 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Transmissible Spongiform Encephalopathy (TSE) agents are defined by their virulence for particular species, their spread in the population, their incubation time to cause disease, and their neuropathological sequelae. Murine adapted human agents, including sporadic CJD (sCJD), New Guinea kuru, and Japanese CJD agents, display particularly distinct incubation times and maximal infectious brain titers. They also induce agent-specific patterns of neurodegeneration. When these TSE agents are transmitted to cultured hypothalamic GT1 cells they maintain their unique identities. Nevertheless, the human kuru (kCJD) and Japanese FU-CJD agents, as well as the sheep 22L and 263K scrapie agents display doubling times that are 8x to 33x faster in cells than in brain, indicating release from complex innate immune responses. These data are most consistent with a foreign viral structure, rather than an infectious form of host prion protein (PrP-res). Profound agent-specific inhibitory effects are also apparent in GT1 cells, and maximal titer plateau in kCJD and FU-CJD differed by 1,000-fold in a cell-based assay. Remarkably, the lower titer kCJD agent rapidly induced de novo PrP-res in GT1 cells, whereas the high titer FU-CJD agent replicated silently for multiple passages. Although PrP-res is often considered to be toxic, PrP-res instead may be part of a primal defense and/or clearance mechanism against TSE environmental agents. Limited spread of particular TSE agents through nanotubes and cell-to-cell contacts probably underlies the long peripheral phase of human CJD.
Introduction
At the present time, textbooks often state with certainty that a misfolded form of the mammalian host prion protein (PrP), without any nucleic acid, is the infectious agent that causes Transmissible Spongiform Encephalopathies (TSEs). Misfolded PrP accumulates as amyloid deposits in brain, and on western blots is identified by its more proteinase K resistant bands (PrP-res). Although PrP-res is believed to be infectious, it does not fulfill Koch's postulates, nor can it explain agent strains.Citation1 Different TSE agents cause sheep scrapie, human Creutzfeldt-Jakob Disease (CJD), “mad cow disease” (BSE) and related diseases of cervids. Even though TSE agents are quite distinct, they typically induce identical PrP-res bands in wild-type (wt) mouse brain. In contrast, β-amyloid-rich Alzheimer Disease (AD) brains are not infectious experimentally, nor do they produce a facsimile of AD neurodegeneration. In TSEs there is a long silent phase when the infectious agent replicates exponentially in the brain until it finally induces PrP-res, clinical disease and spongiform neurodegeneration.Citation2 Although host PrP is clearly involved in susceptibility to infection and pathogenesis, many experimental facts strongly suggest the infectious pathogen contains a nucleic acid genome that can encode different agent strains. Human TSE agents isolated from specific geographic regions show the most dramatic differences in incubation time and brain lesions.Citation3,Citation4
Many different TSE agents are incompletely characterized, and our group has been identifying individual agent features through combined transmission, cell culture and molecular studies. We also developed stable cell-based assays for TSE infectivity that can minimize the expensive and extraordinarily long animal assays.Citation5 It is now possible to rapidly follow infectivity in cells and subcellular fractions,Citation6 an essential parameter for any impartial analysis of the structure of TSE agents. Infection of a single cell line by many different TSE agents can also clarify agent-encoded differences. We therefore propagated the BSE (human vCJD) agent, the New Guinea kuru agent (kCJD) and a variety of other distinct CJD and scrapie agents, including 263K hamster scrapie, in murine GT1 hypothalamic cells.Citation3,Citation4,Citation7 TSE agents in these monotypic cells revealed surprising additional characteristics, such as virus-like interference without PrP-res.Citation7 In sum, although pathologic PrP is generally a good surrogate marker for infection, it rarely discriminates disparate agents.
To appreciate the variety of TSE agents it is essential to keep the host constant. One can completely change the PrP folding and glycosylation patterns by growth in monotypic cells, yet the infectious agent retains its original individual characteristics of incubation time and regional neuroanatomical pathology, as shown by re-inoculation of animals.Citation8 The agent, but not the cell type-specific PrP-res folding pattern, breeds true. Thus PrP-res does not even fulfill Koch's first postulate that “the pathogen is invariably present in a characteristic and constant form.”Citation1 Transmission of a TSE isolate to different species further shows that the agent, but not the species PrP, is preserved. For example, a cow brain infected by the UK BSE agent and human brain infected by the same agentCitation9 show different species-dependent PrP-res bands, yet both brains yield the same agent-specific disease when transmitted to wt mice.Citation3 Moreover, two agents can infect a single animal or cell and show no intermediate agent forms or “chimeric” PrP-res molecules as proposed in the prion model.Citation7,Citation10 Consistent with accumulating evidence, host PrP more likely functions as a receptor that is required for the replicative life cycle of a TSE virus.Citation11,Citation12 Thus PrP knockout mice resist infection,Citation13 and merely increasing PrP enhances susceptibility and accelerates disease. Indeed, cross-species transmissions of the human vCJD and kuru agents are enhanced by using Tga20 mice with 8–10× the normal murine PrP copies.Citation3,Citation4 These positive and unambiguous transmissions clearly do not fit the PrP species-specific “self-catalytic infectious PrP conversion” prion model.Citation14–Citation16
TSE agents also display conventional biological characteristics of covert viruses. These include (1) an initial spread through blood and the lymphoreticular system,Citation17–Citation19 (2) epidemic disease that has been halted by removal of contaminated material and (3) the geographic isolation of distinct TSE agents that implicate both an environmental source and a foreign nucleic acid genome. Geographic TSE agents can rapidly spread worldwide, as in the export of contaminated BSE meat from the UK. Other TSE agents can remain localized, as with the unique human kuru agent of New Guinea that was transmitted during ritual cannibalism.Citation20 The known gastrointestinal route of TSE infection has become particularly problematic for the prion hypothesis because alimentary fluids digest PrP and PrP-res, but infectivity is not reduced in either sheep or hamster TSE models.Citation21,Citation22 The retained TSE infectivity implicates classical viral structures that are known to resist digestion. Furthermore, as shown in a variety of experiments from many laboratories, PrP-res does not correlate quantitatively with infectious titers,Citation1 and isolated microglia that express only tiny amounts of normal PrP (and no detectable PrP-res) contain very high levels of infectivity.Citation23 Hence, although PrP-res is often a reliable indicator of infection, it probably is not an intrinsic agent molecule. Instead, as supported by temporal studies,Citation1 PrP-res appears to be a late pathological response to the infectious agent. Finally, physical and biochemical analyses of infectious particles are most consistent with a viral structure. As one purifies homogeneous 25 nm dense particles by field flow fractionation or sucrose sedimentation, infectivity is completely recovered but abnormal PrP is markedly reduced.Citation24,Citation25 Moreover, when these infectious particles are disrupted, they lose their infectivity and release nucleic acids of viral size, as well as protective nucleic acid binding proteins.Citation24,Citation26 The recent discovery of new circular DNAs, designated Sphinx elements in many different infectious samples that can encode agent-specific properties,Citation27 further undermines the presumption of a protein-only agent.
We here sought to define the most fundamental replication properties of two very different TSE agents in brain and in GT1 cells in culture. We determined doubling times, end-point titration curves, and the induction of PrP-res bands after infection by each agent. Such studies can clarify fundamental agent-host PrP interactions at the single cell level and also establish intrinsic agent properties. We sought to find if unique and large differences in replication and maximal titer plateaus would be maintained in monotypic cells. Serial dilution studies would also yield useful agent-specific reference titration curves that can facilitate purification of different agents.
The advantages of GT1 infectivity assays are manifold. Notably, GT cells are genetically stable and susceptible to a diversity of human (CJD) and sheep/goat (scrapie) agents grown in wt mice. These include eight distinct CJD and scrapie isolates that induce dramatically different patterns of brain lesions, as well as minimal incubation times of 120 to 440 d.Citation3,Citation4 The commonly used N2a neuroblastoma line is susceptible to only to one or two less distinctive scrapie agents. N2a tumor cell clones are genetically unstable and often lose infectivity.Citation8,Citation28,Citation29 Thus profound differences in agent virulence cannot be reliably compared. In contrast, GT1 cell infections have been verified for >1 year in vitro.Citation5,Citation8 De novo production of PrP-res by GT1 cells, the surrogate marker of infection, can also be apparent by passage 1 (p1) on western blots.Citation28 Moreover, although PrP-res band patterns in wt mouse brains are indistinguishable for different geographic agents, such as those causing sporadic CJD (sCJD), Japanese CJD and New Guinea kuru,Citation3,Citation7 these agents can induce agentdiagnostic bands in GT1 cells. This is useful for uncovering cross-contamination, and can be exploited to follow co-infection of a cell by two agents.Citation7 Finally, western blot GT1 PrP-res assays were used because they gave no background or false positives with mock controls whereas published PrP-res cell spot assays show significant background positives.Citation29,Citation30 We here demonstrate 3–4 logs of infectivity can be reliably discriminated.
Different agents would be expected to express their specific intrinsic replication properties during serial end-point dilution studies in GT1 indicator cells. If an agent has an intrinsic replication character or “clock” it should continue to express its unique clock and/or spreading capacities in monotypic cells. A covert TSE virus that is recognized by the host as foreign (unlike host PrP) should also achieve a faster, uninhibited replication rate in cells after it is removed from the complex innate defenses of the animal. On the other hand, in the prion model, the identical PrP in monotypic cells should self-convert to PrP-res at a similar rate regardless of an agent's strain. PrP-res levels should also correlate with infectious titers for all agents grown in GT1 cells. Because specific TSE agents can produce 10,000-fold different maximal titers in brain,Citation4,Citation31 it was of interest to find if these plateaus were an intrinsic, agent-determined property that continued to be expressed in GT1 cells.
The GT1 infectivity assay approach was previously verified with a standard 22L scrapie agent and end-point titers were reproducible, and only slightly lower than mouse end-point titers.Citation5 We here compare the Japanese FU-CJD agent with the New Guinea kuru (kCJD) agent. FU-CJD rapidly induces widespread brain lesions in 110 days (d) whereas kCJD requires >420 d to induce more limited lesions in wt mice.Citation4,Citation10 These studies demonstrate profound differences in the effective replication of these two agents as well as other TSE agents in brain and GT1 cells. There are few reports of end-point titers of different agents in cultured cells, and review of scrapie cell models has generally shown low infectivity, equivalent to only ∼1 LD50 per 1,000 to >10,000 cells.Citation8 We found FU-CJD agent titers in GT1 cells were >3,000-fold higher than those scrapie cultures, and remarkably, were also 10-fold higher than FU-CJD infected mouse brain. The kCJD samples had significantly lower titers than FU-CJD samples, and were limited to 1,000-fold lower titer plateaus in GT1 cells. We here show vastly different agent-specific replication rates and maximal titer plateaus. These intrinsic agent properties are difficult to explain by PrP misfolding.
Results
High titer FU-CJD brain homogenates induce PrP-res slowly in GT1 cells.
All inocula are calculated as cell equivalents (CE) so that infectivity can be consistently expressed on a per cell basis. This allows standard animal assay comparisons where brain contains 109 cells/gm.Citation5 While PrP-res can be absent in infectious samples, its presence is diagnostic for infection. Because PrP-res may not correlate quantitatively with infectivity, accurate agent titers require serial dilutions to determine an end-point (highest dilution at which any PrP-res is eventually induced). GT1 cells exposed to large doses (106 CE) of FU-CJD brain homogenate rapidly express their maximal level of PrP-res (∼25% of total PrP) as previously depicted.Citation8 This maximal PrP-res value in GT1 cells is lower than that found in infected CD-1 mouse brain homogenates (∼55% PrP-res) and thus brain should be more infectious than GT1 cells if PrP-res is the infectious agent. Previous studies showed FU-CJD infected GT1 cells were at least as infectious as mouse brain by animal titration.Citation8 To rigorously determine if GT1 cells infected by FU-CJD agent consistently developed high titers, we titrated several FU-CJD infected brains by serial end point dilution. In this assay, dilutions that are infectious should induce PrP-res, the surrogate marker of infection, until their tissue culture end point is reached (TCID, ½ wells with PrP-res). At low input titers, it may take >5 passages for de novo PrP-res to appear. More virulent, high-titer agents will produce a PrP-res signal at low input CE, i.e., they have high infectivity per cell.
shows a representative western blot of undigested total PrP (−PK lanes) and digested PrP-res (+PK lanes) sampled at p5 after GT1 indicator cells were exposed to FU-CJD mouse brain homogenates. Note the decreasing %PrP-res at dilutions from 3e5 to 3e2 CE. At input dilutions of 30 and 3 CE, PrP-res was not detectable at p5 despite the high 10x amounts loaded in the PrP-res lanes. The diagnostic PrP marker band at 13 kd (arrows), specific for Japanese CJD isolates in GT1 cells, is apparent in all the proteinase K digested lanes. Parent mouse FU-CJD brain homogenates do not show this band.Citation4,Citation31 As would be expected for a high titer, rapidly virulent agent, later passages of cells exposed to lower CE eventually developed positive PrP-res signals. The FU-CJD agent in brain replicated and induced PrP-res down to an input dilution of 3 CE by p13 (). With the lower input of 30 and 3 CE, it was also apparent that cells were infected for at least 20 d (5 passages) without inducing PrP-res. High titers of FU-CJD agent were therefore required to induce PrP-res. This long asymptomatic PrP-res stage of exponential agent infection in cultured cells resembles the 90 d silent PrP-res phase in mice inoculated intracerebrally with maximal LD50.Citation32 Increased dilutions to 0.3 CE, as well as uninfected mouse brain control homogenates were evaluated for additional passages (up to 18), and none showed a positive PrP-res signal. In sum, the GT1 assay for FU-CJD brain revealed 1 TCID per 3 cells with a significant PrP-res silent phase during exponential agent replication. This GT1 titer was comparable to the conventional 500 d mouse assay that showed only a 3-fold higher endpoint of 1 LD50 per brain cell.Citation5
Progressive PrP-res accumulation with FU-CJD titer.
Once FU-infected GT1 cells began to produce PrP-res, it accumulated rapidly, and at a constant rate. This rate of PrP-res accumulation was apparent at each of the 5 log dilutions (3e5 to 30 CE) as graphed in . This plot represents four independent experiments with two different FU-CJD brain homogenates. When PrP-res in GT1 cells reached ∼25% of total cellular PrP, it remained at this plateau during further passages.
Serial dilution data allowed us to construct a standard graph for determining FU-CJD end point tissue culture infectious doses (TCID) from the %PrP-res induced at progressive times after infection. This graph shows the FU-CJD agent titer increases exponentially while PrP-res increases are linear (, p5, p9, p13, p15). There is a 12% increase in PrP-res per log increase of FU-CJD agent in the range of 4–28% PrP-res. The slopes are much steeper between 0.5–4% PrP-res. These lower PrP-res levels are less dependable for accurate titers, but can be confirmed simply by further cell passages where PrP-res progressively accumulates. A span of 4 logs titer was easily discriminated with an input of 30-3e5 CE (, from 10 to 105 TCID). also shows a mean of 3 passages (12 d difference) between each log dilution (compare points at 12–16% PrP-res). Although a 3-fold titer difference could be discriminated at these PrP-res levels (), a 4-fold difference in titer is completely unambiguous. This is clearly a conservative estimate because GT1 infectivity assays consistently revealed close to 100% recoveries of agent in previous subcellular 22L scrapie fractionations.Citation5,Citation6
Effective doubling times (ti) of TSE agents in animals and GT1 cells.
The above culture incubation time assays of FU-CJD brain homogenate allowed us to uncover profound agent doubling time differences between mouse brain and GT1 cells. Cell replication and complex multicellular clearance mechanisms need to be considered when quantifying an agent's effective doubling time. In brain, non-dividing neurons are most commonly associated with high agent titers, whereas infected GT1 cells are continuously dividing. In previous studies of hamster brain, we compared the replication of two relatively fast agents, 263K scrapie and sCJD; i.e., only the agent was changed for controlled comparison. This hamster brain data provides a useful reference standard for comparison of FU-CJD agent replication in mouse brain, and in murine GT1 cultures. The 263K hamster adapted scrapie agent has the shortest known TSE incubation time per log titer rise in brain. This represents the maximal in vivo agent “clock” and yields an effective doubling time (ti) of ∼3.5 d.Citation2 In contrast, the sporadic CJD (sCJD) agent has a >2-fold longer ti of 7.4 d. A species change can make a dramatic difference in agent replication. In wt mice the sCJD agent requires 350 d for disease expression, yet still retains its ability to produce rapid disease in hamsters.Citation33 The murine-adapted sCJD agent also produces a 10,000-fold lower terminal titer than found in hamsters, and thus has a very long doubling time (ti of ∼26 d, see calculation below). This indicates very different species controls of the same agent.
Other TSE agents in mice, such as the Chandler-RML and 22L scrapie agents do not display similarly retarded replication times () even though they induce the same murine PrP-res pattern as sCJD. The murine brain PrP-res pattern in sCJD is also the same as that found with the much faster FU-CJD agent. Moreover, these two CJD models have a 10,000-fold titer difference with only a 10-fold difference in PrP-res.Citation31 These combined data suggests unusually strong inhibitory responses of normal mice to the sCJD agent that cannot be based on the host's PrP or PrP-res folding patterns. Agent-specific clock differences, along with host recognition and defense mechanisms, were further substantiated in studies detailed below.
Pertinent doubling times for exemplary TSE agents in brain and in GT1 cells are summarized in . The calculated ti is a consequence of both agent replication and clearance during the log phase of asymptomatic infection, and has been solved mathematically where:
In animal models, ∼98% of the original intracerebral inoculum is cleared during the first 24 h, and sequential titrations show a constant exponential replication of agent until clinical disease begins.Citation2 Thus mice inoculated with 2e5 LD50 of the FU-CJD agent retain only 2e3 LD50 of the agent in brain for replication. It requires 110 ± 5 d to achieve the original high terminal FU-CJD brain titer of 109 LD50/gm. This represents a 3e5 agent increase, equivalent to a ti doubling time of 6.7 d for this agent in wt mouse brain. In contrast, the same FU-CJD agent in GT1 cells takes only 12 d (3 passages) to achieve a 1 log titer increase (ti = 3.61) or a 2-fold faster rate, as can be seen from inspection of , and from the best fit infectivity slopes in (all R2 = 0.99–1.0). Moreover, the FU-CJD agent doubling time in GT1 cells is actually dramatically shorter, because unlike brain cells, the GT1 cells double every 24 h (1 log per 3.322 d). Thus, in order for the FU-CJD agent simply to remain at a constant level in GT1 cells it would have to double as fast as the cells. The FU-CJD agent is actually replicating even faster because its titer is increasing more than GT1 cells. In fact, FU-CJD is replicating at the combined cell and agent rates, i.e., 1 log every 2.607 d (i.e., ti = ln (2)/0.88323 = 0.79 d). This calculation demonstrates a dramatic 8.4-fold faster FU-CJD agent doubling time in GT1 cells as compared to brain. In murine brain, innate immune responses of many different cell types to a foreign FU-CJD agent can be elicited that can retard or prevent rapid agent accumulation. In fact, a number of innate immune responses have been found in early stages of FU-CJD infection, well before PrP-res is induced.Citation32,Citation34 TSE agents may also be cleared more effectively from brain as affected neurons progressively drop out, further limiting rapid agent accumulation. Although it is often assumed that pathologic PrP is toxic, infected GT1 cells with large amounts of PrP-res showed no discernible toxic changes or cell loss.
A second related feature of GT1 infection was the ability of the FU-CJD agent to spread rapidly to many cells. When only 100 TCID (3e2 brain CE) was applied to 105 GT1 indicator cells (an MOI of 0.001), the infected cells at p2 produced no PrP-res. However, most of the GT1 cells eventually became infected, and by p15 GT1 cells developed their maximal PrP-res level (∼25%). Prior in-situ studies showed that 30–100% of 2 µm thin sections of FU-CJD cells at this maximal PrP-res level also accumulated abundant cytoplasmic PrP amyloid.Citation8 Thus there was a clear and progressive cell-to-cell spread of the FU-CJD agent between GT1 cells, probably via nanotubes and PrP-rich specialized junctions that include viral and neuronal synaptic specializations involved in exchanges of cytoplasmic material between cells.Citation35
FU-CJD agent achieves higher titers in GT1 cells than in brain.
Infectivity studies provided an additional independent opportunity to test if PrP-res is proportional to titer. According to prion theory, FU-CJD infected GT1 cells with 25% PrP-res should have less infectivity than brain with 50–60% PrP-res, yet FU-CJD infected GT1 cells were clearly more infectious than brain. shows a representative western blot assay after exposure to FU-CJD infected GT1 cells. Interestingly, GT1-FU CJD cell homogenates induced PrP-res faster than brain homogenates at the same input CE. The earlier appearance of PrP-res was due to the 10-fold higher titer of the infected cell homogenates. Inspection of the graphs show the same specific, and relatively high level of the FU-CJD agent was needed to induce PrP-res in the indicator cells. shows PrP-res is induced at p7 by as few as 3 CE, and as little as 0.3 CE induced a positive PrP-res signal at p13. The end-point infectivity of FU-CJD agent in GT1 cells was 0.3 TCID per cell, a 10-fold greater titer than FU-CJD brain (3 TCID/cell). Comparison of the time to achieve 8% PrPres at p2 further showed that a brain input of 3e5 CE induced the same amount of PrP-res as 3e3 CE of GT1 cells (compare with ), indicating infection was faster with cell homogenates. However, intrinsic FU-CJD agent clock was not detectably altered by in vitro growth in GT1 cells.
There was only a marginal reduction in the FU-CJD doubling time in GT1 cells versus brain. shows a mean of 2.7 passages/log agent increase, equivalent to 10.9 d/log for FU-CJD-infected GT1 cells. In FU-CJD brain it is 12 d/log and this <10% faster rate is not meaningful in view of the underlying cell replication rate, i.e., 2.551 d/log for FU-CJD culture homogenates versus 2.607 d/log for brain homogenates. These data implicate a fundamental and intrinsic replication clock of the FU-CJD agent that is preserved and cell environment-specific. Finally, the FU-CJD agent in brain, and in GT1 cells, provoked the same % rise in PrP-res per log, further demonstrating that this agent in brain and in cells conserved its fundamental characteristics. The slope for both GT1 cells and brain shows an ∼11% PrP-res change per log titer across a 3 log span ( and ). In sum, (1) GT1 cells produced 10-fold higher amounts of FU-CJD agent than brain, (2) in cultured cells this agent's doubling time was increased >8-fold and (3) the FU-CJD agent maintained a consistent stable phenotype in both environments.
kCJD agent maintains a different plateau and clock in mice and in GT1 cells.
The New Guinea geographic kCJD agent induces a markedly different brain lesion pattern than Japanese CJD isolates such as FU-CJD. It also has a much longer incubation time in wt mice.Citation4 Others have concluded that kCJD, on the basis of this long incubation, is equivalent to sCJD isolates.Citation36 However, simple inspection of coronal mouse brain sections shows kCJD affects extensive thalamic and amygdala regions whereas sCJD shows comparable pathology only in a tiny focal thalamic region. The kCJD agent, unlike sCJD, also elicits abundant PrP-res deposits in spleen,Citation4 and also markedly reduces the PrP shadow (shadoo) protein.Citation37 Moreover, kCJD agent reproducibly induces PrP-res in GT1 cellsCitation4 whereas sCJD infections are silent in that they fail to induce detectable PrP-res in chronically infected GT1 cells.Citation7,Citation8 The inability of the sCJD agent to induce PrP-res in GT1 cells has been verified with four different mouse adapted sCJD agent isolates (reviewed in ref. Citation4 and unpublished data). Another unique feature of the kCJD agent is its extraordinarily shortened incubation time in Tga20 mice with ∼10× normal murine PrP levels. The kCJD agent has an incubation time of 154 d in Tga20 mice versus 440 d in wt mice (). This 286 d incubation time difference is unique in comparison to all other TSE agents, including sCJD and various scrapie agents as determined here. For example, in sCJD the incubation time is only 50 d shorter in Tga20 mice, a much smaller percent change for a very long incubation period.
summarizes the obviously different doubling time calculated here for kCJD as compared to other TSE agents. The ti of kCJD is >3-fold faster in Tga20 than in wt mice, whereas in sCJD it was <1.1-fold faster in the Tga20 mice. Because of these marked incubation differences, we assayed kCJD infectivity of two end-stage Tga20 brains for comparison with kCJD infected wt mouse brains. The kCJD-infected Tga20 and wt mouse brains all showed comparable end-point titers. This demonstrated that the final low infectivity plateaus in brain were ultimately dependent on this agent's intrinsic properties. Normal and Tga20 mice also showed the same %PrP-res difference per log titer on assay and are graphed together in .
Kuru brain contained significantly lower agent titers than FU-CJD brain, even though both agents induced equivalent levels of PrP-res (∼55%) and band patterns on blots.Citation4,Citation37 In GT1 cells, the kCJD agent induced more PrP-res than FU-CJD (∼35% versus 25% respectively) when exposed to ≥10e6 CE. demonstrates the abundant PrP-res in the input kCJD-infected brain (46%, lane 2). Note the new GT1-specific PrP-res band pattern is already visible by p1 at input dilutions of 3e5 and 3e4 CE (). Thus de novo induction of PrP-res by the kCJD agent is very rapid, especially as compared to FU-CJD. The minor 13 kd diagnostic FU-CJD PrP-res band is not seen in kCJD-infected GT1 cells. High CE inputs of kCJD brain were required to induce high levels of PrP-res, and in accord with this observation, serial dilutions confirmed kCJD brains had signifi- cantly lower titers than FU-CJD brains. The lowest kCJD input that induced PrP-res was 3e2 CE. As shown in , by p13 there was only a very faint signal with a 17× load in one of the duplicate 300 CE kCJD input wells. This positive lane contained 0.5% PrP-res. Thus the TCID of kCJD-infected mouse brain is 100-fold less infectious than FU-CJD mouse brain. Even more outstanding is the finding that the kCJD brain contains 1,000-fold less infectivity than FU-CJD-infected GT1 cells.
Unique patterns of PrP-res accumulation in kCJD-infected cells.
Striking differences in the progressive accumulations of PrP-res also differentiated the kCJD and FU-CJD agents in GT1 assays. In kCJD, PrP-res was very high by p1 (20% PrP-res) with an input of 3e5 CE, as shown in . This PrP-res induction contrasts with FU-CJD where it took 4 passages to reach the same high PrP-res level at the same 3e5 CE input (). Notably, kCJD-infected brain was more effective in inducing a host PrP-res response even though it contained 100-fold less agent than FU-CJD brain. An enhanced PrP amyloid response to the kCJD agent may be part of a more efficient host cell defense mechanism involving agent-specific recognition and clearance pathways. This suggests that PrP misfolding can have a cell-protective function, rather than a toxic effect.
Graphic analyses of PrP-res versus input CE demonstrates other dramatic differences of kCJD- and FU-CJD-infected brains. shows that at each serial dilution of kCJD brain input, PrP-res barely increases between p1 and p7 (24 d in culture). Even though PrP-res barely increased with sequential cell passages, the PrP-res marker still reflects kCJD agent titers. The graph in quantifies the relationship of PrP-res to kCJD titer. There is an 8% PrP-res difference per log input kCJD brain titer. This is clearly different from the 11% PrP-res difference per log FU-CJD agent titer. Furthermore, unlike FU-CJD, where each input dilution yielded a separate parallel line, essentially the same single line is seen at different passages over 3 logs of kCJD input.
kCJD-infected cells maintain a constant, low titer plateau at different passages.
To further validate that infectivity, and not just PrP-res, was being maintained with passage, we evaluated the titer of kCJD infected cells at representative long term passages. Three independent cell infections from different kCJD brain homogenates were sampled. At p30, one group of kCJD infected cells showed 3–4-fold more TCID than brain, not a meaningful difference, especially for a single sample. At cells at p51 these cells also contained slightly more infectivity than brain (110%). In the latter example an input of 3e2 cells yielded a faint positive signal by p7 whereas it took six additional passages for brain to induce a comparable signal (, p13). In another independent kCJD infection, cells frozen at p80, and then thawed, also had a TCID that was indistinguishable from brain homogenates (60% of brain TCID). Thus kCJD infectivity was consistently preserved on long-term culture. These data further demonstrated a constant relatively low titer plateau that was specific for the kCJD agent and remarkably was the same for kCJD infected brain and cells. Titer of kCJD cells taken at early passages after high input infection also showed the same titer as brain; at p7 the kCJD titer of GT1 infected cells was 120% of brain infectivity, i.e., 1 TCID per ∼300 cells, in accord with all the other determinations. In contrast to FU-CJD agent propagated in GT1 cells, there was no significant increase of the kCJD agent during early passages or even after continuous multiple passages for more than 200 d in culture. Given the identical monotypic cell background, this stable 1,000-fold difference in maximal titer plateaus of the FU-CJD and kCJD agents must depend on intrinsic agent properties. Either a GT1 cell response to each specific agent, or some form of agent self-inhibition, is likely to underlie these profoundly different plateaus of infectivity.
The kCJD data also revealed an intrinsic kCJD agent clock that was different than that of FU-CJD, not only in brain, but also in GT1 cells. The doubling time of both agents was markedly enhanced by growth in culture. Because the titer remained essentially the same for each kCJD dilution during sequential cell passages, the kCJD agent replication would have to keep up only with ongoing cell replication in chronic cell infections. Thus the kCJD agent had a ti of 1 in culture, or only marginally, but not significantly faster than the GT1 cell doubling time (). Clearly the kCJD agent was not lost in culture, but did alter its rate of replication. The kCJD doubling time in GT1 cells was 33-fold and 10-fold faster than in wt and Tga20 mouse brain respectively (), a remarkable and much greater change than found in the transmissions of FU-CJD brain to GT1 cells. In contrast, the FU-CJD agent released from brain inhibitory mechanisms was only 6.2- to 8.4-fold faster in the monotypic GT1 cell environment. The rapid induction of high levels of PrP-res by the low titer kCJD agent also shows that infectious agent replication and PrP amyloid formation are each governed by independent clocks.
Persistent low kCJD plateaus implicate limited cell-to-cell spread.
If cells take up close to input infectivity applied, and the agent replicates in those cells but it does not spread, then infectivity will remain at a plateau. A total of ∼105 GT1 cells were exposed to the maximal applied dose of ≤0.1% kCJD brain homogenate (106 brain cells/ml). These cells would be exposed to only 3e3 TCID (agent at 1/300th per brain cell). This represents an MOI of 1 TCID per 30 GT1 cells, only 10x more than the infectious particles that can be incorporated initially, as by endocytosis. The kCJD target cells in each dilution persistently maintained close to the applied titer in continuous passages. Moreover, even high titer FU-CJD GT1 cells release very few infectious particles into the supernatant, and efficient infection requires cell-to-cell contact as demonstrated previously in reference Citation7. The continued persistence of kCJD at close to input homogenate titers suggests that once the kCJD infectious particle acquires an intracellular location it can replicate, but is unable to infect new adjacent cells though contact. Thus the kCJD agent keeps up with GT1 cell replication, but is transmitted only to a limited progeny population by cell division.
Discussion
Monotypic GT1 cultures that support a variety of distinct TSE agents offer new avenues for understanding intrinsic agent-specific properties. The GT1 culture model provides an inexpensive and stable source of unique TSE agents that can be rapidly titered with only ∼3-fold less sensitivity than 500 d mouse assays. Infectivity assays are essential for discovery of agent-specific molecules,Citation6,Citation27 and in analyzing various TSE agents we uncovered profound agent differences by serial end-point titrations. The current extensive end-point determinations for many mouse brains and infected GT1 cells was not practical by animal assay, and GT1 assays showed reproducible end-point titers for independent samples. The New Guinea kCJD agent has not been transmitted to cultured cells elsewhere, and it showed striking intrinsic agent differences from both the Japanese FU-CJD agent and the sporadic CJD agents. Each agent had vastly different replication and PrP-res inducing properties in monotypic GT1 cells. Differences in PrP-res accumulation in kCJD- and FU-CJD-infected cells point to a protective role for this host protein, a finding not in accord with the common assumption that PrP-res is toxic.Citation38,Citation39 Other TSE agents, such as the unique vCJD (UK BSE) agent, also propagated in stable GT1 cells, may reveal additional, and previously unappreciated distinctions among TSE agent clocks and their control. The biological GT1 titrations here also added to the many known discrepancies between PrP-res and TSE infectious titers. Titers based only on PrP-res levels, even in this simplified cell culture model, were incorrect by a factor of 100–1,000×, i.e., kCJD induced the same maximal levels of PrP-res as FU-CJD in culture even though kCJD infected cells contained 1,000-fold fewer infectious particles. Using PrP-res as a valid measure of titer can thus be problematic in test tube “PMCA” amplificationsCitation40,Citation41 where large amounts of PrP are converted to PrP-res, but little or no infectivity is typically generated.Citation1
More importantly, the above studies brought out agent-defined replication times and maximal agent plateaus in both brain and infected monotypic cells. FU-CJD- and kCJD-infected wt mouse brains were 100-fold discordant at terminal stages of disease when agent titers are maximal. Although kCJD yielded a uniquely large 240 d shorter incubation time in high PrP Tga20 mice as compared to wt mice, the final brain titer in both mouse genotypes was the same. Scrapie agent titers in Tga20 brain have been the same in wt mice or 1 log lower,Citation42 and specific agents may behave differently in Tga20 mice. In kCJD, the maximal plateau titers in brain define an intrinsic agent property that is not controlled by PrP availability. Complex host innate immune responses have been identified that can be differentially induced by TSE agents, and these are likely to limit the replication of particular agents in brain.Citation23,Citation32,Citation34,Citation43 Many of the observed molecular host responses to these foreign agents are not induced by host PrP-res, and sets of specific innate immune transcripts are highly induced very early in infection, well before any PrP-res can be detected.Citation23,Citation34 Furthermore, when the kCJD and FU-CJD agents were released from complex host controls by passage in GT1 cells, both agents dramatically accelerated their doubling times, a finding consistent with release from inhibitory innate immune molecules and processes in brain. The kCJD agent in culture achieved a 33-fold faster rate than found in wt mice. We are not aware of any observed PrP or thermodynamic host protein self-conversion properties that can account for this strain-specific slow, and then markedly accelerated agent replication in culture. Many viruses, such as poliovirus, show enhanced replication in cultured cells.
The FU-CJD agent achieved higher titers in cells than in brain, consistent with previous studies.Citation8 In contrast, the kCJD agent maintained the same titer in cells and brain. Moreover, FU-CJD yielded a 1,000-fold higher titer at its maximal GT1 plateau than kCJD, a remarkable agent-specific difference. This titer plateau difference was not due to a large variation in effective doubling time of these two agents (ti of 0.8 and 1 respectively), but instead demonstrated dramatic differences in intrinsic agent virulence, even in GT1 neuronal cells that lack adaptive immune defenses. The basis for this observed replication difference probably involves several mechanisms. Since TSE infectious agents, like viruses, are obligate intracellular pathogens, they will be subject to cell controls that may limit final agent plateaus even in monotypic cells. Plateaus in monotypic cells were as agent-specific as in mice. It is of general, as well as therapeutic interest, to understand the molecular basis of these host controls.
The rapid accumulation of PrP-res in kCJD-infected GT1 cells may limit agent titers, as suggested by the data here. Although the kCJD agent was constantly replicating at the same rate as cells (ti = 1, i.e., doubling every day) the agent did not progressively accumulate. If agent is restricted to the few cells initially infected, then only daughter cells will be infected during cell division, while the rest of the parallel-dividing population will remain uninfected. Thus the proportion of infected cells will remain the same in sequential passages. In serial dilution challenges, only the initial kCJD input infectivity was maintained at a steady state, in accord with this prediction. Additionally, cell PrP rapidly converted to its misfolded amyloid form with kCJD infection, even though input samples had relatively low titers of only 1 TCID per 300 cells (∼7 logs LD50/gm by mouse assay). In contrast, in FU-CJD-infected cells (1 TCID per 0.3 cells) agent progressively accumulated much higher titers, yet had a long silent phase of exponential agent replication when no PrP-res was induced. Thus GT1 cells may respond to kCJD infectious particles more vigorously than to FU-CJD particles, with rapidly accumulating PrP-res that forms insoluble amyloid aggregates inside the cell.Citation12 These aggregates may prematurely trap the kCJD agent and enhance its clearance, a mechanism previously suggested.Citation25 Agent trapped within amyloid aggregates may also retard agent dispersal to other cells, or enhance agent removal through efficient endosomal-lysosomal pathways. An abundance of lysosomes is an ultrastructural characteristic of CJD infected brainCitation44 and is known to be pathognomonic for many neurodegenerative diseases including AD.
Additional non-PrP mechanisms can also be involved in limiting agent infection. In wt mice, a classical vaccination strategy showed the low virulence sCJD agent prevented superinfection by the more virulent FU-CJD agent, even though sCJD brain showed no detectable PrP-res.Citation45 Furthermore, the sCJD agent itself can achieve only very low titers in both mice and GT1 cells, indicating a low titer plateau is not dependent on PrP-res induction. The sCJD interference effect was additionally demonstrated in GT1 cells, and completely prevented superinfection by FU-CJD as well as more virulent scrapie agents.Citation7 Surprisingly, virulent agents that induced abundant PrP-res could still be doubly infected with a second strain. Agent-specific control mechanisms, rather than PrP host cell changes, can independently limit replication as well as the final plateau titer of specific agents. This indicates general viral mechanisms of inhibition, such as small RNAs directed against TSE agent sequences, and/or the production of defective interfering particles by less virulent strains, as previously suggested in reference Citation7.
The current results also present another strong possibility not previously considered: less virulent agents may have a limited capacity to spread from cell to cell, and thus attain only a low end-titer plateau. Homogenates used here to infect GT1 cells should be taken up by endocytotic-endosomal structures that incorporate and process a variety of material with little specificity. Therefore incorporated infectious particles should approximate the number of such particles applied, if uptake is completely efficient. Indeed, kCJD infected cells persistently maintained close to the applied input infectivity throughout continuous passages. The continued persistence of kCJD at close to homogenate titers suggested that kCJD infectious particles are taken up non-specifically to an intracellular location, but are then unable to infect other cells. Spreading infections are most efficient though direct cell-to-cell contacts since few infectious particles are released into the supernatant, even with high titer FU-CJD.Citation7 In summary, the kCJD agent keeps up with GT1 cell replication, but is transmitted only to a limited progeny population by cell division. This can represent a symbiotic latent state, and is relevant for low level persistent TSE infections of the lymphoreticular system that can be quiescent for >10 years in humans, as known from iatrogenic peripheral infections.Citation24,Citation25
We have found that PrP is markedly increased in neuronal cells that are terminally differentiated by arresting cell division under physiological conditions. With arrest, nanotubes and PrP-rich plasma membrane cell-to-cell contacts are rapidly increased. Thus the normal function of PrP likely contributes to the elaboration of cell contacts that are used for transfer of viral particles as well as neuronal communication.Citation35 Since PrP appears to act as a required receptor for TSE agent infection and replication, it is possible that less virulent agents such as kCJD and sCJD are transferred less effectively through host membrane PrP during their natural mode of cell-to-cell spread. Interestingly, a previous study showed that PrP-res aggregates in vCJD human brain homogenates were acutely taken up by human Caco-2 cells, and these amyloid collections rapidly spread across nanotubes to adjacent cells.Citation46 This spread, however, may not represent transfer of vCJD infectious particles, because we were repeatedly unable to infect those Caco-2 cells (gift of Monique Rousset) with veri- fied vCJD human homogenates that had transmitted to mice, i.e., no de novo PrP-res was detectable after exposure, even after many cell passages.
On a broad epidemiological scale, the relative virulence of TSE agents can be defined in terms of their geographic spread and their ability to infect many species.Citation33 We here found that agent-specific replication, virulence, and cell-to-cell spread are additional important parameters uncovered by cell culture studies. TSE agent strains appear to be quite stable because even after they are transmitted to a different species and then transmitted back to the first species, the agent again recreates its initial species-specific disease of incubation time and regional neuropathology.Citation4 This implies TSE agents have a genome with limited mutations, unlike rapidly mutating RNA viruses that stochastically would be unable to reverse all their many mutations. TSE agent phenotype stability also contrasts with recombinant amyloid yeast “prions” that do not breed true, but rather produce many heterogeneous prion variants on “infection.”Citation47 The current agent-specific replication and plateau characteristics in monotypic cells further indicates the infectious particle contains a stable foreign nucleic acid genome that is protected from destruction in the geographic environment, and as it transits through the gastrointestinal tract. We have discovered several new circular DNAs of viral size (1.8 kb and 2.4 kb) in various TSE infectious particle preparations that resist detergent lysis and nuclease digestion.Citation27 Interestingly, these DNA sequences have links to environmental endosymbiotic pathogens that are widespread in soil and water. Such prevalent and possibly ancient agents may infrequently cause disease. The remarkably long silent exponential phase of FU-CJD agent replication in GT1 cells is most germane for in vivo latency. Moreover, the limited cell-to-cell spread of kCJD is also relevant for the enormously long peripheral phase of disease, where PrP-res can be contained in a few cells of the lymphoreticular system for many years.Citation25
Materials and Methods
Brains and cell dilutions for rapid endpoint titers.
A minimum of three complete end-point dilution assays were done using serially passaged mouse brains that had been infected with either FU-CJD or kCJD.Citation4,Citation10 Brains from uninfected controls and infected CD-1 mice at terminal stage of disease were frozen at −80°C. Tga20 mice that express murine PrP at 8–10-fold normal levels have a >200 d shorter kCJD incubation time than CD-1 mice,Citation4 and both mouse genotypes were evaluated by serial dilution to determine end-point titers. Brains were rapidly thawed and homogenized in cold PBS (10% wt/vol). Brain contains ∼109 cells/gm and thus a 10% brain homogenate contains 108 cells equivalents (CE) per ml.Citation5 CE calculations facilitated standardized comparisons of infected brain and cultured cells. Serial dilutions of infected brain were made as previously in medium containing 0.01% normal CD-1 mouse brain as a carrier.Citation5 This minimized losses at high dilution, and ensured that components in all inocula were essentially the same. For dilution of cultures, aliquots of newly infected as well as previously infected GT1 cells propagated for short and long times (8–80 passages), were counted, and cell numbers verified independently by protein assay, and frozen at −80°C. Thawed cells were syringed ×10 through a 27G needle, vortexed for 10 s, and bath sonicated ×10 for 10 s before being serially diluted with carrier as above. Identical CE dilutions of brain and cultures were applied to replicate wells for generating the standard reference titration curve of each agent. Only two 6-well plates are needed to test six serial dilutions in duplicate. This format produced rapid and reproducible data from many independent samples.
GT1 infectivity assays of kCJD and FU-CJD agents.
GT1 neuronal indicator cells, maintained in MEM with 10% serum were seeded in 6-well plates as previously for 22L-scrapie agent.Citation5,Citation6 Briefly, replicate wells were plated with 105 uninfected GT1 cells in 2 ml medium. After ∼36 h (30–40% confluence), the medium was replaced with 1 ml of medium containing infectious or uninfected material. The next day another ml of medium was added, and the cells were incubated for 2 additional d. The infectious medium was then removed and the cells washed ×3 with serum-free medium, ×1 with complete medium, and then incubated with 2 ml of fresh medium for 2 more d (90–95% confluence). This protocol maximizes the time of exposure to infectious inocula for very diluted samples, and incorporated suf- ficient time for cell recovery and undisturbed growth after rigorous washing to minimize non-specific retention of inocula and false positives at passage 1 (p1). For subsequent passages, cells were split at 1:4 every 4 d; 105 cells were seeded in each new well and the remaining cells seeded in 25 cm flasks for PrP and PrP-res analyses by western blotting. Flask cells were collected in parallel and analyzed for as long as 15–18 passages to determine endpoint tissue culture infectious doses (TCID). GT1 cells exposed to uninfected brain or cells never gave any positive PrP-res signal on western blots, even after 15 passages. In fact, since dilutions were always applied with a high amount of normal brain homogenate (0.01%), this negative control was always present in every experiment. Additional controls included uninfected GT1 cells (all negative).
Western blot quantification of PrP and PrP-res was performed with standard electrophoresis and blotting methods as previously described in reference Citation8, using a goat anti-PrP antibody (M20, Santa Cruz Biotechnology, Santa Cruz, CA). Limited digestions with 25 µg/ml proteinase K (PK) for 30 min at 37°C were optimized as previously for maximal PrP-res. For normalization of loads, tubulin and other filament antibodies were detected on the same blot,Citation35 and peroxidase labeled secondary antibody signals were developed with chemiluminescent substrate (ChemiGlow, Alpha Innotech, San Leandro, CA). The %PrP-res (of total PrP) was determined in the >3 log linear signal range with Fluorochem software.
Calculation of agent replication times.
Effective doubling times (ti) were calculated according to a previously derived formula.Citation2 In dividing cells, unlike brain, an agent's doubling time is the product of the exponential growth rate of cells multiplied by the exponential growth rate of agent. The following alternate calculation, provided by Michael Frame at Yale, yielded the same numerical results as described in the Results using ti calculations. The following formulas are valid for agent replicating in a single cell, as well as in a population of cells in which the agent is spreading. They are also valid for a population of cells with variable amounts of agent (full proof by MF available on request). Most simply, for a log (10-fold) change:
ln(10)/days to increase 10 fold = ex days; for doubling use ln(2)/days = ex
The following is an example alternately calculated in the Results with the same answer:
Let Cells at days t = C(t) = C(0) × e(k × t) for some constant k
If cells double each day then k = ln(2) = 0.693147 and C(t) = C(0) × e(0.691347 × t)
thus a 10 fold cell increase = ln(10) = 0.693147 × t or 3.3219 days/log Cell increase
Let Agent at days t = A(t) where the m constant applies, A(t) = A(0) × e(m × t)
If A increases 10 fold in 12 days then
10 × A(0) = A(0) × e(m × 12) and m= ln(10)/12 = 0.191882
Finally, the total rate of agent increase = (increase in cells) × (increase in agent)
= C(0) × e(0.693147 × t) × A(0) × e(0.191882 × t). Thus, ln(10)/0.885029 = t = 2.6017 days/log agent increase.
Figures and Tables
Figure 1 Infectivity assays of FU-infected brain homogenates. (A) Western blots of GT1 indicator cells exposed to serial FU-CJD brain dilutions (3e5 to 3 CE) at p5; with dilution decreasing amounts of PrP-res are induced. Proteinase K treatment (PK) and relative protein load indicated in each lane are indicated, as well as the %PrP-res (of total PrP) beneath the +PK lanes. Note the FU-CJD diagnostic 13 kDa PrP-res band (arrow). (B) With further passages, PrP-res signals are detectable with an input of only 3CE as shown at p13. Since additional passages did not show a PrP-res signal with lower input CE, FU-CJD homogenates have 1 TCID per 3 brain cells. (C). Graph of %PrP-res induced by each serial CE dilution at progressive passages. Note that the incubation time curves are roughly parallel for each dilution, i.e., the PrP-res rate of increase is similar for each input dilution. PrP-res in FU-CJD cells became maximal at ∼25%, and minor PrP-res variations between 25–31% were due to variable breakdown during cell collections; when cellular lysosomal enzymes digest PrP during collection, total PrP will be reduced, and thus the %PrP-res falsely increased. (D) Plots the log agent TCID to the linear accumulation of %PrP-res at different passages. More extensive passages with 3 CE input (p15) confirmed a 4 log discrimination of the FU-CJD agent by this assay. This as well as other western blots shown here revealed no additional small bands of <12 kd as previously depicted in references Citation3 and Citation4.
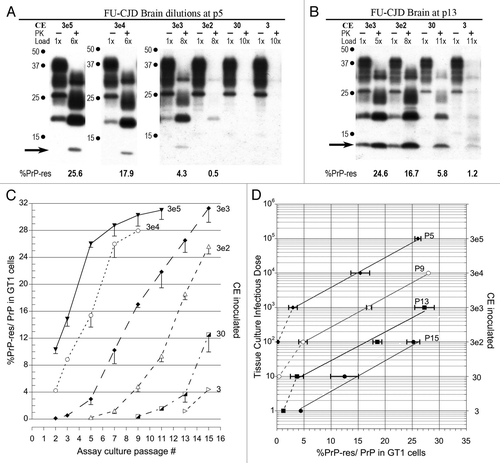
Figure 2 Infectivity of GT1 cells chronically infected with FU-CJD. (A) Representative western blots of GT1 homogenate dilutions at p7 and p13. Progressive passages show infectivity in as little as 0.3 CE by p13. This is a 10 fold higher TCID than FU-CJD brain (). PK treatment, CE applied, %PrP-res induced with each serial dilution, and the diagnostic 13 kDa band are indicated. Note the GT1 cells exposed to FU-infected cell homogenate dilutions accumulate PrP-res more rapidly than those exposed to FU-infected brain homogenates. (B) Graph of progressive PrP-res accumulation at each input CE dilution. (C) Standard plots of log TCID to %PrP-res shows the same slope in FU-CJD infected cells and brain; the only difference is that fewer infected cells were required to induce the same %PrP-res, i.e., the cells were consistently more infectious.
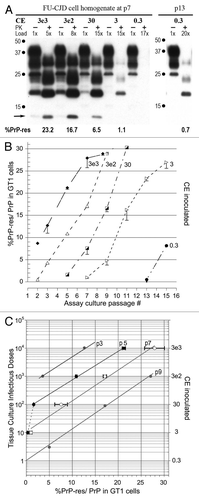
Figure 3 Western blot TCID assays of kCJD-infected brain. (A) Passage number, input cell equivalents (CE) and relative protein loads applied to each lane are indicated. The reference brain-specific PrP-res band pattern loaded with 3e5 brain homogenate CE is not visible in the GT1 indicator cells exposed to kCJD brain. Instead, a de novo GT1-specific PrP-res pattern is already seen at p1. A series of input dilutions, including lower CE are shown for p7 and p13 after kCJD brain infection. Note at p1 and p7 the %PrP-res is the same and does not rise, e.g., compare 3e4 and 3e3 CE inputs at p1 and p7. The %PrP-res in GT1 cells exposed to 300 CE was detectable on only very dark films at p7 but was clearly positive at p13 with a 17× load. Thus kCJD brain contained maximally 1 TCID per 300 cells, a 1,000-fold lower titer than FU-CJD-infected cells. The mouse brain PrP-res pattern was the same for kCJD and FU-CJD as shown previously in references Citation3 and Citation4 and in those transmissions, no bands of <15 kd were seen in kCJD infected GT1 cells. (B) Plot of %PrP-res exposed to CE dilutions at sequential passages confirms the lack of increase in %PrP-res with time. Nevertheless, the different levels of %PrP-res accurately discriminate different input CE s (and titer). There is a significant difference in the %PrP-res from 300 to 3e5 CE at p7, yielding a 3 log TCID span for kCJD. (C) Standard kCJD plot for %PrP-res to infectious titer. Note that only the same slope, but the same line is seen with different inputs at p5 and p7.
Table 1 TSE agents display very divergent replication “clocks” when studied in the same host background as seen in the above numbers
Acknowledgments
This work was supported by NIH grants NINDS ROI 012674 and NIAID grant R21 A1076645.
References
- Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem 2007; 100:897 - 915
- Manuelidis L, Fritch W. Infectivity and host responses in Creutzfeldt-Jakob Disease. Virology 1996; 215:46 - 59
- Manuelidis L, Liu Y, Mullins B. Strain-specific viral properties of variant Creutzfeldt-Jakob Disease (vCJD) are encoded by the agent and not by host prion protein. J Cell Biochem 2009; 106:220 - 231
- Manuelidis L, Chakrabarty T, Miyazawa K, Nduom NA, Emmerling K. The kuru infectious agent is a unique geographic isolate distinct from Creutzfeldt-Jakob disease and scrapie agents. Proc Natl Acad Sci USA 2009; 106:13529 - 13534
- Liu Y, Sun R, Chakrabarty T, Manuelidis L. A rapid accurate culture assay for infectivity in transmissible encephalopathies. J Neurovirol 2008; 14:352 - 361
- Sun R, Liu Y, Zhang H, Manuelidis L. Quantitative recovery of scrapie agent with minimal protein from highly infectious cultures. Viral Immunol 2008; 21:293 - 302
- Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 2005; 310:493 - 496
- Arjona A, Simarro L, Islinger F, Nishida N, Manuelidis L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc Natl Acad Sci USA 2004; 101:8768 - 8773
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 1997; 389:498 - 501
- Manuelidis L, Lu ZY. Attenuated Creutzfeldt-Jakob Disease agents can hide more virulent infections. Neurosci Lett 2000; 293:163 - 166
- Manuelidis L. Beneath the emperor's clothes: the body of data in scrapie and CJD. Annales de L'Institute Pasteur 1997; 8:311 - 326
- Manuelidis L, Yu ZX, Barquero N, Mullins B. Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25 nm virus-like particles. Proc Natl Acad Sci USA 2007; 104:1965 - 1970
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Auget M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner S, Baldwin M, Collinge J, DeArmond S, Marsh R, Tateishi J, et al. Prions 1995; Wien Springer Verlag
- Legname G, Baskakov I, Nguyen H, Riesner D, Cohen F, DeArmond S, et al. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infection in the mouse. J Infect Dis 1967; 117:15 - 22
- Manuelidis EE, Gorgacz EJ, Manuelidis L. Viremia in experimental Creutzfeldt-Jakob disease. Science 1978; 200:1069 - 1071
- Pincock S. Government confirms second case of vCJD transmitted by blood transfusion. Brit Med J 2004; 329:351
- Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science 1977; 197:943 - 960
- Jeffrey M, Gonzalez L, Espenes A, Press C, Martin S, Chaplin M, et al. Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 2006; 209:4 - 14
- Scherbel C, Pichner R, Groschup MH, Mueller-Hellwig S, Scherer S, Dietrich R, et al. Infectivity of scrapie prion protein (PrPSc) following in vitro digestion with bovine gastrointestinal microbiota. Zoonoses Public Health 2007; 54:185 - 190
- Baker CA, Martin D, Manuelidis L. Microglia from CJD brain are infectious and show specific mRNA activation profiles. J Virol 2002; 76:10905 - 10913
- Manuelidis L, Sklaviadis T, Akowitz A, Fritch W. Viral particles are required for infection in neurodegenerative Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 1995; 92:5124 - 5128
- Manuelidis L. Transmissible encephalopathies: speculations and realities. Viral Immunol 2003; 16:123 - 139
- Sklaviadis T, Akowitz A, Manuelidis EE, Manuelidis L. Nucleic acid binding proteins in highly purified Creutzfeldt-Jakob disease preparations. Proc Natl Acad Sci USA 1993; 90:5713 - 5717
- Manuelidis L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 2011; 17:131 - 145
- Vorberg I, Raines A, Priola S. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J Biol Chem 2004; 279:29218 - 29225
- Mahal S, Baker C, Demczyk C, Smith E, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 2007; 104:20908 - 20913
- Li J, Browning S, Mahal S, Oelschlegel A, Weissmann C. Darwinian evolution of prions in cell culture. Science 2010; 327:869 - 872
- Manuelidis L. Vaccination with an attenuated CJD strain prevents expression of a virulent agent. Proc Natl Acad Sci USA 1998; 95:2520 - 2525
- Baker C, Lu Z, Manuelidis L. Early induction of interferon-responsive mRNAs in Creutzfeldt-Jakob disease. J Neurovirol 2004; 10:1 - 12
- Manuelidis L. Transmissible encephalopathy agents: Virulence, geography and clockwork. Virulence 2010; 1:101 - 104
- Lu ZH, Baker C, Manuelidis L. New molecular markers of early and progressive CJD brain infection. J Cell Biochem 2004; 93:644 - 652
- Miyazawa K, Emmerling K, Manuelidis L. Proliferative arrest of neural cells induces prion protein synthesis, nanotube formation and cell-to-cell contacts. J Cell Biochem 2010; 111:239 - 247
- Wadsworth J, Joiner S, Linehan J, Desbruslais M, Fox K, Cooper S, et al. Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc Natl Acad Sci USA 2008; 105:3885 - 3890
- Miyazawa K, Manuelidis L. Agent-specific shadoo responses in transmissible encephalopathies. J Neuroimmune Pharmacol 2010; 5:155 - 163
- Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris D. Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J Neurosci 2008; 28:13258 - 13267
- Sandberg M, Al-Doujaily H, Sharps B, Clarke A, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 542
- Chen B, Morales R, Barria M, Soto C. Estimating prion concentration in fluids and tissues by quantitative PMCA. Nat Method 2010; 7:519 - 520
- Shi S, Dong C, Wang G, Wang X, An R, Chen J, et al. PrP(Sc) of scrapie 263K propagates efficiently in spleen and muscle tissues with protein misfolding cyclic amplification. Virus Res 2009; 141:26 - 33
- Karapetyan Y, Saß P, Mahal S, Sferrazza G, Sherman A, Sales N, et al. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS ONE 2009; 4:5730
- Baker C, Lu ZY, Zaitsev I, Manuelidis L. Microglial activation varies in different models of Creutzfeldt-Jakob disease. J Virol 1999; 73:5089 - 5097
- Manuelidis EE, Gorgacz EJ, Manuelidis L. Interspecies transmission of Creutzfeldt-Jakob disease to Syrian hamsters with reference to clinical syndromes and strains of agent. Proc Natl Acad Sci USA 1978; 75:3432 - 3436
- Manuelidis L, Lu ZY. Virus-like interference in the latency and prevention of Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 2003; 100:5360 - 5365
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman D, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 2009; 11:328 - 336 11
- Brachmann A, Baxa U, Wickner R. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 2005; 24:3082 - 3092