Abstract
Since tetanus was first described by Hippocrates, the devastating diseases caused by pathogenic members of the Gram-positive, anaerobic sporeforming genus Clostridium have ranked among the most dreaded afflictions of humans and domestic animals. The quintessential hallmark of all clostridial diseases is the involvement of potent protein toxins. However, except for some foodborne botulism cases, clostridial diseases are not intoxications involving preformed toxins; rather, these illnesses are true infections involving toxin production by bacteria growing inside the host.
Introduction
Since tetanus was first described by Hippocrates, the devastating diseases caused by pathogenic members of the Gram-positive, anaerobic sporeforming genus Clostridium have ranked among the most dreaded afflictions of humans and domestic animals. The quintessential hallmark of all clostridial diseases is the involvement of potent protein toxins. However, except for some foodborne botulism cases, clostridial diseases are not intoxications involving preformed toxins; rather, these illnesses are true infections involving toxin production by bacteria growing inside the host.
Among the most important pathogenic clostridial species is Clostridium perfringens. This bacterium causes a spectrum of human and veterinary infections ranging from wound infections (most notably traumatic gas gangrene) to enteric disease and enterotoxemias. C. perfringens is a prolific bacterial toxin producer, reportedly producing >17 different toxins. However, individual isolates never produce this entire toxin arsenal, providing the basis for a widely-used classification scheme assigning C. perfringens isolates to one of five types (A–E) depending upon their production of four typing toxins ().
Each C. perfringens type is associated with particular diseases,Citation1 suggesting that certain toxins, or combinations of toxins, are important contributors to those diseases. However, since each C. perfringens isolate produces multiple toxins, it can only be conclusively determined whether a specific toxin(s) is important for virulence by fulfilling Molecular Koch's postulates.Citation2 This classical approach involves comparing, in appropriate animal models, the pathogenicity of a wild-type C. perfringens parent isolate vs. a series of isogenic toxin null mutants. For those toxin mutants exhibiting reduced pathogenicity, complementation to return normal toxin production should restore virulence.
By fulfilling Molecular Koch's postulates, it has now been established that C. perfringens type A isolates must produce α-toxin and, to a lesser extent, perfringolysin O (PFO) to cause traumatic gas gangrene in mice.Citation3–Citation5 Similar analyses demonstrated that expression of C. perfringens enterotoxin (CPE) is necessary for type A human gastrointestinal disease strains to induce intestinal pathology in rabbit ileal loops.Citation6 Recent progress in dissecting the virulence of C. perfringens type C isolates is described in the following section.
Clostridium perfringens Type C: Associated Diseases and Virulence
In humans, C. perfringens type C isolates cause enteritis necroticans, also known as pigbel or darmbrand.Citation1 This illness occurs in individuals with low intestinal trypsin activity due to diet or disease (trypsin is an important host defense mechanism against type C infection since this enzyme can inactivate toxins, particularly β toxin). Human enteritis necroticans involves intestinal necrosis, bloody diarrhea and abdominal pain and can also be swiftly fatal, particularly when toxemia occurs. This disease is endemic throughout Southeast Asia/Oceania, particularly Papua New Guinea, although some cases occur in developed countries, mainly in diabetics.
Type C isolates also cause economically-important disease in domestic animals, especially sheep.Citation1 These sheep infections involve sudden death or acute neurologic signs, which may be accompanied by bloody diarrhea. Therefore, type C infections of sheep are often enterotoxemias, where the associated neurologic signs result from toxins that are produced in the intestines but then absorbed into the circulation to affect internal organs such as the brain.
With respect to their virulence, type C isolates typically produce, at minimum, β-toxin, α-toxin and PFO.Citation7 In recent molecular Koch's postulates studies,Citation7 we assessed the contributions of these toxins to the pathogenicity of type C disease isolate CN3685 (). Single and double isogenic toxin null mutants of CN3685 were constructed using efficient Targetron® mutagenesis approaches newly developed for C. perfringens.Citation9 Each mutant was then virulence-tested in a rabbit ileal loop model where (in the presence of trypsin inhibitor, TI) wild-type CN3685 causes necrotizing enteritis. These experiments revealed that inactivating the cpb gene renders CN3685 avirulent for causing enteric disease, i.e., in contrast to wild-type CN3685, the cpb null mutant caused neither intestinal necrosis nor bloody fluid accumulation in rabbit ileal loops. Furthermore, complementing the cpb null mutant to restore β-toxin production significantly increased intestinal virulence, confirming that the isogenic cpb mutant had specifically lost virulence due to cpb inactivation. However, a CN3685 double mutant that did not produce α-toxin or PFO remained fully virulent in rabbit ileal loops. This studyCitation7 further determined that, in the presence of TI, purified β-toxin induces hemorrhagic necrosis in rabbit ileal loops. Collectively, these results indicated that β-toxin is necessary and sufficient for the enteric virulence of type C isolate CN3685 in rabbit ileal loops.
As mentioned, type C infections often involve enterotoxemias, where fatal internal organ damage results from systemically circulating toxins absorbed from the intestines. Therefore, the CN3685 toxin null mutants were also recently tested in a mouse intraduodenal challenge model that, in the presence of TI, reproduces the lethal aspects of type C enterotoxemias.Citation10 In this assay, wild-type CN3685 was nearly 100% lethal for mice, but an isogenic cpb null mutant was strongly attenuated for lethality. In contrast, an isogenic CN3685 double mutant unable to produce α-toxin or PFO showed only a small decrease in lethality. Together, the rabbit ileal loop and mouse lethality studies demonstrated that β-toxin is mainly responsible for the ability of type C isolate CN3685 to cause both necrotizing enteritis and lethal enterotoxemias.
Host Cell Contact Induces a Rapid Upregulation of Toxin Production by Type C Isolates
In 2006, we had determined that type C isolates growing in bacterial culture media begin producing β-toxin during mid-log phase growth, with peak β-toxin production occurring in the late-log/earlystationary growth phase.Citation7 However, since type C disease involves toxin production in the intestines, we recently examinedCitation11 whether contact with human enterocyte-like Caco-2 cells might affect the kinetics of toxin secretion or production by type C isolates.
These studies by Vidal et al.Citation11 revealed that β-toxin, PFO, α-toxin and beta2 toxin, but not TpeL toxin, accumulate very rapidly in culture supernatants of type C strains growing in tissue culture medium containing Caco-2 cells ( and data not shown). In contrast, there was little or no accumulation of those four toxins in supernatants of the same type C strains growing for similar timepoints in tissue culture medium (no Caco-2 cells) or bacterial media. This rapid host cell-induced toxin accumulation in culture supernatants did not involve host cells stimulating type C isolate growth or stimulating secretion of preformed toxins from type C isolates. Instead, RT-PCR analyses demonstrated that host cell contact stimulated a rapid onset of toxin gene transcription by these bacteria.
Pathogenic bacteria commonly sense environmental changes using two component regulatory systems and quorum sensing systems in order to optimize their production of virulence factors such as toxins. Earlier studiesCitation12–Citation14 had shown that, for C. perfringens type A strain 13 growing in bacterial media, expression of α-toxin and PFO is regulated by both the VirS/VirR two component regulatory system and the LuxS quorum sensing system. Therefore, we investigatedCitation11 whether those systems might also be involved in the host cell-induced upregulation of toxin production by type C isolates. After confirming the presence of virS/virR and the luxS genes in type C isolates, virR and luxS null mutants were prepared in type C isolate CN3685. While the luxS null mutant still rapidly upregulated toxin production in the presence of Caco-2 cells, the virR null mutant lost its responsiveness to the presence of host cells. Confirming that the VirS/VirR system is involved in rapid host cell-induced upregulation of toxin expression by type C isolates, complementation to restore VirR production allowed the virR mutant to regain the ability to upregulate toxin production in the presence of host cells.
Our recent studyCitation11 then explored the nature of the host cell signaling that causes type C isolates to rapidly produce toxins. Several other mammalian cells lines, in addition to Caco-2 cells, were able to induce the quick increase in toxin production by type C isolates. This rapid host cell-induced upregulation of toxin production was found to require close contact between the type C isolates and host cells, although tight adherence of the bacteria to the host cell surface did not occur. Pretreating Caco-2 cell cultures with trypsin or phospholipase C had no effect on the subsequent ability of host cells to signal type C strains to upregulate their toxin production. However, Pronase pretreatment of Caco-2 cells increased toxin expression levels following type C infection, suggesting proteins on the Caco-2 cell surface might partially mask the host cell signaling molecule or block secretion of the host signal.
Host Cell-Induced Upregulation of Toxin Production Increases the Cytotoxic Activity of Type C Isolates
Our recent studyCitation11 also asked whether the rapid host cell-induced upregulation of toxin production described above has cytotoxic consequences for Caco-2 cells. This experiment involved growing type C isolates for 1 h in tissue culture medium that did or did not contain Caco-2 cells. Sterile supernatants from those cultures were then separately applied to Caco-2 cell cultures. Demonstrating that host cell-induced upregulation of toxin production increases type C isolate cytotoxicity, extensive Caco-2 cell morphologic damage was only observed in wells receiving supernatants from type C cultures grown in the presence of Caco-2 cells.
Posssible Significance of these Findings for Understanding Type C Isolate Pathogenesis
Dissecting the virulence of type C isolate CN3685Citation3 has helped to explain why C. perfringens produces so many different toxins. While α-toxin and PFO are important contributors to histotoxic disease,Citation3–Citation5 it is now apparent that other toxins play the major role in C. perfringens enteric disease or enterotoxemias. Besides our type C isolate data, this conclusion is supported by, (1) an α-toxin mutant of a type A avian necrotic enteritis strain retaining virulence in a chicken intestinal disease model,Citation15 while a NetB toxin null mutant of that isolate was avirulentCitation16 and (2) CPE mutants of two type A strains, which still produced α-toxin and (for one strain) PFO, causing no pathologic effects in rabbit ileal loops.Citation5
Perhaps the most novel observation provided by the Vidal et al. studyCitation11 was, to our knowledge, the first direct evidence for host: Clostridium crosstalk that could influence pathogenesis. Specifically, our new observationsCitation11 indicate that contact between host cells and type C isolates causes signaling in C. perfringens that affects the kinetics of clostridial toxin gene transcription and production. This toxin upregulation could help to explain the rapid death associated with many type C infections.Citation1
Finally, the Vidal et al. studyCitation11 also showed that the rapid host cell-induced upregulation of toxin production involves some, but not all, systems previously implicated in regulation of toxin gene transcription by type A isolates growing in bacterial media. Notably, the same VirS/VirR two component regulatory system previously shown to be important for toxin production regulation by type A isolates growing in bacterial mediaCitation13,Citation14 has now also been implicated in type C isolate sensing the presence of host cells and responding by upregulating their toxin production. It remains to be determined why toxin production kinetics by type C isolates are so much faster in the presence of host cells. However, these findingsCitation11 imply some need for caution in extrapolating disease implications from toxin regulation results obtained from studies using C. perfringens growing in bacterial medium.
Questions for Future Research
Does contact with host cells upregulate toxin production by other C. perfringens strains or other clostridial spp.? Our recent studyCitation11 partially addressed this question by showing that some type D isolates exhibit Caco-2 cell-induced upregulation of epsilon toxin production. Whether C. perfringens type A, B and E isolates, or other clostridial species, show altered toxin production kinetics in the presence of host cells remains to be investigated.
How do type C isolates activate VirS/VirR during host cell-induced upregulation of toxin production? Two component regulatory systems often use quorum sensing systems to respond to environmental signals. While the LuxS quorum sensing system does not seem to be important for host cell-induced upregulation of toxin production, two studies recently showed that inactivating the agrB gene in C. perfringens type A strain 13 delays the onset of α-toxin and PFO production when this strain grows in bacterial media.Citation17,Citation18 Those findings open the possibility that the Agr quorum sensing system might also be involved in host-cell induced toxin upregulation by type C isolates.
What host signal activates early toxin gene transcription by type C isolates? The unknown host signal appears to be ubiquitously present in cultured cells. We observedCitation11 type C isolates upregulate their toxin production in the presence of cultured cells from several mammalian spp., and with different tissue origins (i.e., enterocyte-like cells, kidney cells and fibroblasts), suggesting that the signal is widely distributed among host cells. Whether the host cell signal is a labile secreted factor or a molecule present on the mammalian cell surface remains to be determined.
Does contact with host cells induce type C isolates to upregulate transcription of other potential virulence genes? Since the pathogenicity of type C isolates likely involves poorly studied nontoxin virulence factors such as adhesions or the capsule, studies should explore whether host cell contact also affects the transcription of potential nontoxin virulence genes.
Figures and Tables
Figure 1 Molecular Koch's postulates dissection of toxin contributions to the virulence of type C isolate CN3685 in rabbit ileal loops. In this model, wild-type CN3685 causes necrotizing enteritis, with an accumulation of bloody fluid inside the ileal loops. Similar pathology was observed for isogenic mutants unable to produce perfringolysin O (pfoA), α-toxin (plc), or lacking both perfringolysin O and α-toxin (plc/pfoA). In contrast, an isogenic mutant (cpb) unable to produce β-toxin was completely attenuated for virulence; this virulence was restored by complementation (not shown in ). also shows that purified β-toxin alone can reproduce the pathology caused by wild-type CN3685 infection. Modified and used with permission from Sayeed et al.Citation3
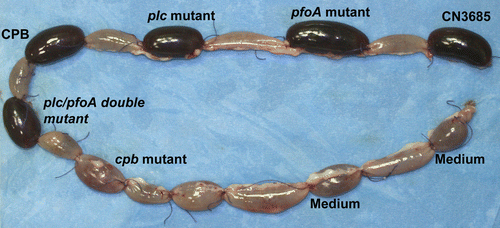
Figure 2 The presence of host cells affects the kinetics of β-toxin accumulation in supernatants of type C isolate JGS1495. Equal number of JGS1495 cells were inoculated into a tissue culture dish containing bacterial culture media [either fluid thioglycollate broth (FTG) or tryptic soy broth-glucoseyeast extract (TGY)] tissue culture medium [minimal essential medium (MEM)], or MEM containing Caco-2 enterocyte-like cells. After 3 h incubation at 37°C, each culture was harvested and the culture supernatant was subjected to western blotting using an anti-β-toxin monoclonal antibody. Used with permission from Vidal et al.Citation11
![Figure 2 The presence of host cells affects the kinetics of β-toxin accumulation in supernatants of type C isolate JGS1495. Equal number of JGS1495 cells were inoculated into a tissue culture dish containing bacterial culture media [either fluid thioglycollate broth (FTG) or tryptic soy broth-glucoseyeast extract (TGY)] tissue culture medium [minimal essential medium (MEM)], or MEM containing Caco-2 enterocyte-like cells. After 3 h incubation at 37°C, each culture was harvested and the culture supernatant was subjected to western blotting using an anti-β-toxin monoclonal antibody. Used with permission from Vidal et al.Citation11](/cms/asset/e6cd6588-4d34-4375-b540-96044c609fbb/kvir_a_10910679_f0002.gif)
Table 1 Toxintyping classification of C. perfringens isolates
References
- McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins T. Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E. The enterotoxic clostridia. The prokaryotes 2004; New York Springer 698 - 752
- Falkow S. Molecular Koch's postulates applied to microbial pathogenicity. Rev Infect Dis 1988; 10:5274 - 5276
- Awad MM, Bryant AE, Stevens DL, Rood JI. Virulence studies of chromosomal alpha-toxin and theta toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol 1995; 15:191 - 202
- Awad MM, Ellemor DM, Boyd RL, Emmins JJ, Rood JI. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect Immun 2001; 69:7904 - 7910
- Hickey MJ, Kwan RYQ, Awad MM, Kennedy CL, Young LF, Hall P, et al. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathogens 2008; 4:1000045
- Sarker MR, Carman RJ, McClane BA. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol Microbiol 1999; 33:946 - 958
- Fisher DJ, Fernandez-Miyakawa MF, Sayeed S, Poon R, Adams V, Rood JI, et al. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect Immun 2006; 74:5200 - 5210
- Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, et al. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol 2008; 67:15 - 30
- Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl Environ Microbiol 2005; 71:754207
- Uzal F, Saputo J, Sayeed S, Vidal JE, Fisher DJ, Poon R, et al. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C enterotoxemias. Infect Immun 2009; 77:5291 - 5299
- Vidal JE, Ohtani K, Shimizu T, McClane BA. Contact with enterocyte-like Caco-2 cells induces rapid upregulation of toxin production by Clostridium perfringens type C isolates. Cell Microbiol 2009; 11:1306 - 1328
- Ohtani K, Hayashi H, Shimizu T. The luxS gene is involved in cell-cell signaling for toxin production in Clostridium perfringens. Mol Microbiol 2002; 44:171 - 179
- Lyristis M, Bryant AE, Sloan J, Awad MM, Nisbet IT, Steven DL, Rood JI. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol Microbiol 1994; 12:761 - 777
- Shimizu T, Ba-Thien W, Tamaki M, Hayashi H. The virR gene, a member of a class of two component response regulators, regulates the production of perfringolysin O, collagenase and hemagglutinin in Clostridium perfringens. J Bacteriol 1994; 176:1616 - 1623
- Keyburn L, Sheedy SA, Ford ME, Williamson MM, Awad MM, Rood JI, Moore RJ. Alpha-toxin of Clostridium perfringens is not an essential virulence factor in necrotic enteritis in chickens. Infect Immun 2006; 74:6496 - 6500
- Keyburn AL, Boyce JD, Vaz P, Bannam TL, Ford ME, Parker D, et al. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathogens 2008; 8:26
- Ohtani K, Yuan Y, Hassan S, Wang R, Wang Y, Shimizu T. Virulence gene regulation by the agr system in Clostridium perfringens. J Bacteriol 2009; 191:3919 - 3927
- Vidal JE, Chen J, Li J, McClane BA. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS One 2009; 4:6232