Abstract
Type I and II interferons (IFNs αβ and γ) have opposing effects on immune resistance to certain pathogenic bacteria. While IFNγ generally plays a protective role, IFNαβ exacerbates Listeria monocytogenes and Mycobacterium tuberculosis infections. Our findings provided evidence that this increased susceptibility reflects a novel antagonistic cross talk between IFNαβ and IFNγ. Macrophages infected with L. monocytogenes strains that induce IFNαβ production responded poorly to IFNγ, as measured by reduced phosphorylation of STAT1 and reduced IFNγ-dependent gene expression. The impaired responsiveness to IFNγ correlated with reduced expression of its receptor, IFNGR, by both infected and bystander macrophages. Down regulation of IFNGR was dependent on responsiveness to IFNγ and mimicked by recombinant IFNβ. Mice lacking responsiveness to IFNαβ (IFNAR1-/-) retained high IFNGR expression, developed higher expression of MHC-II on macrophages and DCs, and were more resistant to systemic L. monocytogenes infection - but only in the presence of IFNγ. Thus, the ability of IFNαβ to down regulate IFNGR provides an explanation for its ability to reduce responsiveness to IFNγ and to increase host susceptibility to bacterial infection. It remains to be determined whether and how such antagonistic interferon crosstalk benefits the host.
Recognition of microbial products by mammalian Toll like, Nod-like and other receptors leads to the rapid synthesis and secretion of diverse immune regulatory cytokines and chemokines. Amongst the cytokines produced at early times after intracellular bacterial infection are the type I and II interferons IFNαβ and IFNγ. IFNαβ and IFNγ regulate the activation of macrophages, DCs, natural killer (NK) cells and T cells. The interferons also influence chemokine production by immune and somatic cell types and thus the recruitment of neutrophils and other immune and inflammatory cells to sites of infection. IFNγ plays a crucial role in immunity to a variety of intracellular parasites, which is consistent with its known roles in activating macrophage anti-bacterial effector mechanisms and promoting DC and Th1-type T-cell activation. Mice lacking responsiveness to IFNγ fail to contain systemic infection by numerous pathogens, including the intracellular bacteria L. monocytogenes and M. tuberculosis.Citation1–Citation4
Cellular responsiveness to IFNγ requires the expression of a functional heterodimeric cell surface receptor complex, IFNGR. The proteins comprising the IFNGR complex, IFNGR1 and IFNGR2, are products of distinct genes that reside on different chromosomes in both mouse and man. The dimeric IFNγ cytokine binds to the IFNGR1 subunit. Aggregation of IFNγ-IFNGR1-IFNGR2 complex triggers the activation of a canonical signaling cascade involving Janus kinases (JAKs) 1 and 2 and the signal transducer and activator of transcription (STAT) 1 protein (). Since both receptor subunits are required for the activation of signaling cascades, mice or human cells with deficiency in either subunit are non-responsive to IFNγ.Citation1,Citation5–Citation8 Polymorphisms in the ifngr1 promoter region are also associated with susceptibility or resistance to certain infectious and inflammatory diseases, suggesting that the level or pattern of ifngr1 expression may have important effects on the immune response.Citation9–Citation11 IFNαβ bind to a distinct receptor, IFNAR, comprised of the IFNAR1 and IFNAR2 subunits. Signaling through the IFNAR elicits activation of JAK1 and TYK2, which subsequently activate STAT1 and STAT2. In response to both IFNγ and IFNαβ, complexes of STAT proteins undergo dimerization and post-transcriptional modifications that activate their ability to enter the cell nucleus, bind specific elements on DNA and regulate the expression of target genes.
Type I interferon production is regulated at the transcriptional level and transcription is induced during infections by a variety of viral and bacterial pathogens. Transcription of IFNαβ genes can be induced by recognition of microbial products through toll like receptors (TLRs) or via other pattern recognition receptors (PRRs) present in the host cell cytosol. For L. monocytogenes and M. tuberculosis, data suggest that the presence of bacteria or bacterial products in the host cell cytosol is a requirement for IFNαβ production. However, until recently, the ligands and receptors contributing to the IFNαβ production in response to these pathogens has remained elusive. Several PRRs were found to be dispensible for type I IFN production in response to L. monocytogenes infection, including TLRs 2 and 4, Nods 1 and 2, RIG-I and MAVS/VISA. Activation of IFNαβ production by M. tuberculosis was also found to be TLR-independent.Citation12,Citation13 With regards to additional bacterial components required for in the triggering of IFNαβ production, work with M. tuberculosis has implicated the early secreted antigen 6 kilodaltons (Esat-6) secretion system 1 (ESX-1).Citation13 The ESX1 system can disrupt phagosomal membranes and is thus thought to allow release of bacterial components into the cytosol where they may trigger host receptors to initiate IFNαβ production. Similarly, multidrug efflux pumps (MDRs) potentially allow cytosolic release of small molecules essential for type I interferon induction by L. monocytogenes infection.Citation14 What is released by these secretion systems has remained more elusive. Work by Stetson and Medzhitov indicated that transfection of L. monocytogenes DNA into the cytosol of host cells could trigger IFNαβ production independently of TLRs and NODs1 and 2.Citation15 However, it is not clear to what extent bacterial DNA actually accesses the cytosol during infection. It is also unclear how the host cell senses such cytosolic DNA and distinguishes this from cellular DNA. More recently, McWhirter et al. found that cyclic-di-guanosine monophosphate (c-di-GMP), a low molecular weight bacterial second messenger, can trigger IFNαβ production.Citation16 Woodward et al. subsequently showed that cyclic diadenosine monophosphate (c-di-AMP) also induces IFNαβ.Citation17 Mutant L. monocytogenes strains that overexpress MDRs release higher amounts of c-di-AMP. Whether the M. tuberculosis ESX1 secretion system also triggers IFNαβ by permitting appropriate release of cGMP or cAMP remains to be seen.
In contrast to IFNγ, IFNαβ is not required for defense against a growing list of bacterial pathogens. IFNAR1-/- mice, which lack expression of the receptor for IFNαβ, are more resistant to L. monocytogenes and M. tuberculosis infections.Citation13,Citation18–Citation22 The mechanisms by which IFNαβ increase host susceptibility to L. monocytogenes, M. tuberculosis and other bacterial pathogens are not entirely clear. However, several correlations have been made. For example, during L. monocytogenes infection IFNαβ production and responsiveness correlate with: (1) increased serum or splenic concentrations of IFNγ, MCP-1 and IL-6 at 1–3 dpi, reduced serum concentrations of IL-12p70 at 2 dpi and a reduced proportion of splenic TNFα + CD11b+ splenic macrophages at 2 dpi.Citation20,Citation21,Citation23,Citation24 (2) increased apoptosis of lymphocytes,Citation19 and macrophages.Citation20 Spleens of the infected IFNAR1-/- mice were also shown to harbor ∼2-fold more splenic macrophages and neutrophils and a higher percentage of TNFα + CD11b+ cells.Citation20,Citation21 Such increases in myeloid cells may reflect reduced apoptosis or increased cellular recruitment in the IFNAR1-/- animals. With regards to lymphocyte apoptosis, Unanue and colleagues later showed that as for IFNAR1-/- animals, Rag-/- and IL-10-/- hosts were more resistant than wildtype hosts at early stages of L. monocytogenes infection.Citation24 They also showed that IL-10 production was increased in the spleens of infected wildtype mice when compared to IFNAR1-/- or Rag-/- mice. Although this increase in IL-10 correlated with increased lymphocyte apoptosis, serum IFNγ levels were also higher in the wildtype animals at days 1–3 post-infection.Citation20,Citation21 Thus, it was not clear that the increased IL-10 seen in the IFNAR1-/- mice was sufficient to functionally suppress IFNγ production or activity. Nonetheless, the available data hinted that the ability of IFNαβ to promote L. monocytogenes replication might be associated with impaired cellular immune responses, including macrophage activation.
With this previous work in mind, our study initially investigated whether infection with L. monocytogenes suppressed the ability of macrophages to respond to IFNγ treatment.Citation25 We were curious whether such suppression might occur during L. monocytogenes infection, in part because of prior work by Ernst, Harding and colleagues with M. tuberculosis.Citation26–Citation29 They showed that M. tuberculosis infection suppressed macrophage upregulation of IFNγ-inducible gene products such as CIITA and class II MHC. We also initially investigated upregulation of MHC-II expression by infected or uninfected bone marrow derived macrophages (BMM) in response to stimulation with IFNγ. Similar to M. tuberculosis, wildtype L. monocytogenes infection suppressed MHC-II upregulation. Using stably transfected RAW264.7 macrophages, we next used luciferase reporter constructs to measure transcription from IFNγ-inducible the c2ta-p4 promoter and a GAS reporter construct. In both cases, and using multiple independent cell lines, infection by wild type bacteria suppressed the induction of gene expression by IFNγ. Moreover, when we investigated earlier events in the signaling pathway, phosphorylated STAT1 levels following IFNγ treatment were diminished in infected cells. These data suggested that L. monocytogenes infection suppressed the IFNγ signaling pathway at a very early stage. With regards to the bacterial requirements for suppression, we noted that cytosolic escape of L. monocytogenes was essential for the reduced responsiveness to IFNγ. Infection of macrophages with Hly-deficient L. monocytogenes mutant strains, which are unable to escape the phagosome, failed to dampen IFNγ responses.
To learn which upstream host factors were affected by cytosolic L. monocytogenes infection, we evaluated the expression of genes known to be involved in IFNγ responses using Affymetrix gene chips. The array analyses and subsequent quantitative PCR revealed that ifngr1 gene expression was significantly reduced in wt Lm infected cells within a few hours of infection. Antibody staining revealed that cell surface and total cellular IFNGR1 protein levels correlated with the gene expression data. Furthermore, cell surface staining for IFNGR2 was also reduced, demonstrating that the entire IFNGR complex is downregulated in response to infection of cultured macrophages with L. monocytogenes. We subsequently confirmed these in vitro data using systemic L. monocytogenes infection of mice. These in vivo studies showed that IFNGR1 surface expression was significantly reduced within 1 dpi in macrophages, DCs and B cells, but not T cells or NK cells. Since downregulation of IFNGR affected the entire population of susceptible cells, we suspected that infected cells might release a soluble factor to mediate downregulation. Indeed, this was shown to be the case using a variety of methods. Ultimately, we discovered that the responsible factor was IFNαβ and that cells or mice lacking expression of IFNAR1-/- failed to downregulate IFNGR1 expression.
Our in vitro findings suggested that failure to downregulate cell surface IFNGR expression might contribute to the increased resistance of IFNAR-/- mice to bacterial infections. To test this, we asked whether IFNγ was required for the increased resistance of IFNAR1-/- mice to systemic L. monocytogenes infection. Using antibody depletion of IFNγ, we found that the bacterial burdens at 3 dpi in IFNAR1-/- mice depleted of IFNγ were indistinguishable from those seen in IFNAR+ mice, whereas burdens in unmanipulated IFNAR1-/- mice were 1,000-fold lower. Furthermore, the substantial reduction in bacterial burdens in the IFNAR1-/- mice correlated with increased expression of MHC-II by macrophages and DCs when compared to that seen on cells from the IFNAR+ mice. The increase in MHC-II expression by the IFNAR1-/- myeloid cells required IFNγ and correlated with increased expression of IFNGR1, but not increased IFNγ production. Indeed, consistent with their higher bacterial burdens, serum IFNγ concentrations are higher in the IFNAR+ mice at this stage of infection.Citation21 Therefore, our findings supported the notion that the increased sensitivity of IFNAR1-/- myeloid cells to IFNγ is due to their failure to downregulate the IFNGR and enhances their ability to eradicate L. monocytogenes during systemic in vivo infection. This model is illustrated in and provides a simple explanation how IFNαβ can suppress macrophage activation. This mechanism may permit IFNαβ to suppress macrophage activation by IFNγ,Citation30–Citation34 and increase susceptibility to diverse bacterial infections.Citation13,Citation18–Citation22,Citation35–Citation39 We further speculated that these effects of might account for the benefits of IFNβ in treatment of relapsing-remitting multiple sclerosis (MS).
Two studies published since ours have provided additional correlations between IFNαβ responsiveness and increased susceptibility to bacterial infection: Sher and colleagues reported that the induction of IFNαβ by stabilized Poly-IC treatment increased susceptibility to acute and chronic M. tuberculosis infection.Citation40 These effects required expression of IFNAR and correlated with increased ccl2 expression and increased infiltration of a CCR2-dependent myeloid cell population that was highly permissive to bacterial replication. Consistent with our data, macrophages in the poly-IC treated mice showed an IFNAR-dependent reduction in expression of MHC-II and the IFNGR. Interestingly, the reduced expression of IFNGR was seen both in populations of myeloid cells that showed increased susceptibility to M. tuberculosis as well as populations that did not. This observation suggests that factors in addition to IFNGR downregulation may govern whether a myeloid becomes more susceptible to bacterial infection in response to IFNαβ. A second study by Monack and colleagues correlated IFNAR expression and suppression of IL-17 production by a subset of γδT cells.Citation39 In the absence of IFNAR, IL-17 production was increased in mice infected with either Francisella tularensis or L. monocytogenes. Increased IL-17 correlated with a ∼2-fold increase in neutrophil accumulation in the spleens of these infected mice. However, it was not clear whether the recruitment of these neutrophils actually benefits the host response. Indeed, Ernst and colleagues recently found that increased IL-17 production and neutrophil recruitment increased susceptibility of mice to aerosol infection with M. tuberculosis.Citation41 If suppression of neutrophil recruitment were the major effect of IFNαβ in this setting, one might thus expect that IFNAR expression would benefit the host. Yet, as mentioned above, IFNαβ seems to have a net negative effect in the context of M. tuberculosis infection.Citation13,Citation22 Perhaps any potential positive or negative effects of IL-17 suppression by IFNαβ are superseded by their effects on IFNGR expression. In this light, we note that in recent studies with the mouse model of MS, EAE, it was recently shown that IFNβ treatment benefits the host only when disease was induced with IFNγ-producing Th1-type T cells.Citation42 When EAE was instead induced with Th17 cells, IFNβ treatment actually exacerbated disease, despite its ability to suppress IL-17 production.
In summary, there is a rapidly growing list of associations between IFNαβ and the dysregulation of immune responses during bacterial infections as well as inflammatory diseases such as MS. Our findings have revealed a novel mechanism by which IFNαβ can suppress host responsiveness to IFNγ. Although the existing data strongly support the notion that such suppression negatively impacts host resistance to bacterial infection, the precise consequences of such antagonistic crosstalk during infection, inflammation and clinical use of IFNα and IFNβ to treatment human disease, remain to be determined experimentally. Towards this end, it will be important to better understand the mechanism by which IFNαβ mediates downregulation of the IFNGR. Our initial studies revealed that IFNαβ treatment reduces steady-state amounts of ifngr1 transcripts and we have more recently found that such reductions are due to a very rapid block in transcription of ifngr1 rather than to reductions in ifngr1 mRNA stability (unpublished data). The mechanistic basis for this block is a subject of current investigation. Ultimately, such studies may reveal novel approaches to treat bacterial infections and inflammatory diseases associated with type I and II interferons.
Figures and Tables
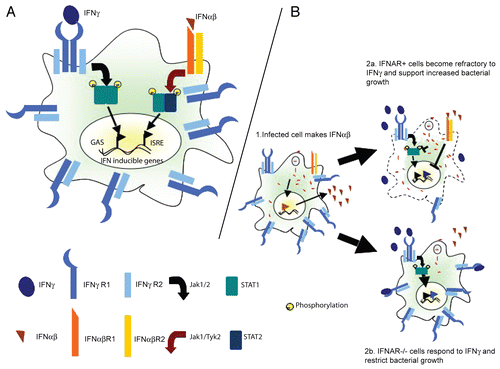
Figure 1 Antagonistic interferon cross talk and increased susceptibility to bacterial infection. (A) Schematic showing the canonical JAK-STAT signaling pathways involved in responses to IFNγ and IFNα/β. IFNγ activates gene expression through gamma activated sequences (GAS). IFNαβ modulates gene expression through interferon stimulated regulatory elements (ISRE). (B) IFNα/β produced by Listeria monocytogenes infected cells signals through the IFNAR on infected or bystander cells to block transcription of the ifngr1 gene and consequently IFNGR expression by macrophages or other antigen presenting cells (APCs). APCs with intact IFNα/β signaling are therefore less responsive to IFNγ when bacterial pathogens induce IFNαβ and hence more permissive to bacterial growth and replication.
Acknowledgements
Studies in our lab were funded by NIH grants #AI065638, AI055701 and AI065357-NO#2 to L.L.L. J.H. received support from NIH training grants #AI075-05 and AI52066-06. S.K. received support from the Cancer Research Institute.
Addendum to:
References
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, et al. Immune response in mice that lack the interferon-gamma receptor. Science 1993; 259:1742 - 1745
- Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med 1993; 178:2243 - 2247
- Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferongamma genes. Science 1993; 259:1739 - 1742
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 1993; 178:2249 - 2254
- Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996; 335:1941 - 1949
- Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med 1996; 335:1956 - 1961
- Lu B, Ebensperger C, Dembic Z, Wang Y, Kvatyuk M, Lu T, et al. Targeted disruption of the interferon-gamma receptor 2 gene results in severe immune defects in mice. Proc Natl Acad Sci USA 1998; 95:8233 - 8238
- Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest 1998; 101:2364 - 2369
- Koch O, Kwiatkowski DP, Udalova IA. Context-specific functional effects of IFNGR1 promoter polymorphism. Hum Mol Genet 2006; 15:1475 - 1481
- Bulat-Kardum L, Etokebe GE, Knezevic J, Balen S, Matakovic-Mileusnic N, Zaputovic L, et al. Interferon-gamma receptor-1 gene promoter polymorphisms (G-611A; T-56C) and susceptibility to tuberculosis. Scand J Immunol 2006; 63:142 - 150
- Cooke GS, Campbell SJ, Sillah J, Gustafson P, Bah B, Sirugo G, et al. Polymorphism within the interferon-gamma/receptor complex is associated with pulmonary tuberculosis. Am J Respir Crit Care Med 2006; 174:339 - 343
- Shi S, Blumenthal A, Hickey CM, Gandotra S, Levy D, Ehrt S. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-alphabeta receptor and STAT1. J Immunol 2005; 175:3318 - 3328
- Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 2007; 178:3143 - 3152
- Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky T Jr, et al. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci USA 2008; 105:10191 - 10196
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006; 24:93 - 103
- McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med 2009; 206:1899 - 1911
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science 2010; 328:1703 - 1705
- Fehr T, Schoedon G, Odermatt B, Holtschke T, Schneemann M, Bachmann MF, et al. Crucial role of interferon consensus sequence binding protein, but neither of interferon regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J Exp Med 1997; 185:921 - 931
- Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to listeria infection. J Exp Med 2004; 200:535 - 540
- O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I Interferon Production Enhances Susceptibility to Listeria monocytogenes Infection. J Exp Med 2004; 200:437 - 445
- Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice Lacking the Type I Interferon Receptor Are Resistant to Listeria monocytogenes. J Exp Med 2004; 200:527 - 533
- Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci USA 2001; 98:5752 - 5757
- Jia T, Leiner I, Dorothee G, Brandl K, Pamer EG. MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J Immunol 2009; 183:1271 - 1278
- Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med 2006; 203:933 - 940
- Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med 2010; 207:327 - 337
- Fortune SM, Solache A, Jaeger A, Hill PJ, Belisle JT, Bloom BR, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol 2004; 172:6272 - 6280
- Pai RK, Pennini ME, Tobian AA, Canaday DH, Boom WH, Harding CV. Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect Immun 2004; 72:6603 - 6614
- Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium tuberculosis 19 kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 2006; 176:4323 - 4330
- Benson SA, Ernst JD. TLR2-dependent inhibition of macrophage responses to IFN-gamma is mediated by distinct, gene-specific mechanisms. PLoS One 2009; 4:6329
- Ling PD, Warren MK, Vogel SN. Antagonistic effect of interferon-beta on the interferon-gamma-induced expression of Ia antigen in murine macrophages. J Immunol 1985; 135:1857 - 1863
- Inaba K, Kitaura M, Kato T, Watanabe Y, Kawade Y, Muramatsu S. Contrasting effect of alpha/beta- and gamma-interferons on expression of macrophage Ia antigens. J Exp Med 1986; 163:1030 - 1035
- Fertsch D, Schoenberg DR, Germain RN, Tou JY, Vogel SN. Induction of macrophage Ia antigen expression by rIFN-gamma and downregulation by IFN-alpha/beta and dexamethasone are mediated by changes in steady-state levels of Ia mRNA. J Immunol 1987; 139:244 - 249
- Fertsch-Ruggio D, Schoenberg DR, Vogel SN. Induction of macrophage Ia antigen expression by rIFN-gamma and downregulation by IFN-alpha/beta and dexamethasone are regulated transcriptionally. J Immunol 1988; 141:1582 - 1589
- Yoshida R, Murray HW, Nathan CF. Agonist and antagonist effects of interferon alpha and beta on activation of human macrophages. Two classes of interferon gamma receptors and blockade of the high-affinity sites by interferon alpha or beta. J Exp Med 1988; 167:1171 - 1185
- Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, Jiao L, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol 2008; 181:2092 - 2102
- Nagarajan UM, Prantner D, Sikes JD, Andrews C Jr, Goodwin AM, Nagarajan S, et al. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun 2008; 76:4642 - 4648
- Martin FJ, Gomez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest 2009; 119:1931 - 1939
- Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 2009; 119:1910 - 1920
- Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 2010; 184:3755 - 3767
- Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest 2010; 120:1674 - 1682
- Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 2009; 31:974 - 985
- Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 2010; 16:406 - 412