Abstract
Uropathogenic Escherichia coli (UPEC) produces ~80% of community-acquired UTI, the second most common infection in humans. During UTI, UPEC has a complex life cycle, replicating and persisting in intracellular and extracellular niches. Host and environmental stresses may affect the integrity of the UPEC genome and threaten its viability. We determined how the host inflammatory response during UTI drives UPEC genome instability and evaluated the role of multiple factors of genome replication and repair for their roles in the maintenance of genome integrity and thus virulence during UTI. The urinary tract environment enhanced the mutation frequency of UPEC ~100-fold relative to in vitro levels. Abrogation of inflammation through a host TLR4-signaling defect significantly reduced the mutation frequency, demonstrating in the importance of the host response as a driver of UPEC genome instability. Inflammation induces the bacterial SOS response, leading to the hypothesis that the UPEC SOS-inducible translesion synthesis (TLS) DNA polymerases would be key factors in UPEC genome instability during UTI. However, while the TLS DNA polymerases enhanced in vitro, they did not increase in vivo mutagenesis. Although it is not a source of enhanced mutagenesis in vivo, the TLS DNA polymerase IV was critical for the survival of UPEC during UTI during an active inflammatory assault. Overall, this study provides the first evidence of a TLS DNA polymerase being critical for UPEC survival during urinary tract infection and points to independent mechanisms for genome instability and the maintenance of genome replication of UPEC under host inflammatory stress.
Introduction
Urinary tract infections (UTIs) are among the most common bacterial infections in humans, affecting about 10 million people annually in the US.Citation1,Citation2 Understanding conserved molecular mechanisms of UTI pathogenesis may help to identify key bacteria pathways necessary for virulence that, in turn, may provide the basis for the development of new therapeutics, crucial to renew the battle against UTI.
Uropathogenic Escherichia coli (UPEC) is the leading etiology of UTIs.Citation1 UPEC adheres to the urinary tract epithelium, and within the bladder, invades into the epithelium through multiple endocytic pathways and the invagination of fusiform vesicles.Citation3,Citation4 In some circumstances UPEC rapidly replicates in intracellular bacterial communities (IBC), a biofilm-like state observed in animal and human infections.Citation3,Citation5 UPEC may also chronically persist in the epithelium (16).
In most hosts, a robust innate immune response is initiated upon the earliest stages of UPEC UTI. During cystitis, the interaction between host toll-like receptor 4 (TLR4) of urinary epithelium and lipopolysaccharide (LPS) of UPEC results in a pro-inflammatory cytokine response with IL-6 and IL-8.Citation6,Citation7 Antimicrobial peptides and complement may contribute to cell wall stress, while oxidative radicals damage cell structures such as the outer membrane, critical enzymes and the genome of UPEC.Citation8,Citation9
The oxidative components of the innate immune response are numerous. Nitric oxide (NO·) produced by inducible nitric oxide synthase (iNOS) in response to UTI inhibits bacterial growth by modifying DNA and proteins.Citation10 Oxidation may directly damage genomic DNA as well as the dNTPs pool, leading to increased lesions and errors during DNA synthesis.Citation11,Citation12 These alterations may result in stalling of DNA replication, immediate termination of replication and subsequent cell death. Alternatively, repair of these lesions and errors may require low fidelity bypass synthesis, resulting in mutations, which in turn may promote selective changes such as increased virulence or antibiotic resistance.Citation13,Citation14
The most important components of mutational level control entail critical DNA synthesis and DNA repair processes are DNA polymerases. There are five known E. coli polymerases where the high fidelity DNA polymerase III (Pol III) and DNA polymerase I (Pol I) are essential and responsible for genome replication and Okazaki fragment processing, respectively.Citation15 DNA polymerases Pol II, Pol IV and Pol V serve as accessory enzymes.Citation16–Citation18 The primary function for Pol II is remains unclear, but it has been suggested that Pol II may serve as a backup enzyme for Pol IIICitation16 and re-initiates DNA synthesis in UV-irradiated E. coli cells.Citation19 Like Pol III and Pol I, Pol II is considered a high fidelity enzyme due to its proofreading exonuclease activity.Citation15 On the other hand, Pol IV, encoded by dinB (annotated in some UPEC genomes as dinP) and Pol V (UmuD'2C) are highly conserved Y-family translesion synthesis (TLS) polymerases with well-established low fidelity synthesis in vitro under laboratory conditions, due, in part, to lack of exonuclease proofreading function.Citation20,Citation21
Pol II, Pol IV and Pol V have been previously shown to be SOS-inducible, with increases in the number of molecules upon SOS conditions from 50 to 300, 25 to 2,500 and 0 to 200, respectively.Citation21,Citation22 In addition to host components and environments that may induce an SOS response, Perez-Capilla and colleagues demonstrated that fluoroquinolones used in the treatment of UTI also induced the expression of Pol IV.Citation23
Pol IV and Pol V are necessary for bypassing some errors in the genome that stall replication, but bypass of errors stalling replication can be mutagenic.Citation24 Overexpression of Pol IV and Pol V significantly increases the number of spontaneous mutations.Citation25,Citation26 Genetic deletion of dinB was shown to result in a modest decrease in the mutation frequency when the target gene was located on episomal DNA (F'), while deletion of umuDC coding for Pol V produces no significant decrease in mutation rate under non-stressed in vitro laboratory growth.Citation18 In vitro, Pol IV has a known role in error-free bypass of cytotoxic alkylating DNA lesions and in error-free translesion synthesis through 8-oxo-guanine lesions, as produced by reactive oxygen species and nitrofuran antibiotics.Citation27,Citation28 Pol V has a known essential function in the survival of UV irradiation through its synthesis over thymidine dimer photoproducts and abasic sites.Citation29 However, the relevant in vivo functions of Pol IV and Pol V within a host remain unclear.
Mismatch repair (MMR) and RecA-dependent homologous recombination are additional major mechanisms described in E. coli to process damaged DNA, repair errors and repair gaps. MMR is a highly conserved post-replicative system designed to recognize and repair DNA errors as base mispairs, frameshifts and deletions that occur during DNA synthesis and recombination. Depletion, saturation or loss of MMR components in vitro results in increased mutagenesis.Citation30,Citation31 MMR may be suppressed or saturated under certain physiological conditions such as stationary phase growth and chemical stress.Citation31,Citation32 These scenarios produce an MMR-deficient phenotype and are associated with increased mutagenesis.Citation31 Homologous recombination is activated when chromosomal damage cannot be bypassed by synthesis and viability is threatened. DNA lesions blocking DNA replication may lead to the uncoupling of leading and lagging strand synthesis, resulting in exposed single stranded regions of DNA. These regions may be covered with the nucleofilament RecA, which simultaneously cleaves LexA, activating the SOS response and de-repressing a myriad of LexA repressed targets, including three DNA polymerases.Citation33
How the integrity of the genome is managed under the relevant physiologic and inflammatory stress conditions present during infection is unclear. In our prior work, we demonstrated that UPEC undergoes an SOS-response during infection.Citation34 The SOS-response has also been demonstrated to be important for UPEC to persist in the inflamed host,Citation35 strongly suggesting that host stress components damage the genome and that induction of RecA-dependent SOS pathways was critical for survival during infection.
Here, our aim was to test the hypothesis that inflammation during UTI destabilizes the UPEC genome resulting in: (1) increased acquisition of mutations and (2) a requirement for the TLS DNA polymerases to bypass DNA errors/adducts and assure survival during the host inflammatory response. Through a combination of in vitro (outside the host) and in vivo (inside the host) studies, we demonstrate that the mutability of UPEC is strongly enhanced in vivo during experimental UTI and that genome instability is driven by physical stress in the urinary tract as well as inflammatory stress induced through a TLR4-dependent mechanism. We further demonstrate that during experimental UTI the UPEC TLS DNA polymerase Pol IV is essential for virulence, but despite its lack of proofreading functions, does not contribute to genome instability during infection. Together, these data suggest that UPEC genome instability affects a large proportion of the population during infection and that in vitro studies do not predict the roles of the genome repair processes under physiological host stress.
Results
Mutability of UPEC is strongly enhanced during occupation of the host.
To investigate the genome instability of UPEC under host stress conditions, the prototypic UPEC strain UTI89, a K1 human cystitis isolate, was used.Citation3,Citation36 A series of forward mutagenesis assays were employed to determine in vitro and in vivo mutation frequencies at unlinked, independent alleles (detailed in Materials and Methods). First, the mutation frequency of UTI89 was determined to assess if its constitutive mutation frequency was comparable to that of other E. coli strains. It has been shown that some of the natural isolates of E. coli have an enhanced mutator phenotype.Citation37 To exclude UTI89 as a constitutive hypermutator, we compared the in vitro mutation frequencies under non-stress growth conditions of the prototypic UPEC strains UTI89 and NU14Citation3 as well as the comparator strain MG1655,Citation38 a prototypic commensal isolate and found that they were all similar (). Variation in the mutation frequencies at different alleles ranged from ∼0.3/108 for nalidixic acid (nal) resistance, up to 310/108 for lacI mutant formation (p-gal growth test).
Next, we determined the frequency of mutations during UTI where UPEC is subjected to host physical and inflammatory stresses. We anticipated that infection would produce a marked increase in genome instability. We infected female C3H/HeN mice via transurethral catheterization to initiate cystitis. At 48 h post-inoculation (hpi), bladder homogenates were plated on both non-selective and selective media. UTI89 efficiently infected the bladders, reaching an average number of ∼106 CFU/organ (). Bacteria isolated from the infections where also immediately tested (without intermediate culturing) for a mutability phenotype using three of the forward mutagenesis assays. The mutation frequency was calculated for lacI mutants, for 6-azauracil (6-aza) and 5-fluorocytosine (5-FC) resistance ( and respectively). For all of the markers, we observed that the mutation frequencies were significantly elevated, up to 100-fold compared to the frequencies in the inocula. To determine if the frequency and degree to which genome instability was observed in vivo was specific to one host, the experiments were replicated in female BALB/c mice. As shown in , the bacterial burden and mutation frequencies for UTI89 at 48 hpi were equivalent in the two mouse strains. These data demonstrate that UTI produces significant genome instability for UPEC.
Induction of the host innate immune response significantly increases the genome instability of UPEC UTI89.
We considered that the host environment, which may include physical, chemical and metabolic stresses, may promote mutagenesis, while inflammatory stress could be additive. To model one aspect of physical and metabolic stress for UPEC during its persistence in the urinary tract, UPEC was grown in urine that was collected and pooled from healthy human donors. Overnight growth of UTI89 in human urine resulted in an increase in the mutation frequencies relative to the levels observed in bacteria grown in Luria-Bertani broth (LB) (). Of the multiple phenotypes assessed as measures of mutation frequencies, nalidixic acid resistance was the most dramatically altered, with ∼20-fold increase in urine over LB broth. For rifampicin (rif) and 6-aza resistance, we observed 5- and 8-fold increases in mutagenesis, respectively. To exclude the possibility that this phenotype is unique to UTI89, we cultured 12 independent E. coli strains (10 UPEC and 2 commensal) in pooled human urine and LB broth. As shown in , all of the strains had similar mutation frequencies relative to UTI89. In each case, the mutation frequencies were enhanced significantly by growth in human urine. Thus, UTI89 proved to be a representative strain in terms of mutability and did not have a hyper- or hypo-mutable phenotype relative to other E. coli in these assays.
Next, we sought to determine the contribution of the innate immune response and induction of inflammation to the development of UPEC genome instability. Female C3H/HeN and C3H/HeJ mice were infected for 48 hpi with UTI89 introduced by transurethral catheterization. C3H/HeJ mice that are hyporesponsive to LPS-TLR4 signaling were used as an inflammationattenuated strain to assess the role of inflammation as a driver of UPEC genome instability in vivo.Citation39 As shown in , infection of C3H/HeJ mice resulted in a mutation frequency approximately 1-log lower than the frequency in C3H/HeN mice. The frequency of mutation for UTI89 in the C3H/HeJ mice animals was similar to the elevated frequency in pooled human urine relative to the inoculum (), again suggesting that the host environment, even in the absence of a strong inflammatory response, still produces significant stress and genome instability for UPEC relative to in vitro growth. However, the combination of host environment and innate immune-driven inflammation produced the greatest degree of genome instability.
MMR is not a major contributor to genome instability or fitness during acute UPEC UTI.
The MMR system, a post-replicative repair system, was investigated as a potential source of the elevated in vivo mutation frequencies observed. MMR detects and repairs most of the typical replication errors and, therefore, was considered a potentially important source of spontaneous mutagenesis in vivo. We hypothesized that, during UTI, the UPEC MMR system may be suppressed or saturated by damage induced through host inflammatory mediators, leading ultimately to high mutation rates. To address this hypothesis, we constructed an MMR-deficient strain of UTI89 carrying a complete deletion of mutL. In vitro analyses showed that UTI89 ΔmutL strain had an expected strong mutator phenotype (). The largest increase in mutation frequency was observed for rif and nal markers (130- and 60-fold increases in mutation frequency compared to the wt parent, respectively).
We determined the contribution of MMR to UPEC mutability and fitness in vivo by infecting C3H/HeN mice with the mutL deletion and wt parent strains. Consistent with a prior study evaluating the fitness of an MMR-deficient UPEC due to inactivation of mutS,Citation40 we found that elimination of MMR did not significantly alter the fitness of UTI89 by 48 hpi (). As shown in , UTI89 ΔmutL, isolated from the bladder at 48 hpi, had a significantly higher frequency of mutation than MMR-proficient wt isogenic parent. In vivo and in vitro data showed the same pattern and degree of increased mutability in the MMR-deficient strain compared to wt, suggesting that MMR is not suppressed or saturated during UTI and that a loss of capacity by MMR is not the major factor driving high mutability in vivo. Together, these data suggest that MMR does not have a major role in genome instability or in counteracting the host response during acute UTI.
Pol IV is essential for full virulence during UTI but does not drive genome instability during infection.
After determining that MMR was not a major contributor to the mutability of UPEC in vivo, we assessed if the accessory DNA polymerases, known to perform critical bypass synthesis on genomic lesions and errors but lacking proofreading functions, may be involved in producing the >100-fold increase in mutation frequency observed in vivo. We also hypothesized that the TLS DNA polymerases may be essential for fitness and virulence during infection to bypass lesions and errors produced by host inflammatory mediators. We focused on the roles of the TLS polymerases Pol IV and Pol V. Each has be en previously shown to be highly induced during the SOS response,Citation41,Citation42 and prior work has shown the SOS response to be activated during UTI.Citation34
To test the role of Pol IV and Pol V in the mutability and survival of UTI89 in vivo, we created single and double deletion mutants in the dinB and umuDC genes coding for Pol IV and Pol V, respectively. In addition, we created a deletion in impCA alone or in combination with other polymerase deletion mutations. The impCA open reading frames are present in a 1,911 nt region (sequence 54,600–56,511) of the UTI89 plasmid, pUTI89 and the predicted proteins present some homology to UmuD′2C (1e-30; coverage 61%).
First, we confirmed that all of the single and multi-gene deletion mutants did not have growth defects in vitro (data not shown). We subsequently tested the mutation frequencies of the polymerase mutants in vitro. The mutants were grown in vitro under non-stressed (LB broth) and stressed (human urine) conditions. We observed that the mutation frequencies for all of the dinB deficient strain grown in LB broth media were significantly decreased, compared to the isogenic wt parent, as measured by 6-aza resistance (). No alteration in the mutation frequencies was observed due to deletions in umuDC or impCA alone (data not shown). Furthermore, deletion of umuDC or impCA in the ΔdinB background did not produce any significant alteration in the mutation frequencies of the single ΔdinB mutant. Although strains carrying the single dinB deletion or in combination with other mutations grown in vitro in urine trended toward lower mutation frequencies, these changes were not significant (wt vs. ΔdinB, p = 0.2). Thus, under laboratory, non-stressed growth conditions, UPEC Pol IV alone produces a modest but significant increase in the mutant frequency.
To investigate the role of the accessory TLS DNA polymerases in the survival and mutability of UTI89 during UTI, we conducted 48 h infections of C3H/HeN and C3H/HeJ mice with the series of UTI89 derivatives carrying single and combination mutations in the TLS DNA polymerase genes. As shown in , the loss of Pol IV (dinB deletion) alone or in combination with other mutations resulted in a significant loss of fitness relative to the isogenic wt strain. In contrast, mutations in umuDC or impCA without mutation in dinB did not significantly alter the fitness of UPEC during experimental UTI. Single copy chromosomal complementation of ΔdinB restored the in vivo loss of fitness ().
Next, the mutation frequencies of the mutant and wt strains recovered from infected bladder homogenates at 48 hpi were measured. The overall mutation frequency for all of the strains was significantly increased during infection relative to the inoculum (). However, single and combined mutations in the polymerase genes did not produce statistically significant alterations in the mutation frequencies relative to wt during experimental UTI. There was a trend toward increased mutagenesis among all three strains carrying a deletion in dinB. In contrast, the single copy dinB complemented strain had a frequency of mutation restored to wt levels (Sup. Fig. 1). These data suggest that although Pol IV is required for the full fitness of UPEC during UTI, Pol IV, despite lacking proofreading function, produces high-fidelity repair of the genome during infection. This may suggest that the specific lesions for which Pol IV is required to bypass are either correctly repaired or are otherwise lethal. Alternatively, Pol IV may be required to stall the replication complex, allowing other factors sufficient time for DNA replication and repair.
To confirm that the role of Pol IV in UPEC virulence during UTI was not bacterial or mouse strain specific, several additional experiments were performed. First, we constructed a complete deletion of the dinB open reading frame in the prototypic and well-characterized urosepsis isolate CFT073.Citation43 C3H/HeN mice were infected by transurethral catheterization, and at 48 hpi, bacterial CFU were measured in bladder homogenates. As shown in Supplemental Figure 2, CFT073 ΔdinB had a significant loss of fitness relative to the wt parent strain, thus demonstrating that Pol IV is important for virulence in independent UPEC isolates. Second, C57BL/6 mice were infected by transurethral inoculation with wt UTI89 and the ΔdinB isogenic derivative. As shown in Supplemental Figure 3A, UTI89 ΔdinB retained the significant loss of fitness previously observed in C3H/HeN mice and further showing that the effect is not host-dependent. The mutation frequency for the 6-aza marker was increased relative to in vitro levels, further confirming our observations from C3H/HeN mice (Sup. Fig. 3B). Together, these data demonstrate that Pol IV is important for survival for UPEC in the host environment during experimental UTI, while not contributing to genome instability.
Pol IV is required to maintain UP EC virulence under TLR4-dependent inflammatory stress in vivo.
We hypothesized that Pol IV would be required under inflammatory stress and that, with reduced inflammatory stress, a Pol IV mutant would not be significantly attenuated. To address this hypothesis, isogenic derivatives of UTI89 carrying combinations of mutations in dinB, umuDC and impCA were used to infect LPS hypo-responsive mice, C3H/HeJ. As shown in , each of the strains had no loss of fitness in this host after 48 hpi, in sharp contrast to the attenuation of ΔdinB in the C3H/HeN background where TLR4 signaling is intact. As anticipated, the frequency of 6-azauracil resistance was lower in the C3H/HeJ mice for all strains, measured at approximately 1-log higher than the frequency in vitro () and approximately the same frequency as observed after growth in human urine from non-infected individuals ().
Discussion
Our work on the genome instability of UPEC during UTI provides new insights into the plasticity of the pathogen genome under host stress and essential mechanisms required to maintain DNA integrity during infection and, thus, virulence. First, we demonstrate that the urinary tract environment, even in the absence of a significant innate immune response, stimulates a marked increase in mutation frequency. Our data indicate that growth in urine alone is a significant stimulus for mutagenesis. Urine has high osmolarity, elevated urea, low pH and low nutrient availability, each of which may contribute to the stress that ultimately drives pathways involved in UPEC mutagenesis. Preliminary studies supplementing urine with a simple carbon source such as glucose only partially lowers the frequency of mutations for 6-aza (∼0.5-log; Gawel and Seed, unpublished), suggesting that nutrient deprivation is only partially causal for inducing mutagenic stress pathways.
We also have demonstrated that the innate immune response, as triggered by the LPS-TLR4 pathway, results in an environment that significantly increases mutagenesis above the level of urine alone. The innate immune response to UPEC UTI is complex and differs in the bladder and kidneys. In the bladder, the major site of infection in our experimental model, a TLR4 response upregulates iNOS expression, resulting in increased nitric oxide.Citation10,Citation44 Concurrently, IL-6 and IL-8 are produced to recruit neutrophils into the urinary tract. Neutrophils have been previously shown to hone in on extracellular UPEC while also migrating to cells containing intracellular UPEC.Citation45 Phagocytosis of UPEC may provide a physical stress on UPEC that promotes mutagenesis while the neutrophil oxidative burst produces oxidative radicals with well-known consequences on DNA modification, introducing errors and lesions.Citation12 In the subepithelium, mast cells and macrophages are recruited, providing additional inflammatory and phagocytic challenges for UPEC. These current studies are not yet sufficient to determine if the measured genome instability arises because of enhanced expression of bacterial mutagenic factors, suppression of bacterial error-correcting mechanisms in the presence of host-induced DNA damage, or employment of low-fidelity DNA repair mechanisms to bypass host-incited DNA damage other than tested thus far. It seems likely that host factors such as oxidative radicals directly produce genome damage, necessitating repair and resulting in the introduction of mutations.
We hypothesized that one principle source of mutations in the UPEC genome during host stress may arise from suppression or saturation of MMR. Experimental testing of this hypothesis, however, demonstrated that MMR is active during infections, significantly reducing the frequencies of mutations incurred to a degree similar to non-stress UPEC in vitro. Based on the >1-log increase in mutation frequency in the MMR mutant (ΔmutL), we conclude that numerous post replication errors require MMR for correction during UTI. However, the relative increase in frequency of mutations between wt and the MMR mutant is not greater in vivo than in vitro. Thus, while the MMR system appears to be important for maintaining genome stability in vitro and in vivo, the host environment and inflammatory response does not necessitate exceptional requirements for the MMR system.
Previously, SulA, a component of the SOS regulon, was shown to play an essential role in UPEC virulence in the mouse model of human UTI.Citation34 More recently, Li et al. confirmed the importance of the SOS response for UPEC during UTI by showing the attenuation a RecA-deficient strain of UTI89 during murine UTI.Citation35 Expression of the TLS polymerases IV and V is greatly enhanced as part of the SOS response,Citation41,Citation42 and the polymerases have important roles in long-term survival and stationary phase mutagenesis in vitro,Citation22,Citation46 raising the possibility that these polymerases were not only responsible for enhanced mutation frequencies measured during UTI but also were critical factors necessary for mitigating genomic injury sustained during infection. Numerous prior studies have demonstrated that each of these polymerases yield low fidelity synthesis during processing undamaged and damaged templates due to that lack of exonuclease functions.Citation24 In addition to evaluating the potential roles of Pol IV and Pol V in genome instability and fitness of UPEC during UTI, we considered the possibility that additional homologs may be present and thus responsible for any observed phenotypes. In Salmonella typhimurium, a plasmid-borne homolog of Pol V called SamAB has been described.Citation47 A BLAST analysis of the UTI89 genome for Pol V homologs suggested such a factor, annotated as encoded by the open reading frames impCA, may be present on the megaplasmid. We did not find any in vitro and in vivo genetic evidence for an active role of Pol V or ImpC/ImpA proteins in pathogenicity and mutability of UPEC UTI89 strain. While no major role for Pol V was observed, we cannot fully exclude that substitution for Pol V by another accessory polymerase does mask any observation of an in vivo function.
In contrast to the lack of an apparent requirement for Pol V and ImpCA in vivo during UTI, we discovered a novel in vivo phenotype for Pol IV during UTI. The loss of Pol IV in UTI89 and CFT073 resulted in the attenuation of both strains during UTI in multiple host backgrounds. Interestingly, the mutation frequencies of Pol IV mutant were not significantly different than wt. The Pol IV-deficient strain trended toward having higher mutation frequencies than wt UTI89, opposite of what was expected were Pol IV contributing to mutagenesis during UTI. In our in vitro studies, we also observed a modest anti-mutagenic effect with the loss of dinB in UTI89 when grown in rich LB medium. However, this phenotype was abolished when the mutant was propagated in human urine, yielding the same mutation frequencies as wt (). This may indicate that Pol IV is recruited for translesion synthesis over different errors or lesions in vitro and in vivo and that the bypass of these damages is low and high fidelity in vitro and in vivo, respectively. As working model we propose, that certain lesions or errors introduced into the genome during host initiate stress conditions may predominate and not only require Pol IV for bypass but result in error-free synthesis. This may indicate the nature of the host response and lesions induced in vivo for which Pol IV is so important in maintaining viability and thus virulence. Consistent with this idea, we found that the Pol IV mutant was no longer attenuated in mice with an LPS hypomorphic phenotype, whereby LPS-TLR4 signaling is known to induce the innate immune response of which an oxidative stress response is part. These data together indicate a unique and important role for Pol IV in the maintenance and fidelity of DNA synthesis of UPEC under inflammatory stress.
The alternative explanation for the loss of fitness in the dinB mutant fitness under inflammatory stress conditions during UTI may be due to a requirement for DinB as a brake on DNA synthesis, allowing other repair factors sufficient time to resolve DNA damage and errors, thus serving as a specific checkpoint to secure genome stability.Citation48 Thus, the loss of dinB would be expected to have the phenotypes we observed in vivo, namely a survival defect and enhanced mutagenesis. This is also consistent with our measurements of DinB-independent mutagenesis during UTI that was enhanced with inflammation.
Parallel to our report, Gutierrez et al. recently showed that the absence of DinB resulted in loss of fitness in competive infections with wt UPEC CFT073 in a murine model of bacteremia,Citation49 consistent with our demonstration of a role of DinB in UTI pathogenesis. In that report, the authors demonstrate that the attenuated phenotype of the dinB mutant was restored when the yafP gene, encoding for an N-acetyltransferase, was also deleted. Guttierez et al. concluded, that YafP enzyme may play a role in the activation of numerous mutagenic compounds for which DinB is required to process the lesions resulting from these derivatives. However, it must be pointed that the dinB-yafP two gene organization in the UPEC strain CFT073 is rather unique to only about 40% of studied by them E. coli strains. Nevertheless, majority of E. coli strains (including UPEC UTI89 strain and the commensal K-12 MG1655 strain) have a four gene operon structure (dinB-yafN-yafO-yafP). Therefore we may expect that in our experimental system the phenotype for yafP may differ from the prior report due to (1) the differencies in type of stress associated with bladder infection vs. extraintestinal and (2) the structure of the yaf operon in used model isolates CFT073 and UTI89. Future studies will elucidate the broader role of the yaf operon in extraintestinal virulence among diverse strains and the interactions of its gene products with DinB.
If Pol IV, Pol V, ImpCA or the loss of MMR functionality do not act to enhance in vivo mutagenesis, under what processes does enhanced mutagenesis arise? There several additional processes through which their imbalance, dysfunction and inactivation may result in mutagenesis. One may predict the source is in one of three systems: (1) alterations in 3′→5′ proofreading by the major and accessory polymerases; (2) inhibition or failure of base-excision repair (BER) or nucleotide-excision repair (NER); and (3) alteration in dNTP pools such as inhibition of MutT, involved in sanitation of damaged dNTPs.Citation50 Future studies will determine the relative activities of these systems in vivo and their contributions to mutagenesis and pathogen adaption.
One intriguing idea is that under inflammatory stress, UPEC engages essential systems to manage specific genome integrity, without which viability is threatened, while concurrently activating mutagenesis as an evolved stress response through which adaptive mutations may be generated. This concept may imply that mutagenesis is not necessarily the essential consequence of repairing otherwise fatal damage. Instead, stress-induced mutagenesis may be activated through a portion of the population resulting in an infrequent selective advantage through rare beneficial mutations that arise to improve the phenotype of UPEC either as a pathogen or as a commensal, the state in which the organisms most commonly exists. In terms of adaptation, prior studies of UPEC have demonstrated pathoadaptive changes among UPEC in the E. coli pangenome. One example is the FimH lectin adhesin of type 1 pili, a critical virulence factor for UPEC. FimH of UPEC has an increased affinity for monomannosylations present on apical proteins of the urinary tract such as uroplakins while FimH of commensal bacteria have greater affinity for tri-mannosylations more likely to be present in nonurinary tract niches.Citation13,Citation14,Citation51
In conclusion, we have shown that the interaction of UPEC with the host results in highly enhanced genome instability, independent of the TLS polymerases and the post-replicative MMR system. Our data suggest that the UPEC genome has significant plasticity during infection and the host, particularly during inflammatory responses, may be a major driver in diversity among these pathogen-commensal organisms. Furthermore, we have demonstrated a major role for the TLS DNA polymerase IV in mitigating host inflammatory stress during UTI. Given the conservation of Pol IV among prokaryotes in particular, the TLS DNA Polymerases may have important global roles in the maintenance of genome integrity and tolerating host inflammatory assaults.
Materials and Methods
Strains.
Bacterial strains and oligonucleotides used in this study are listed in and Supplemental Table 1, respectively. All deletion mutants were constructed by the red recombinase method entailing direct replacement of gene(s) of interest with an antibiotic resistant cassette flanked by FLP recombinase recognition sequencesCitation52 or by generalized transduction using P1 phage. The chromosomal complementation of the dinB deletion was performed using a two-step procedure. First, a complete deletion of proAB, a linked locus to dinB, was made in UTI89 (wt) and UTI89 ΔdinB using red recombinase and resulting in proAB::cat. Next, each strain was subjected to generalized transduction using P1 phage particles derived from wt UTI89 and selecting for proAB+ on MM medium lacking proline. The reconstitution of dinB was confirmed by PCR.
Media.
Minimal medium (MM) and Luria-Bertani (LB) broth solid and liquid media were standard recipes as described.Citation53 Human urine was collected and pooled from at least 3 healthy donors. Minimal medium (MM) with Vogel-Bonner saltsCitation54 was supplemented with 5 µg/ml of nicotinic acid and 0.4% glucose. Solid media contained 1.5% agar. Where required, antibiotics and/or selection agents were added to the following final concentrations: ampicillin (amp), 100 µg/ml; chloramphenicol (cam), 20 µg/ml; kanamycin (kan), 50 µg/ml; rifampicin (rif), 100 µg/ml; 6-azauracil (6-aza), 40 µg/ml; 5-fluorocytosine (5-FC), 40 µg/ml; nalidixic acid (nal), 40 µg/ml. P-gal solid MM media contained 1.5% agar supplemented with 5 µg/ml of nicotinic acid and 800 µg/ml phenyl-β-D-galactopyranoside as a carbon source.
Experimental murine UTI.
Mice (6–8-week-old female) were obtained from Charles River Laboratories through the NCI—Frederick resource. Bacterial strains were prepared for infection as previously described.Citation61 Mice were inoculated with 1–2 × 107 CFU by transurethral catheterization in a volume of 50 µl.
Mutation frequency measurements.
For in vitro tests, we used 6–12 independent LB or urine cultures (3 ml each) initiated from single colonies (one colony per tube) and grown over night to saturation at 37°C while rotating on a rotator wheel. The 100 µl of 106-fold diluted cultures were plated on LB and MM plates to determine the total cell count. The 100 µl of the undiluted cultures were plated on selective plates to determine the number of resistant (rifr, nalr, 6-azar, 5-FCr, respectively) or lacI mutant colonies. UTI89 mutL (MMR-mismatch repair deficient) cultures were 10-fold diluted prior plating on LB/rif or MM/6-aza plates due to high mutation rates observed in MMR deficient background.
In vivo mutation frequencies were determined by homogenizing bladders in PBS containing Triton-X 100 (0.02%) and plating homogenate on MM/6-aza, MM/5-FC or MM/p-gal plates. Total cell counts were determined on LB or MM plates in parallel. Cultures were growth at 37°C. Mutation frequencies were calculated as the number of mutants per plate divided by the total number of cells.
The forward mutagenesis assays employed differ in reporter/marker gene and its location on chromosome. The rifampicin resistant and nalidixic acid resistant mutants result from forward mutation(s) occurred in rpoB and gyrA marker genes, respectively. The 6-azauracil resistant and 5-fluorocytidine resistant mutants arise due to mutation(s) in upp or/and codA genes of pyrimidine salvage pathway. Colonies growing on P-gal as a carbon source were formed due to forward mutation(s) appeared in lacI repressor allele of lac operon.
Statistical analyses.
The statistic analyses (average frequencies; standard error (SE) or non parametric Mann-Whitney U-test) were determined by using the statistical software program Prism (GraphPad). Statistical significance was defined by attaining p-value ≤ 0.05. Horizontal lines present on the graphs indicate the geometric mean for each group.
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal studies were performed under protocols reviewed and approved by the Duke University Institutional Animal Care and Use Committee (Protocol #: A143-09-05). Urethral catheterization of the animals and the initiation of the infections were carried out under isoflurane anesthesia to minimize trauma to the animals.
Urine specimens from adults providing written informed consent were de-identified and collected in accordance with Protocol #Pro00026252, reviewed and approved by the Duke University Institutional Review Board.
Figures and Tables
Figure 1 UTI promotes higher mutation frequencies in UTI89. (A) UTI89 infection rates (CFU/Bladder × 104 at 48 hpi). (B–D) mutation frequency per 108 cells measured for: 6-aza (B), lacI (C) and 5-FC (D) markers.
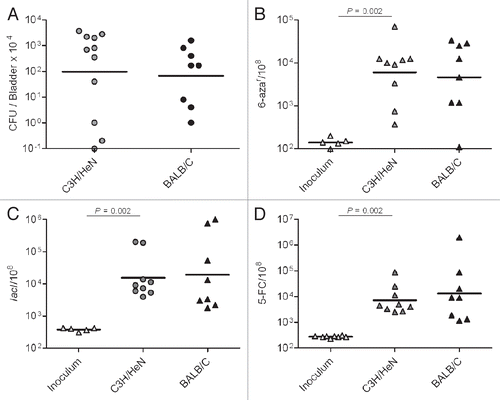
Figure 2 Growth under urine stress increases mutability for multiple E. coli strains. E. coli strains UPEC : CI5, E80, DS17, CFT073, GR12, PY2, UTI89, r-UTI89, NU14, EC45 and K-12: MG1655 and KA796 were grown overnight in LB broth and urine, in parallel, at 37°C. Mutation frequencies plotted as resistant colonies per 108 cells. Each plotted point is based on three independent cultures.
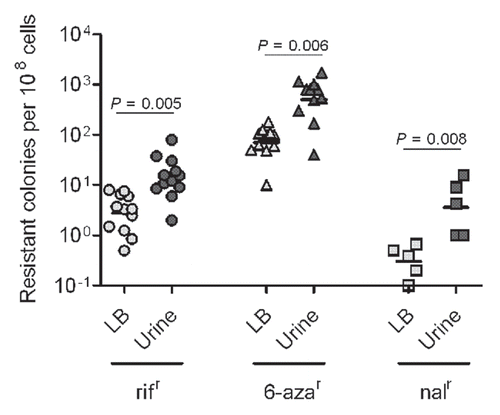
Figure 3 Host environment and innate immune response stimulate progressive increases mutation frequencies. Mutation frequencies are shown as number of 6-azar colonies per 108 cells.
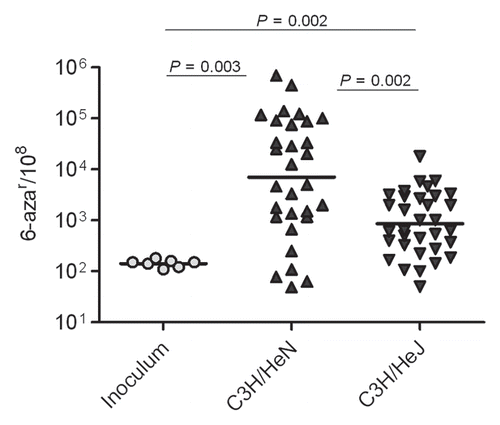
Figure 4 MMR is not suppressed or saturated during UTI. (A) Infection rates (CFU/Bladder × 104) of UTI89 and UTI89 ΔmutL strains. (B) Mutation frequencies per 108 cells measured for 6-azar marker.
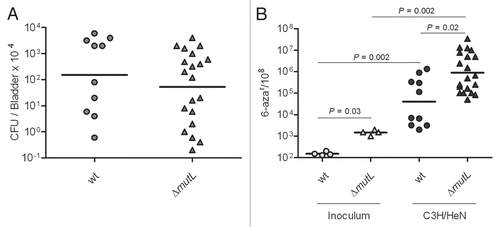
Figure 5 Mutation frequencies of UPEC UTI89 derivatives lacking genes encoding for TLS DNA polymerases. Strains were grown over night in LB broth and urine, in parallel, at 37°C. Mean mutation frequencies for 6-azar are based on eight independent cultures. Error bars show standard error (SE). Statistically significant differences (p < 0.05) in mutation rates frequencies compared to wt UTI89 are indicated by a star (*). p values were calculated using the non-parametric Mann-Whitney test.
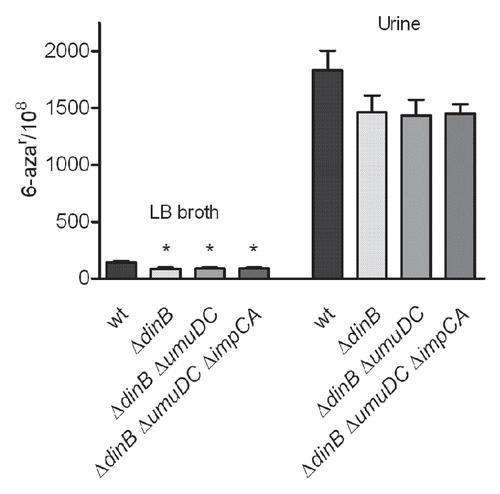
Figure 6 DNA polymerase IV improves survival of UTI89 strain in vivo but does not contribute to genome instability. (A) Infection rates (CFU/Bladder × 104) of UTI89 and its mutant derivatives. (B) Mutation frequencies per 108 cells measured for the 6-azar marker. The mutation frequencies were statistically different (p < 0.05) between the inocula and the bacteria recovered from infected bladders for all comparisons.
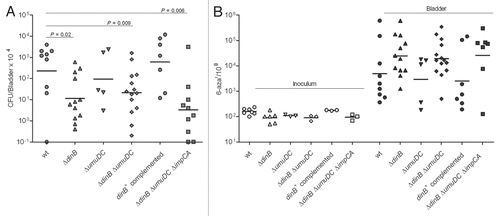
Figure 7 The ΔdinB mutant is not attenuated in the LPS hyporesponsive TLR4-host. (A) Infection rates (CFU/Bladder × 104) of UTI89 and its mutant derivatives. (B) mutation frequency per 108 cells measured for 6-azar marker. The mutation frequencies were statistically different (p < 0.05) between the inocula and the bacteria recovered from infected bladders for all comparisons.
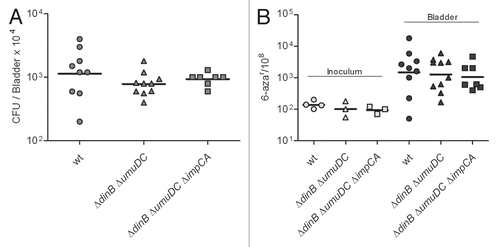
Table 1 In vitro mutation frequencies for UPEC UTI89Table Footnotea
Table 2 Growth in normal human urine increases the mutability of UPEC UTI89 in vitroTable Footnotea
Table 3 The UTI89 MMR mutant has enhanced mutability in vitroTable Footnotea
Table 4 E. coli strains used in this study
Additional material
Download Zip (197.8 KB)Acknowledgments
The authors thank Drs. Joseph St. Geme, Ravi Jhaveri and Roel M. Schaaper for their thoughtful comments and critiques. Support for this work was partially provided by K08DK074443 and ORWH SCOR P50DK064540.
References
- Foxman B. Epidemiology of urinary tract infections: incidence, morbidity and economic costs. Dis Mon 2003; 49:53 - 70
- Nicolle LE. Uncomplicated urinary tract infection in adults including uncomplicated pyelonephritis. Urol Clin North Am 2008; 35:1 - 12
- Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998; 282:1494 - 1497
- Bishop BL, Duncan MJ, Song J, Li G, Zaas D, Abraham SN. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med 2007; 13:625 - 630
- Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 2003; 301:105 - 107
- Hang L, Wullt B, Shen Z, Karpman D, Svanborg C. Cytokine repertoire of epithelial cells lining the human urinary tract. J Urol 1998; 159:2185 - 2192
- Schilling JD, Martin SM, Hung CS, Lorenz RG, Hultgren SJ. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc Natl Acad Sci USA 2003; 100:4203 - 4208
- Kurutas EB, Ciragil P, Gul M, Kilinc M. The effects of oxidative stress in urinary tract infection. Mediators Inflamm 2005; 2005:242 - 244
- Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB Jr, Ganz T. Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Invest 1998; 101:1633 - 1642
- Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2004; 2:820 - 832
- Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 2004; 423:12 - 22
- Russo MT, Blasi MF, Chiera F, Fortini P, Degan P, Macpherson P, et al. The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol Cell Biol 2004; 24:465 - 474
- Chen SL, Hung CS, Pinkner JS, Walker JN, Cusumano CK, Li Z, et al. Positive selection identifies an in vivo role for FimH during urinary tract infection in addition to mannose binding. Proc Natl Acad Sci USA 2009; 106:22439 - 22444
- Sokurenko EV, Courtney HS, Ohman DE, Klemm P, Hasty DL. FimH family of type 1 fimbrial adhesins: functional heterogeneity due to minor sequence variations among fimH genes. J Bacteriol 1994; 176:748 - 755
- Kornberg A, Baker TA. DNA Replication 1992; New York WH Freeman and Company
- Banach-Orlowska M, Fijalkowska IJ, Schaaper RM, Jonczyk P. DNA polymerase II as a fidelity factor in chromosomal DNA synthesis in Escherichia coli. Mol Microbiol 2005; 58:61 - 70
- Fuchs RP, Fujii S, Wagner J. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem 2004; 69:229 - 264
- Kuban W, Jonczyk P, Gawel D, Malanowska K, Schaaper RM, Fijalkowska IJ. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J Bacteriol 2004; 186:4802 - 4807
- Rangarajan S, Woodgate R, Goodman MF. Replication restart in UV-irradiated Escherichia coli involving pols II III, V, PriA, RecA and RecFOR proteins. Mol Microbiol 2002; 43:617 - 628
- Fujii S, Fuchs RP. Interplay among replicative and specialized DNA polymerases determines failure or success of translesion synthesis pathways. J Mol Biol 2007; 372:883 - 893
- Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 2001; 266:207 - 215
- Yeiser B, Pepper ED, Goodman MF, Finkel SE. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA 2002; 99:8737 - 8741
- Perez-Capilla T, Baquero MR, Gomez-Gomez JM, Ionel A, Martin S, Blazquez J. SOS-independent induction of dinB transcription by beta-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J Bacteriol 2005; 187:1515 - 1518
- Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, et al. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 2000; 404:1014 - 1018
- Kuban W, Banach-Orlowska M, Bialoskorska M, Lipowska A, Schaaper RM, Jonczyk P, et al. Mutator phenotype resulting from DNA polymerase IV overproduction in Escherichia coli: preferential mutagenesis on the lagging strand. J Bacteriol 2005; 187:6862 - 6866
- Maliszewska-Tkaczyk M, Jonczyk P, Bialoskorska M, Schaaper RM, Fijalkowska IJ. SOS mutator activity: unequal mutagenesis on leading and lagging strands. Proc Natl Acad Sci USA 2000; 97:12678 - 12683
- Bjedov I, Dasgupta CN, Slade D, Le Blastier S, Selva M, Matic I. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 2007; 176:1431 - 1440
- Hori M, Yonekura S, Nohmi T, Gruz P, Sugiyama H, Yonei S, et al. Error-Prone Translesion DNA Synthesis by Escherichia coli DNA Polymerase IV (DinB) on Templates Containing 1,2-dihydro-2-oxoadenine. J Nucleic Acids 2010; 8075 - 8079
- Pham P, Rangarajan S, Woodgate R, Goodman MF. Roles of DNA polymerases V and II in SOS-induced error-prone and error-free repair in Escherichia coli. Proc Natl Acad Sci USA 2001; 98:8350 - 8354
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem 2005; 74:681 - 710
- Negishi K, Loakes D, Schaaper RM. Saturation of DNA mismatch repair and error catastrophe by a base analogue in Escherichia coli. Genetics 2002; 161:1363 - 1371
- Harris RS, Feng G, Ross KJ, Sidhu R, Thulin C, Longerich S, et al. Mismatch repair protein MutL becomes limiting during stationary-phase mutation. Genes Dev 1997; 11:2426 - 2437
- Cox MM. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol 2007; 42:41 - 63
- Justice SS, Hunstad DA, Seed PC, Hultgren SJ. Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc Natl Acad Sci USA 2006; 103:19884 - 19889
- Li B, Smith P, Horvath DJ Jr, Romesberg FE, Justice SS. SOS regulatory elements are essential for UPEC pathogenesis. Microbes Infect 12:662 - 668
- Chen SL, Hung CS, Xu J, Reigstad CS, Magrini V, Sabo A, et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: A comparative genomics approach. Proc Natl Acad Sci USA 2006; 103:5977 - 5982
- Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, et al. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 1997; 277:1833 - 1834
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science 1997; 277:1453 - 1474
- McAdam KP, Ryan JL. C57BL/10/CR mice: nonresponders to activation by the lipid a moiety of bacterial lipopolysaccharide. J Immunol 1978; 120:249 - 253
- Labat F, Pradillon O, Garry L, Peuchmaur M, Fantin B, Denamur E. Mutator phenotype confers advantage in Escherichia coli chronic urinary tract infection pathogenesis. FEMS Immunol Med Microbiol 2005; 44:317 - 321
- Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001; 158:41 - 64
- Quillardet P, Rouffaud MA, Bouige P. DNA array analysis of gene expression in response to UV irradiation in Escherichia coli. Res Microbiol 2003; 154:559 - 572
- Mobley HL, Green DM, Trifillis AL, Johnson E, Chippendale GR, Lockatell CV, et al. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 1990; 58:1281 - 1289
- Bogdan C, Rollinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol 2000; 12:64 - 76
- Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, et al. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc Natl Acad Sci USA 2004; 101:1333 - 1338
- Foster PL. Adaptive mutation in Escherichia coli. J Bacteriol 2004; 186:4846 - 4852
- Nohmi T, Hakura A, Nakai Y, Watanabe M, Murayama SY, Sofuni T. Salmonella typhimurium has two homologous but different umuDC operons: cloning of a new umuDC-like operon (samAB) present in a 60-megadalton cryptic plasmid of S. typhimurium. J Bacteriol 1991; 173:1051 - 1063
- Uchida K, Furukohri A, Shinozaki Y, Mori T, Ogawara D, Kanaya S, et al. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol Microbiol 2008; 70:608 - 622
- Gutierrez A, Elez M, Clermont O, Denamur E, Matic I. Escherichia coli YafP protein modulates DNA damaging property of the nitroaromatic compounds. Nucleic Acids Res 2011; In press
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis 2005; Washington DC American Society for Microbiology 2
- Weissman SJ, Beskhlebnaya V, Chesnokova V, Chattopadhyay S, Stamm WE, Hooton TM, et al. Differential stability and trade-off effects of pathoadaptive mutations in the Escherichia coli FimH adhesin. Infect Immun 2007; 75:3548 - 3555
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 2000; 97:6640 - 6645
- Miller JH. Experiments in molecular genetics 1972; Cold Spring Harbor NY Cold Spring Harbor Laboratory
- Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem 1956; 218:97 - 106
- Svanborg Eden C, Hull R, Falkow S, Leffler H. Target cell specificity of wild-type E. coli and mutants and clones with genetically defined adhesins. Prog Food Nutr Sci 1983; 7:75 - 89
- Abraham SN, Babu JP, Giampapa CS, Hasty DL, Simpson WA, Beachey EH. Protection against Escherichia coli-induced urinary tract infections with hybridoma antibodies directed against type 1 fimbriae or complementary D-mannose receptors. Infect Immun 1985; 48:625 - 628
- Garofalo CK, Hooton TM, Martin SM, Stamm WE, Palermo JJ, Gordon JI, et al. Escherichia coli from urine of female patients with urinary tract infections is competent for intracellular bacterial community formation. Infect Immun 2007; 75:52 - 60
- Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, et al. The Gal(alpha 1–4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc Natl Acad Sci USA 1994; 91:11889 - 11893
- Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, et al. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 1989; 53:1 - 24
- Schaaper RM, Danforth BN, Glickman BW. Rapid repeated cloning of mutant lac repressor genes. Gene 1985; 39:181 - 189
- Hannan TJ, Mysorekar IU, Chen SL, Walker JN, Jones JM, Pinker JS, et al. LeuX tRNA-dependent and -independent mechanisms of Escherichia coli pathogenesis in acute cystitis. Mol Microbiol 2008; 67:116 - 128