Abstract
Viruses induce an antiviral host response by activating the expression of antiviral host genes. However, viruses have evolved a wide range of strategies to counteract antiviral host responses. One of the strategies used by many viruses is the general inhibition of host gene expression, also referred to as a host shut-off mechanism. Here we discuss our recent findings that influenza virus infection results in the inhibition and degradation of host RNA polymerase II (Pol II) and that the viral RNA polymerase plays a critical role in this process. In particular, we found that Pol II is ubiquitylated in influenza virus infected cells and ubiquitylation can be induced by the expression of the RNA polymerase. Moreover, the expression of an antiviral host gene could be inhibited by the over-expression of the RNA polymerase. Both ubiquitylation and the inhibition of the host gene were dependent on the ability of the RNA polymerase to bind to Pol II. Further studies will be required to understand the interplay between the host and viral transcriptional machineries and to elucidate the exact molecular mechanisms that lead to the inhibition and degradation of Pol II as a result of viral RNA polymerase binding. These findings extend our understanding of how influenza virus counteracts antiviral host responses and underpin studies into the mechanisms by which the RNA polymerase determines virulence.
Influenza virus infection elicits a wide range of defense mechanisms that are triggered by the so-called virus-sensing receptors expressed by the host. These defense mechanisms are coordinated by the expression of signalling molecules, including type I interferons (IFNα and IFNβ). One of the major pathways activated in influenza virus infected cells involves the RNA helicase RIG-I which senses full-length 5′-triphosphate (5′-PPP)-containing viral genomic RNA.Citation1 RIG-I signals via the mitochondrial antiviral signalling protein (MAVS, also known as IPS-1, VISA and Cardif) to cellular kinases which in turn activate transcription factors, including interferon response factor 3 (IRF-3), IRF-7 and nuclear factor κB (NFκB). These transcription factors then coordinate the expression of IFNs which result in the activation of a myriad of antiviral host genes inducing an antiviral state that results in the suppression of viral infection.Citation2
The general inhibition of host gene expression appears to be an efficient way of counteracting such antiviral host mechanisms. For example, the matrix protein of vesicular stomatitis virus was shown to inhibit Pol II activity by inhibiting the TATA-binding protein, a subunit of the basal transcription factor TFIID.Citation3 Bunyaviruses express the NSs protein which was shown to interact with Med8, a component of the mediator complex that is involved in regulating the activity of Pol II.Citation4 This interaction leads to the degradation of Pol II resulting in a general block in the transcription of all host genes including interferon. Influenza virus encodes the NS1 protein that has also been shown to possess an interferon antagonist function.Citation5 Several mechanisms have been proposed as to how NS1 might inhibit interferon expression. For example, NS1 has been shown to interact with CPSF30 leading to a general inhibition of 3′ end formation of Pol II-transcribed host mRNAs and thus to the inhibition of interferon expression.Citation6,Citation7 However, NS1-independent virus-induced host shut-off has also been described.Citation8 More recently, a mechanism for regulating innate immune responses involving the RNA polymerase has been proposed. In particular, overexpression of the viral polymerase subunits was shown to inhibit cellular interferon responses to either viral genomic RNA transfection or infection with an NS1-deficient virus.Citation9 Our recently published data provide a functional insight into the mechanisms of how the RNA polymerase could contribute to the inhibition of antiviral host responses in influenza viruses infected cells.Citation10
The RNA polymerase of influenza virus is a complex composed of three subunits, polymerase basic protein 1 (PB1), polymerase basic protein 2 (PB2) and polymerase acidic protein (PA).Citation11–Citation13 The PB1 subunit shares homology with other RNA polymerases and it contains the polymerase active site. In contrast, the PB2 and PA subunits show little homology with other proteins. Whereas the function of PB2 in binding of the 5′ cap structure of host mRNAs has long been known, the function of PA remained unclear until functional and structural studies revealed its role in endonucleolytic cleavage of host mRNAs.Citation14–Citation19 These functions are required for generating capped RNA fragments which are then used by the viral RNA polymerase as primers for the initiation of viral mRNA synthesis. Thus, influenza virus is absolutely dependent on active host Pol II transcription.Citation20,Citation21 Moreover, ongoing Pol II transcription may be required for the splicing and nuclear export of viral mRNAs.Citation22
This functional association between the viral and host transcriptional machineries prompted us to investigate whether there are any physical links between the two transcriptional machineries. Our group was the first to demonstrate that the influenza virus RNA polymerase associates with the host Pol II transcriptional machinery through the interaction of the viral polymerase with the C-terminal domain (CTD) of Pol IICitation23 (), an observation subsequently confirmed by others.Citation24–Citation26 We found that the viral polymerase preferentially targets the serine-5 phosphorylated form of the CTD that is characteristic of the transcriptionally engaged initiating form of Pol II as opposed to the unengaged non-phosphorylated form and the serine-2 phosphorylated form that has been associated with Pol II engaged in elongation. This finding is supported by co-immunoprecipitation, co-purification and co-localization studies of the viral RNA polymerase with Pol II as well as chromatin immunoprecipitation assays, showing that the viral RNA polymerase associates with the promoter region of actively transcribed Pol II genes through its interaction with the large subunit of Pol II.Citation23,Citation27 Together these findings suggest a mechanism by which influenza virus hijacks the host transcriptional machinery in order to access host functions that are required for viral transcription. Capping as well as splicing and packaging of host mRNAs into ribonucleoprotein complexes take place co-transcriptionally, in close association with Pol II.Citation28 By targeting host Pol II, the viral RNA polymerase could gain access to capped RNA primers as well as downstream RNA processing, linking viral transcription to the cellular mRNA synthetic and nuclear export pathways.
Although, this association might represent first of all a mechanism to aid the transcription of viral genes, more recently we also addressed the question of how this association affects Pol II function. To this end we analysed Pol II expression patterns and functionality during a time course of influenza virus infection.Citation10,Citation27 We found that Pol II elongation is inhibited upon influenza virus infection, resulting in the arrest of Pol II at the promoter region early in infection (). However, later in infection there was a depletion of promoter-associated Pol II. This depletion turned out to be caused by a general reduction in Pol II levels due to the proteolytic degradation of Pol II. Degradation of Pol II by the proteasome has been proposed to act as a safety mechanism to clear arrested Pol II as a result of DNA damage induced by drugor UV-treatment.Citation29,Citation30 The influenza virus polymerase, by binding to and inhibiting Pol II, could induce this host mechanism, resulting in the ubiquitylation and proteasomal degradation of Pol II. Indeed, we observed increased ubiquitylation of the large subunit of Pol II in influenza virus infected cells. We found that ubiquitylation could also be triggered by the overexpression of viral RNA polymerase complex that can bind Pol II, but not by the overexpression of a mutant viral RNA polymerase with reduced Pol II binding activity.Citation10 Thus, these data provide support for the hypothesis that viral RNA polymerase binding to Pol II triggers proteasomal Pol II degradation.
Another recent study linked the degradation of Pol II to the proteolytic activity of the PA subunit of the viral RNA polymerase.Citation26 The PA subunit has been proposed to possess or induce a proteolytic activity resulting in the reduction of co-expressed reporter proteins.Citation31,Citation32 However, the molecular mechanisms of PA-induced proteolytic degradation have not been elucidated and recent crystallographic analysis of an N-terminal domain of PA, previously implicated in the proteolytic activity, has failed to reveal a functional domain that could be linked to proteolytic activity.Citation15,Citation18 In contrast, an endonucleolytic domain has been identified that is involved in the generation of capped RNA primers for viral transcription. This raises the possibility that the previously observed effects on the protein could have been caused at the RNA level. Further studies will need to be performed to address the molecular mechanisms that lead to PA-induced protein level reductions.
The specific inhibition of Pol II and the reduction of its levels in influenza virus infected cells strongly suggest that they are contributing factors to the influenza virus induced host shut-off mechanism. As such they could have far reaching consequences for virulence. Indeed, another recent study correlated the ability of influenza viruses to induce Pol II degradation with virulence and proposed that the ability of an influenza virus to degrade Pol II might be an important factor in determining the virulence of influenza viruses.Citation33 A more complex study involving a wide range of influenza viruses with low and high virulence in various hosts will be required to address this question in more detail.
Overall, our data support the hypothesis that binding of the trimeric viral RNA polymerase complex to the CTD of Pol II is responsible for Pol II inhibition and degradation.Citation10 However, many questions remain to be addressed. The contribution of PA to the proteolytic degradation of Pol II remains unclear and it is also unclear why overexpressed viral RNA polymerase complex that induces Pol II inhibition and ubiquitylation does not trigger an overall reduction of all forms of Pol II. These findings suggest that additional factors might be required. Moreover, the molecular mechanisms of the degradation process itself remain unclear. The E3 ubiquitin ligase Nedd4 was shown to be involved in the ubiquitylation of Pol II in response to UV-induced DNA damage in human cells.Citation34 Nedd4 has previously been identified in a screen for modulators of early stage HIV-1 infection and Nedd4 binding protein 2 (N4BP2) has also been identified as a host factor required for influenza virus infection.Citation35,Citation36 Addressing the question whether Nedd4 is specifically recruited to Pol II in influenza virus infected cells could provide further insight into the molecular mechanisms involved.
The hypothesis that influenza virusinduced inhibition and degradation of Pol II benefits the virus through the inhibition of the expression of antiviral genes is somewhat counterintuitive considering the dependence of influenza virus mRNA synthesis and processing on on-going Pol II transcription. However, the peak of mRNA synthesis is reached relatively early during the course of viral infection, while genome replication predominates late in infection.Citation37,Citation38 This observation resulted in various models to explain the “switch” from transcription to genome replication during the viral life cycle, (1) implicating the nucleoprotein as a switching factor, (2) proposing that a second, non-template associated trans-acting polymerase is required for genome replication or (3) that small viral RNA triggers the switch to replication.Citation39–Citation41 While our results do not exclude the importance of these factors in genome replication, they strongly suggest that the sharp decline in viral mRNA synthesis after the early peak is simply the consequence of Pol II inhibition and degradation. An initial burst in viral mRNA levels might ensure the synthesis of sufficient levels of viral proteins for progeny virions while genome replication, a process independent of Pol II and promoted primarily by viral proteins, can proceed late during infection. Overall, many centuries of host-virus co-evolution probably resulted in a finely balanced outcome which on one hand enables the virus to synthesize sufficient levels of viral mRNAs and on the other, prevents host gene expression that would result in the release of antiviral factors, which could in turn induce an antiviral state in neighboring cells preventing viral spread.
Our study provides the foundation for further studies to dissect the role of the influenza virus RNA polymerase in determining virulence. Although the precise molecular mechanism of Pol II inhibition and degradation remains elusive, our results strongly suggest a role for the viral RNA polymerase in the general host shut-off induced in cell infected by influenza virus.
Figures and Tables
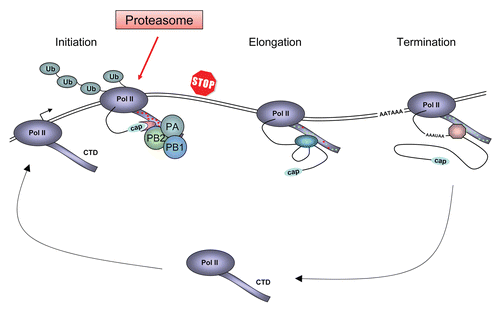
Figure 1 Schematic model for the Pol II transcription cycle in cells infected with influenza virus and the role of the viral RNA polymerase in the inhibition and degradation of Pol II. The model depicts the large subunit of Pol II with the C-terminal domain (CTD) which is differentially phosphorylated during the transcription cycle (indicated by red and green dots). During the transcription cycle cellular factors involved in capping, splicing and polyadenylation associate with the CTD. The polyadenylation signal AATAAA is indicated. In influenza virus infected cells, the viral RNA polymerase (composed of the PB1, PB2 and PA subunits) binds to the serine-5 phosphorylated CTD of Pol II engaged in transcription initiation. On one hand, this association allows the viral RNA polymerase to access the 5′ cap structure of nascent Pol II transcripts and on the other, it results in the premature termination of Pol II. Arrested Pol II is ubiquitylated (Ub) by the cellular machinery and eventually is degraded by the proteasome. Modified from Chan et al.Citation27
Acknowledgements
Research in E.F.'s laboratory is supported by grants from the Medical Research Council and the Wellcome Trust.
Addendum to:
References
- Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative strand RNA virus infection. Cell 2010; 140:397 - 408
- Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity 2007; 27:370 - 383
- Yuan H, Puckett S, Lyles DS. Inhibition of host transcription by vesicular stomatitis virus involves a novel mechanism that is independent of phosphorylation of TATA-binding protein (TBP) or association of TBP with TBP-associated factor subunits. J Virol 2001; 75:4453 - 4458
- Leonard VH, Kohl A, Hart TJ, Elliott RM. Interaction of Bunyamwera Orthobunyavirus NSs protein with mediator protein MED8: a mechanism for inhibiting the interferon response. J Virol 2006; 80:9667 - 9675
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 2008; 89:2359 - 2376
- Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol Cell 1998; 1:991 - 1000
- Noah DL, Twu KY, Krug RM. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 2003; 307:386 - 395
- Zurcher T, Marion RM, Ortin J. Protein synthesis shut-off induced by influenza virus infection is independent of PKR activity. J Virol 2000; 74:8781 - 8784
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009; 139:1255 - 1267
- Vreede FT, Chan AY, Sharps J, Fodor E. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 2010; 396:125 - 134
- Elton D, Digard P, Tiley L, Ortin J. Kawaoka Y. Structure and function of the influenza virus RNP. Influenza Virology: Current Topics 2006; Caister Academic Press 1 - 36
- Neumann G, Brownlee GG, Fodor E, Kawaoka Y. Orthomyxovirus replication, transcription and polyadenylation. Curr Top Microbiol Immunol 2004; 283:121 - 143
- Ruigrok RW, Crepin T, Hart DJ, Cusack S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr Opin Struct Biol 2010; 20:104 - 113
- Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 2002; 76:8989 - 9001
- Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009; 458:914 - 918
- Fechter P, Mingay L, Sharps J, Chambers A, Fodor E, Brownlee GG. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J Biol Chem 2003; 278:20381 - 20388
- Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 2008; 15:500 - 506
- Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009; 458:909 - 913
- Ulmanen I, Broni BA, Krug RM. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc Natl Acad Sci USA 1981; 78:7355 - 7359
- Amorim MJ, Digard P. Influenza A virus and the cell nucleus. Vaccine 2006; 24:6651 - 6655
- Engelhardt OG, Fodor E. Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol 2006; 16:329 - 345
- Amorim MJ, Read EK, Dalton RM, Medcalf L, Digard P. Nuclear export of influenza A virus mRNAs requires ongoing RNA polymerase II activity. Traffic 2007; 8:1 - 11
- Engelhardt OG, Smith M, Fodor E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 2005; 79:5812 - 5818
- Mayer D, Molawi K, Martinez-Sobrido L, Ghanem A, Thomas S, Baginsky S, et al. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J Proteome Res 2007; 6:672 - 682
- Rameix-Welti MA, Tomoiu A, Dos Santos Afonso E, van der Werf S, Naffakh N. Avian Influenza A virus polymerase association with nucleoprotein, but not polymerase assembly, is impaired in human cells during the course of infection. J Virol 2009; 83:1320 - 1331
- Rodriguez A, Perez-Gonzalez A, Nieto A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J Virol 2007; 81:5315 - 5324
- Chan AY, Vreede FT, Smith M, Engelhardt OG, Fodor E. Influenza virus inhibits RNA polymerase II elongation. Virology 2006; 351:210 - 217
- Luna R, Gaillard H, Gonzalez-Aguilera C, Aguilera A. Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma 2008; 117:319 - 331
- Ratner JN, Balasubramanian B, Corden J, Warren SL, Bregman DB. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II. Implications for transcription-coupled DNA repair. Biol Chem 1998; 273:5184 - 5189
- Somesh BP, Reid J, Liu WF, Sogaard TM, Erdjument-Bromage H, Tempst P, et al. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 2005; 121:913 - 923
- Naffakh N, Massin P, van der Werf S. The transcription/replication activity of the polymerase of influenza A viruses is not correlated with the level of proteolysis induced by the PA subunit. Virology 2001; 285:244 - 252
- Sanz-Ezquerro JJ, de la Luna S, Ortin J, Nieto A. Individual expression of influenza virus PA protein induces degradation of coexpressed proteins. J Virol 1995; 69:2420 - 2426
- Rodriguez A, Perez-Gonzalez A, Hossain MJ, Chen LM, Rolling T, Perez-Brena P, et al. Attenuated strains of influenza A viruses do not induce degradation of RNA polymerase II. J Virol 2009; 83:11166 - 11174
- Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell 2007; 28:386 - 397
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008; 135:49 - 60
- Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, et al. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology 2009; 387:473 - 481
- Robb NC, Smith M, Vreede FT, Fodor E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol 2009; 90:1398 - 1407
- Shapiro GI, Gurney Jr, Krug RM. Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus-specific RNAs. J Virol 1987; 61:764 - 773
- Beaton AR, Krug RM. Transcription antitermination during influenza viral template RNA synthesis requires the nucleocapsid protein and the absence of a 5′ capped end. Proc Natl Acad Sci USA 1986; 83:6282 - 6286
- Jorba N, Coloma R, Ortin J. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS pathogens 2009; 5:1000462
- Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, et al. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci USA 2010; 107:11525 - 11530