified by screening selected mutants for their capacity to repress the tRNA genes in response to MMS. The expectation was that repression would be dampened in cells lacking critical components of the checkpoint pathway that triggers repression.
In this study, the necessary control experiment comparing transcription in untreated wild-type and mutant cells revealed a completely unanticipated phenomenon: tRNA gene transcription is induced in some checkpoint mutants during S phase of normal proliferation. Specifically, the tRNA genes were induced in MEC1, MRC1, RAD53 and DUN1 mutants. Since tRNA gene expression in a RAD53-null mutant was inhibited by wild-type but not a kinase-dead Rad53, it is likely that Rad53 is in a signaling pathway that controls tRNA gene expression during unchallenged proliferation. Deletion of TEL1 and RAD9, which encode components of the DNA damage checkpoint pathway, did not affect tRNA gene transcription in normally cycling cells. Based on these results, Nguyen et al. proposed that tRNA gene transcription during normal proliferation is limited by a replication stress checkpoint signaling pathway ().Citation50
The idea that individual replication stress checkpoint proteins are active in normally cycling cells is now well supported by experimental evidence. During unchallenged proliferation, Mec1 phosphorylates serine 129 of histone H2ACitation51 and the Ddc1 subunit of a checkpoint DNA clamp;Citation52 it also promotes fork progression through some “difficult-to-replicate” regions of chromosomes.Citation53 Mrc1 associated with the RPC promotes fork progression (reviewed in ref. Citation46), and a subpopulation of Rad53 molecules is activated.Citation54 Collectively, these results suggest that a replication stress signaling pathway is active in normally cycling cells. And, although signaling by this pathway does not reach a threshold that restrains cell cycle progression, it does set a limit to the rate of tRNA gene transcription.
This is a novel form of regulation amongst yeast genes that respond to structural abnormalities in DNA. That is, while transcription of numerous protein-coding genes is known to be regulated by checkpoint signaling under conditions of genotoxic stress, none of these is controlled by checkpoint signaling during normal cycling.Citation49 Perhaps tRNA genes have special properties that warrant their regulation by replication stress proteins during normal cycling? The obvious possibility is that tRNA genes receive special attention from the replication stress checkpoint pathway because of their capacity to modulate replication fork progression.
The molecular mechanism of repression of tRNA gene transcription by replication stress checkpoint proteins in normally cycling cells remains unknown. Numerous cellular stresses can trigger repression of tRNA gene transcription and, typically, this repression depends on Maf1. Maf1 does not, however, mediate repression by replication stress checkpoint signaling during unchallenged proliferation. Nguyen et al. therefore proposed that replication stress signals directly control a Maf1-independent step in the RNAP III transcription cycle.Citation50 One possibility is suggested by recent work from the Willis lab on “facilitated recycling” of RNAP III. Facilitated recycling promotes reinitiation of RNAP III on the gene it has just transcribed by coupling transcription termination to reinitiation. This process contributes to the high rate of tRNA gene transcription in yeast and human and is not subject to regulation by Maf1 in vitro.Citation55 A step in facilitated recycling therefore might be controlled by replication stress checkpoint proteins in normally cycling cells. An alternative possibility is that tRNA genes are passively repressed during normal S phase by checkpoint-activated recombination proteins that are recruited to them when fork stress reaches a critical threshold. We consider this scenario unlikely because deletion of RRM3 stimulates recombination at tRNA genes but does not affect transcription.Citation30
Replication Stress Checkpoint Proteins Control tRNA Gene Transcription in Genotoxin-Treated Cells
Normally cycling cells exploit the replication stress pathway rather than the DNA damage pathway to control tRNA gene transcription. Does this mean that exogenous replication inhibitors, but not direct inducers of DSBs, can trigger signaling that results in repression of the tRNA genes? Indeed, that seems to be the case.Citation50 Yeast cells do not repress tRNA gene transcription when DSBs are directly induced by either γ-irradiation or a radiomimetic drug. tRNA gene transcription however is repressed when replication is blocked in MMS- and HU-treated cells. Chromatin immunoprecipitation analysis of cross-linking to tRNA genes revealed that repression by HU has all the hallmarks of repression under other conditions of stress, namely reduced association of RNAP III and TFIIIB subunits and increased association of TFIIIC and the Maf1 repressor.Citation50
The idea that replication stress caused by MMS and HU is a trigger for tRNA gene repression was reinforced by the finding that core components of the replication stress signaling pathway—Mec1, Mrc1, Rad53 and Dun1—are required for comprehensive inhibition of tRNA gene transcription by both reagentsCitation50 (). Deletion of RAD9 had no effect on repression by MMS and HU, which is again consistent with the notion that the DNA damage checkpoint pathway is not important for tRNA gene regulation.
The involvement of Dun1 in signaling to the tRNA genes under conditions of replication stress is not surprising in view of the fact that it is necessary for replication checkpoint induction of some mRNA genes.Citation56 Interestingly, Dun1 does not appear to have a role in the regulation of the tRNA genes during normal proliferation. This observation suggests that replication stress control of the tRNA genes, although built around a Mec1 → (Mrc1) → Rad53 pathway, does not involve exactly the same mechanism in normally cycling and genotoxin-treated cells.
Tel1 is implicated in replication stress control of tRNA gene transcription by the finding that repression by HU and MMS is dampened in a mec1Δ tel1Δ. double mutant, but not in either single mutant.Citation50 How Tel1 contributes to tRNA gene regulation is not clear, since it has not been implicated in replication stress checkpoint signaling. One possibility is that in HU- and MMS-treated cells, the formation of DSBs following fork collapse engages a Tel1 → (Rad9) → Rad53 pathway that represses the tRNA genes. The problem with this model is that deletion of RAD9 has no effect on tRNA gene transcription in normally cycling or HU/MMS-treated cells.Citation50 Therefore, we speculate that Tel1 is only involved in repression of the tRNA genes by HU or MMS when Mec1 function is compromised. Tel1-dependent repression of the tRNA genes, for example, might only occur in MEC1 mutants faced with a high burden of replication interference. Activation of Tel1 at tRNA genes is highly plausible. Indeed, Tel1 is localized to tRNA genes in untreated and HU-treated wild-type cells and is weakly active at these genes in a MEC1-null mutant during normally cycling.Citation51,Citation57,Citation58
Checkpoint Control of the Maf1 Repressor in Genotoxin-Treated Cells
The transcription proteins that directly receive inputs from the replication checkpoint pathway in normally cycling cells remain uncharacterized. It is clear, however, that checkpoint repression in cells treated with HU is achieved by mechanisms that directly involve Maf1.Citation50 Specifically, repression in HU is accompanied by Maf1 dephosphorylation and recruitment to tRNA genes, and repression of transcription and Maf1 dephosphorylation both partly depend on Rad53. Collectively, these results lead to a model in which the regulation of Maf1 by Rad53 (and, presumably, upstream components of the replication stress checkpoint pathway) is critical for tRNA gene repression when replication is perturbed by an exogenous reagent (). It is not surprising that repression by HU involves Maf1, since numerous other cellular stresses trigger repression of tRNA gene transcription by a mechanism that depends on Maf1.Citation9
Putting together what has been discovered so far, we suggest a model in which a signaling module comprised of Mec1, Mrc1 and Rad53 contributes to repression of the tRNA genes in normally cycling cells and cells treated with genotoxins that perturb fork movement. When the checkpoint system is fully engaged in genotoxin-treated wild-type cells, repressive signaling to the tRNA genes also involves Dun1 and Maf1 ().
The Nature of Signal Initiation in the Checkpoint Pathway that Represses the tRNA Genes
In the canonical replication stress checkpoint pathway, the chromosome abnormalities that trigger signaling in cells treated with HU or MMS are sensed by Mec1.Citation44,Citation59 Mec1 contributes to tRNA gene repression in HU- and MMS-treated cells.Citation50 Therefore, the known mechanisms of signal initiation at lesions caused by HU and MMS likely trigger signaling that represses tRNA gene transcription, as follows. In HU-treated cells, starvation for dNTPs inhibits replication elongation by a mechanism that generates about 100 nucleotides excess of single strand DNA (ssDNA) at stalled forks. It is the appearance of this excess ssDNA that leads to Mec1 activation.Citation60 In MMS-treated cells, stalling is due to physical interference with replisome progression by a DNA lesion, rather than depletion of dNTPs; none the less, Mec1 activation also depends on the appearance of excess single strand DNA at stalled forks.
Therefore, the appearance of excess ssDNA likely triggers the checkpoint signaling that impinges on the tRNA gene transcriptional machinery. Is chemical interference with fork progression (template DNA modification or starvation for dNTPs) the only mechanism by which this excess ssDNA can be generated? More specifically, is it possible that normal replication of tRNA genes can result in the formation of ssDNA that elicits checkpoint signaling? Several lines of evidence are consistent with this possibility. As described under “Replication interference by tRNA genes during normal S phase,” active tRNA genes themselves interfere with fork movement in the course of unchallenged proliferation. Importantly, Szilard et al. recently demonstrated that tRNA genes are sites of accumulation of serine 129-phosphorylated H2A (γ-H2A) during unchallenged proliferation, and that serine 129 phosphorylation of H2A at tRNA genes heavily depends on Mec1 and replication.Citation51 Since other chromosome structure abnormalities that result in the appearance of excess ssDNA (for example, DSBs) trigger the appearance of γ-H2A, it seems possible that tRNA genes are sites at which Mec1 is normally activated by excess ssDNA generated in the course of their replication. Finally, Mec1 represses the tRNA genes during unchallenged cycling.Citation50 Collectively, these findings support a working model in which the “abnormal” structures formed by normal fork pausing at tRNA genes in budding yeast can locally activate Mec1 for repression of tRNA gene transcription. Pausing at tRNA genes and enrichment of γ-H2A are less prominent features of normal replication in fission yeast than in budding yeast.Citation26,Citation61 These findings could reflect more rapid checkpoint-dependent PIC disassembly in fission yeast (if indeed it occurs in this organism) than in budding yeast.
The Spatial Range of Checkpoint Signals that Repress tRNA Gene Transcription
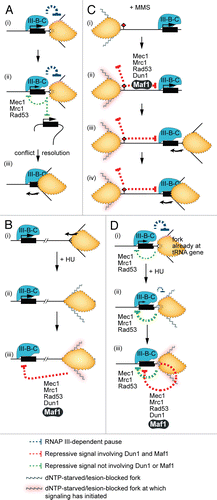
The available evidence is consistent with the existence of a cis-acting signaling mechanism for checkpoint repression of the tRNA genes. Several observations suggest that the tRNA genes might also be subject to transrepression by the replication stress checkpoint signaling pathway. First, fork stalling at early replication origins is known to generate signals that can inhibit firing of late origins. In other words, checkpoint signals can reach origins at which there is no chromosome abnormality. Second, the possible spatial barrier to signaling between tRNA genes is reduced by their clustering in the nucleolus.Citation62 Putting together what is known about signal initiation and the spatial range of checkpoint signaling leads to the working models shown in . We propose that normal pausing at tRNA genes is associated with molecular transactions that generate excess ssDNA; recognition of this ssDNA as ‘damage’ is the first step in activation of replication stress signaling (). A tRNA gene that gives rise to a checkpoint signal would be repressed by that signal, and weakly signal repression in trans. The latter repression of transcription would reduce the likelihood of fork pausing at trans-targets that have not already been replicated. Repression of an individual tRNA gene in an HU/MMS-treated cell could be due to trans signals from forks that have stalled far away from the gene ( and C show this signaling in HU and MMS, respectively). The local signal generated by a fork that was already naturally paused at a tRNA gene when the cell was exposed to genotoxin could also be additive with the signal caused by the effect of the genotoxin on that fork. Consider a tRNA gene at which a cis signal is partly inhibiting transcription and weakening the transcription-dependent pause during normal S phase []. Further fork perturbation owed to treatment with HU might strengthen the local Dun1/Maf1-independent signal for repression [] and trigger repressive signaling that also involves Dun1 and Maf1 []. Collectively, these events would intensify local repression and further dampen fork pausing.
Possible Advantages of tRNA Gene Repression in Genotoxin-treated Cells
In addition to asking mechanistic questions about checkpoint control of the tRNA genes in genotoxin-treated cells, it will be important to determine how this regulation is advantageous to the cell. The manner by which fitness is enhanced probably depends on the underlying cause of the replication stress and the extent of fork perturbation it has caused.
In HU-treated cells, repression of tRNA gene transcription could reduce the likelihood that a fork already stalled at a tRNA gene will collapse (the RNAP III-dependent component of fork perturbation would be lessened; ). We partly base this proposal on studies of chromosome instability reported by the Weinert lab. Admire et al.Citation33 showed that a fragment of ∼4,000 base pairs from chromosome VII of budding yeast confers chromosome instability during normal proliferation. Instability at this site, which contains two tRNA genes that cause fork pausing, is increased in MEC1 and MRC1 mutants. In our speculative model, this increase is at least partly due to failure to repress tRNA gene transcription (and therefore RNAP III loading). The fact that MAF1-null mutants are sensitive to HUCitation50,Citation63 lends further support to the model that repression of tRNA gene transcription increases genome stability.
In MMS-treated cells, tRNA gene repression could promote timely completion of replication. Some forks that have not stalled because of an encounter with a DNA lesion will progress until they meet a tRNA gene []. Completion of replication of DNA between such a gene and a downstream lesion will be accelerated if that tRNA gene has been repressed [].
In the scenarios outlined above, checkpoint-dependent fluctuations in tRNA gene transcription do not influence genome stability by affecting the amount of tRNA available for translation. We do not, however, rule out the possibility that tRNA gene induction and repression cause changes in translation that promote genome stability.
Broader Implications: tRNA Gene Transcription, Replication Stress and Oncogenesis
Abnormally high tRNA gene transcription and replication stress have been separately linked to oncogenesis. It has been clear for some time that the high rate of translation necessary for the aggressive proliferation phenotype of some cancer cell types is achieved partly by induction of tRNA gene transcription.Citation13 Furthermore, the increased capacity of cancer cells for tRNA synthesis can come about by mechanisms that involve tumor suppressors or proto-oncogenes which directly inhibit and activate (respectively) tRNA gene transcription.Citation64
Remarkably, several recent studies support the emerging view that there is a direct causal link between abnormally high tRNA gene transcription and oncogenesis.Citation65,Citation66 Immortalized fibroblasts can be transformed by overexpression of Brf1, a core subunit of TFIIIB (), or a single initiator methionine tRNA.Citation66 In such transformed cells, expression of c-Myc is elevated. Since c-Myc directly and indirectly stimulates RNAP III transcription,Citation65,Citation67 transformation by Brf1 and tRNAiMet is probably associated with pervasive induction of the tRNA genes.
Just as induction of tRNA gene transcription seems to be important for oncogenesis, so is, in some cases, interference with DNA replication. Specifically, there is increasing evidence that an early causal event in the evolution of some sporadic tumors is the development of replication stress. This evidence supports a model that proposes that the frequency of DNA DSBs in precancerous cells is elevated as a result of oncogene-induced collapse of replication forks.Citation68,Citation69 The genetic changes caused by such DSBs are proposed to lead to the chromosome instability phenotype of sporadic tumors.
It is not clear how oncogenes cause replication stress. However, several oncogenes which trigger replication stress, including Ras and c-Myc, are known to induce tRNA gene transcription.Citation12,Citation13 Building on this observation, we propose a speculative model that causally links misregulated tRNA gene transcription to replication-dependent genome instability in precancerous cells. Specifically, under the presumption that active tRNA genes inhibit fork progression in human, we suggest that elevated RNAP III transcription may be a contributing factor to oncogene-induced replication fork collapse in precancerous cells.
During the evolution of cells towards the fully transformed phenotype, the phase of DSB formation that depends on replication stress is followed by a period of development during which the replication stress signaling sensor ATR (Mec1 in yeast) and the tumor suppressor p53,Citation69 are often inactivated. p53 turns out to be a repressor of tRNA gene transcription,Citation70,Citation71 and there are suggestions that tRNA gene transcription is controlled by replication stress checkpoint signaling in human. First, the RNAP III transcriptional machinery, Maf1 and core components of the replication stress checkpoint pathway are conserved in human.Citation43–Citation45,Citation72–Citation74 Second, p53 is an effector of the replication stress checkpoint pathway.Citation75 And, finally, transcription of human tRNA genes is repressed by MMS.Citation42 If tRNA gene transcription during S phase is limited by the replication stress checkpoint pathway in human, then an extension of the model would be that a feed-forward cascade of genetic alterations that leads to increased tRNA gene transcription is a contributing factor to the development of genome instability during tumor formation.
In thinking about how elevated RNAP III transcription might promote oncogenesis, it is important to take into account the properties of the Alu elements derived from tRNA genes. There are more than a million Alu elements scattered throughout the human genome.Citation76 In the cell types so far examined, very few of these short interspersed elements nucleate the assembly of a RNAP III PIC.Citation77 It is intriguing nonetheless that their transcription by RNAP III is dependent on p53,Citation70,Citation71 and is induced in some tumor cells.Citation78,Citation79 Furthermore, Alu elements are important for some replication-dependent genome rearrangement events in preneoplastic lesions.Citation80 Therefore, along with the tRNA genes, the RNAP III-transcribed Alu elements warrant further attention as possible sources of replication stress in precancerous cells.
Figures and Tables
Figure 1 tRNA genes and the transcriptional machinery. (A) Alignment of three of ten wild-type budding yeast genes in the tL(CAA) tRNA gene family (also called the Leu3 family). The leucine anticodon is boxed and the intron is yellow. The genes are identical in their coding sequences. Also shown is the sequence of SUP53, which results from an A to T mutation (orange) in the anticodon of the wild-type tL(CAA)C gene. SUP53 is a leucine-inserting amber suppressor. (B) Sequence of tL(CAA)C showing the locations of the A and B boxes (blue), which are bound by TFIIIC. Transcription initiates at the arrow. The terminator is a run of five T residues (purple) just downstream of the coding region. (C) The core components of the tRNA gene transcriptional apparatus in budding yeast and their human counterparts (tabulated).Citation4–Citation6
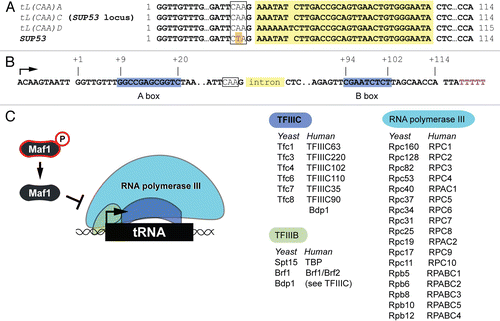
Figure 2 Replication interference by tRNA genes. The straight arrow shows the direction of transcription. The curved arrows show the direction of polymerase movement during nucleic acid synthesis. Curved T icons represent inhibitory effects on replication.
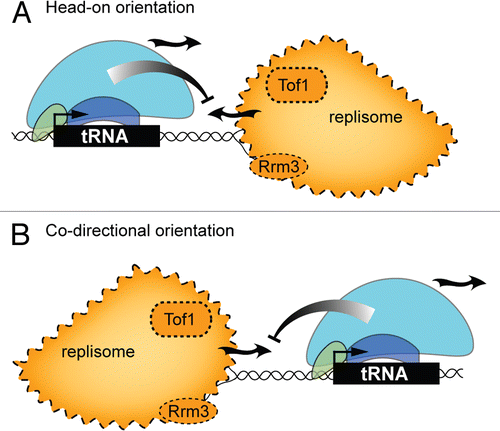
Figure 3 Control of tRNA gene transcription in yeast by replication stress checkpoint signaling molecules. In this model the checkpoint proteins are organized according to dependencies established in the literature. Dashed lines represent signaling steps that are yet to be fully described. ssDNA, single strand DNA.
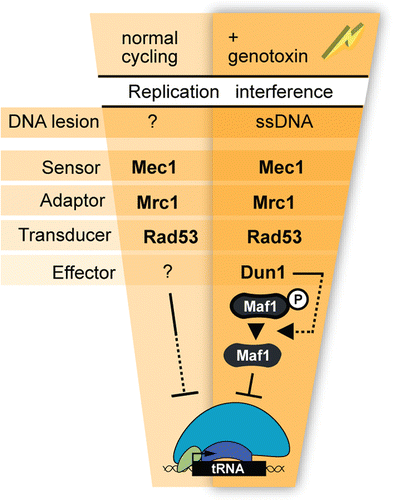
Figure 4 Mechanism of initiation and spatial range of checkpoint signaling to the tRNA genes. The models depict events that occur during normal cycling (A), and under conditions of replication stress induced by HU and MMS (B–D). The scenarios outlined include cis-signaling where a fork is paused at a tRNA gene (A-i and D), trans-signaling from a fork paused at a tRNA gene to another tRNA gene (A-ii), and trans-signaling from a stalled fork to a distal tRNA gene (B and C). The curved arrow represents the direction of fork movement. The PIC on tRNA genes is labeled ”III-B-C.”
Acknowledgements
Work on this topic has been supported by grants to M.C.S. from the National Cancer Institute of Canada (supported by the Canadian Cancer Society), the Canadian Institutes for Health Research, the Cancer Research Society and the Alberta Heritage Foundation for Medical Research. J. Cobb, R. McFarlane, V. Nguyen, P. Pasero, T. Weinert and R. Wellinger are acknowledged for helpful discussions.
References
- McFarlane RJ, Whitehall SK. tRNA genes in eukaryotic genome organization and reorganization. Cell Cycle 2009; 8:3102 - 3106
- Richard GF, Kerrest A, Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 2008; 72:686 - 727
- Marck C, Kachouri-Lafond R, Lafontaine I, Westhof E, Dujon B, Grosjean H. The RNA polymerase III-dependent family of genes in hemiascomycetes: comparative RNomics, decoding strategies, transcription and evolutionary implications. Nucleic Acids Res 2006; 34:1816 - 1835
- Hu P, Wu S, Sun Y, Yuan CC, Kobayashi R, Myers MP, et al. Characterization of human RNA polymerase III identifies orthologues for Saccharomyces cerevisiae RNA polymerase III subunits. Mol Cell Biol 2002; 22:8044 - 8055
- Mertens C, Roeder RG. Different functional modes of p300 in activation of RNA polymerase III transcription from chromatin templates. Mol Cell Biol 2008; 28:5764 - 5776
- Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev 2002; 16:2593 - 2620
- Huibregtse JM, Evans CF, Engelke DR. Comparison of tRNA gene transcription complexes formed in vitro and in nuclei. Mol Cell Biol 1987; 7:3212 - 3220
- Ciesla M, Boguta M. Regulation of RNA polymerase III transcription by Maf1 protein. Acta Biochim Pol 2008; 55:215 - 225
- Upadhya R, Lee J, Willis IM. Maf1 is an essential mediator of diverse signals that repress RNA polymerase III transcription. Mol Cell 2002; 10:1489 - 1494
- Johnson D. New connections identify Sch9 as a central node in ribosome biosynthesis. Cell Cycle 9:26 - 27
- Willis IM, Desai N, Upadhya R. Signaling repression of transcription by RNA polymerase III in yeast. Prog Nucleic Acid Res Mol Biol 2004; 77:323 - 353
- Johnson DL, Johnson SA. Cell biology: RNA metabolism and oncogenesis. Science 2008; 320:461 - 462
- White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol 2005; 6:69 - 78
- Gottesfeld JM, Wolf VJ, Dang T, Forbes DJ, Hartl P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science 1994; 263:81 - 84
- White RJ, Gottlieb TM, Downes CS, Jackson SP. Cell cycle regulation of RNA polymerase III transcription. Mol Cell Biol 1995; 15:6653 - 6662
- Donze D, Kamakaka RT. RNA polymerase III and RNA polymerase II promoter complexes are heterochromatin barriers in Saccharomyces cerevisiae. EMBO J 2001; 20:520 - 531
- Simms TA, Dugas SL, Gremillion JC, Ibos ME, Dandurand MN, Toliver TT, et al. TFIIIC binding sites function as both heterochromatin barriers and chromatin insulators in Saccharomyces cerevisiae. Eukaryot Cell 2008; 7:2078 - 2086
- Hull MW, Erickson J, Johnston M, Engelke DR. tRNA genes as transcriptional repressor elements. Mol Cell Biol 1994; 14:1266 - 1277
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science 1996; 272:1030 - 1033
- Labib K, Gambus A. A key role for the GINS complex at DNA replication forks. Trends Cell Biol 2007; 17:271 - 278
- Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol 2001; 310:1 - 26
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 2003; 12:1525 - 1536
- Bardeleben C, Kassavetis GA, Geiduschek EP. Encounters of Saccharomyces cerevisiae RNA polymerase III with its transcription factors during RNA chain elongation. J Mol Biol 1994; 235:1193 - 1205
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 2009; 34:722 - 734
- Sekedat MD, Fenyo D, Rogers RS, Tackett AJ, Aitchison JD, Chait BT. GINS motion reveals replication fork progression is remarkably uniform throughout the yeast genome. Mol Syst Biol 2010; 6:353
- Pryce DW, Ramayah S, Jaendling A, McFarlane RJ. Recombination at DNA replication fork barriers is not universal and is differentially regulated by Swi1. Proc Natl Acad Sci USA 2009; 106:4770 - 4775
- Brewer BJ, Fangman WL. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 1988; 55:637 - 643
- Linskens MH, Huberman JA. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol Cell Biol 1988; 8:4927 - 4935
- Rothstein R, Michel B, Gangloff S. Replication fork pausing and recombination or “gimme a break”. Genes Dev 2000; 14:1 - 10
- de la Loza MC, Wellinger RE, Aguilera A. Stimulation of direct-repeat recombination by RNA polymerase III transcription. DNA Repair (Amst) 2009; 8:620 - 626
- Giuliodori S, Percudani R, Braglia P, Ferrari R, Guffanti E, Ottonello S, et al. A composite upstream sequence motif potentiates tRNA gene transcription in yeast. J Mol Biol 2003; 333:1 - 20
- Ong WC, Ibrahim M, Town M, Johnson JD. Functional differences among the six Saccharomyces cerevisiae tRNATrp genes. Yeast 1997; 13:1357 - 1362
- Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, et al. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev 2006; 20:159 - 173
- Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet 2008; 4:1000175
- Bessler JB, Zakian VA. The amino terminus of the Saccharomyces cerevisiae DNA helicase Rrm3p modulates protein function altering replication and checkpoint activity. Genetics 2004; 168:1205 - 1218
- Azvolinsky A, Dunaway S, Torres JZ, Bessler JB, Zakian VA. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev 2006; 20:3104 - 3116
- Boule JB, Zakian VA. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res 2006; 34:4147 - 4153
- Mohanty BK, Bairwa NK, Bastia D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2006; 103:897 - 902
- Hodgson B, Calzada A, Labib K. Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol Biol Cell 2007; 18:3894 - 3902
- Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell 2001; 106:575 - 584
- Johnston IM, Allison SJ, Morton JP, Schramm L, Scott PH, White RJ. CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells. Mol Cell Biol 2002; 22:3757 - 3768
- Crighton D, Woiwode A, Zhang C, Mandavia N, Morton JP, Warnock LJ, et al. p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J 2003; 22:2810 - 2820
- Reina JH, Azzouz TN, Hernandez N. Maf1, a new player in the regulation of human RNA polymerase III transcription. PLoS One 2006; 1:e134
- Lambert S, Carr AM. Checkpoint responses to replication fork barriers. Biochimie 2005; 87:591 - 602
- Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys 2009; 53:17 - 31
- Friedel AM, Pike BL, Gasser SM. ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol 2009; 21:237 - 244
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097 - 1106
- Schultz MC. DNA damage regulation of the RNA components of the translational apparatus: new biology and mechanisms. IUBMB Life 2003; 55:243 - 247
- Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell 2001; 12:2987 - 3003
- Nguyen VC, Clelland BW, Hockman DJ, Kujat-Choy SL, Mewhort HE, Schultz MC. Replication stress checkpoint signaling controls tRNA gene transcription. Nat Struct Mol Biol 2010; 17:976 - 981
- Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, et al. Systematic identification of fragile sites via genome-wide location analysis of γ-H2AX. Nat Struct Mol Biol 2010; 17:299 - 305
- Paciotti V, Lucchini G, Plevani P, Longhese MP. Mec1p is essential for phosphorylation of the yeast DNA damage checkpoint protein Ddc1p, which physically interacts with Mec3p. EMBO J 1998; 17:4199 - 4209
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002; 297:602 - 606
- Kats ES, Albuquerque CP, Zhou H, Kolodner RD. Checkpoint functions are required for normal S-phase progression in Saccharomyces cerevisiae RCAF- and CAF-I-defective mutants. Proc Natl Acad Sci USA 2006; 103:3710 - 3715
- Cabart P, Lee J, Willis IM. Facilitated recycling protects human RNA polymerase III from repression by Maf1 in vitro. J Biol Chem 2008; 283:36108 - 36117
- Huang M, Elledge SJ. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol Cell Biol 1997; 17:6105 - 6113
- Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, et al. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr Biol 2008; 18:566 - 575
- Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, Wehr TS, et al. Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 2007; 130:499 - 511
- Tercero JA, Longhese MP, Diffley JF. A central role for DNA replication forks in checkpoint activation and response. Mol Cell 2003; 11:1323 - 1336
- Tourriere H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst) 2007; 6:900 - 913
- Rozenzhak S, Mejía-Ramírez E, Williams JS, Schaffer L, Hammond JA, Head SR, et al. Rad3ATR decorates critical chromosomal domains with γH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet 2010; 6:1001032
- Thompson M, Haeusler RA, Good PD, Engelke DR. Nucleolar clustering of dispersed tRNA genes. Science 2003; 302:1399 - 1401
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 2006; 126:611 - 625
- White RJ. RNA polymerase III transcription—a battleground for tumour suppressors and oncogenes. Eur J Cancer 2004; 40:21 - 27
- Johnson SA, Dubeau L, Johnson DL. Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J Biol Chem 2008; 283:19184 - 19191
- Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell 2008; 133:78 - 89
- Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci USA 2007; 104:14917 - 14922
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220 - 228
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352 - 1355
- Cairns CA, White RJ. p53 is a general repressor of RNA polymerase III transcription. EMBO J 1998; 17:3112 - 3123
- Chesnokov I, Chu WM, Botchan MR, Schmid CW. p53 inhibits RNA polymerase III-directed transcription in a promoter-dependent manner. Mol Cell Biol 1996; 16:7084 - 7088
- Huang Y, Maraia RJ. Comparison of the RNA polymerase III transcription machinery in Schizosaccharomyces pombe, Saccharomyces cerevisiae and human. Nucleic Acids Res 2001; 29:2675 - 2690
- Pluta K, Lefebvre O, Martin NC, Smagowicz WJ, Stanford DR, Ellis SR, et al. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol Cell Biol 2001; 21:5031 - 5040
- Johnson SS, Zhang C, Fromm J, Willis IM, Johnson DL. Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases. Mol Cell 2007; 26:367 - 379
- Demidova AR, Aau MY, Zhuang L, Yu Q. Dual regulation of Cdc25A by Chk1 and p53-ATF3 in DNA replication checkpoint control. J Biol Chem 2009; 284:4132 - 4139
- Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 2002; 35:97 - 112
- Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol 2010; 17:620 - 628
- Panning B, Smiley JR. Activation of expression of multiple subfamilies of human Alu elements by adenovirus type 5 and herpes simplex virus type 1. J Mol Biol 1995; 248:513 - 524
- Tang RB, Wang HY, Lu HY, Xiong J, Li HH, Qiu XH, et al. Increased level of polymerase III transcribed Alu RNA in hepatocellular carcinoma tissue. Mol Carcinog 2005; 42:93 - 96
- Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008; 27:3256 - 3264