Abstract
PB1-F2 is a 90 amino acid protein that is expressed from the +1 open reading frame in the PB1 gene of some influenza A viruses. The PB1-F2 protein has been shown to contribute to viral pathogenicity, but the molecular mechanisms for mediating virulence have been unclear. Previous reports demonstrate that PB1-F2 promotes cell death, causes immunopathology and increases pro-inflammatory responses. Our group has identified a single point mutation from asparagine (N) to serine (S) at position 66 in the PB1-F2 protein that dramatically increases the virulence of highly pathogenic avian H5N1 influenza viruses and of the 1918 pandemic strain. In search for the mechanism by which PB1-F2 N66S increases pathogenicity, we have identified and characterized a novel function of PB1-F2, i.e. interferon antagonism, both in vitro and in the mouse model. Here, we discuss a hypothesis for a possible molecular link between the pro-apoptotic and anti-interferon functions of PB1-F2.
Introduction
Influenza is a global health concern due to its potential to cause pandemics with significant death rates and socio-economic costs. The pandemics that occurred in 1918, 1957 and 1968 were all caused by influenza viruses that express the pathogenicity factor PB1-F2. PB1-F2 is the smallest known influenza virus protein and is encoded by the +1 alternate open reading frame in the PB1 gene of some influenza A virus strains. Since its identification in 2001 by Chen et al., several groups have proposed mechanisms by which the PB1-F2 protein contributes to influenza virus pathogenicity. It was shown that PB1-F2 has pro-apoptotic activity in vitro by interacting with the mitochondrial voltage-dependent anion channel 1 (VDAC1) and the adenine nucleotide translocator 3 (ANT3) proteins.Citation1 Furthermore, PB1-F2 was suggested to induce cell death specifically in immune cellsCitation2,Citation3 in a strain-specific manner.Citation4 Other studies demonstrated that PB1-F2 can induce inflammatory responses resulting in immune cell infiltration, increased cytokine levels and tissue damage in the lung of infected mice.Citation4–Citation8 The pro-inflammatory property of PB1-F2 is thought to promote secondary bacterial infections which caused substantial morbidity during the 1918 pandemic.Citation7 A report by Mazur et al. describes an interaction of PB1-F2 with the PB1 polymerase protein in the nucleus, which leads to an increased polymerase activity in vitro.Citation9 This interaction, however, does not appear to contribute to influenza virus pathogenesis in a mouse model.Citation10 It is surprising that such a small protein as PB1-F2 can carry out multiple functions. Here, we will discuss a possible link between the pro-apoptotic and interferon antagonistic properties of PB1-F2.
PB1-F2 Has Anti-Interferon Activity
We have previously identified a crucial residue in the PB1-F2 protein that is associated with increased virulence of highly pathogenic avian H5N1 influenza viruses and of the 1918 pandemic strain.Citation5 Specifically, we found that a serine (S) at position 66 in the PB1-F2 protein dramatically increases influenza virus pathogenicity compared with isogenic viruses with an asparagine (N) at this position.Citation5 In search for the molecular mechanism by which PB1-F2 N66S increases virulence, we performed microarray analyses on lung homogenates and found an early suppression of interferon-stimulated genes (ISGs) in mice infected with a PB1-F2 N66S expressing virus.Citation6 In our most recent work, we performed in vitro assays to further characterize the newly identified anti-interferon (IFN) activity of PB1-F2.Citation11 We found that the N66S point mutation increases the interferon antagonism function of the PB1-F2 protein in an in vitro transfection system, when expressed from a Newcastle disease virus (NDV) vector and in the context of viral infection. Furthermore, we showed that the anti-IFN activity of PB1-F2 is mediated via its C-terminal region which has previously been shown to also have pro-apoptotic activityCitation1 and that PB1-F2 exerts its anti-IFN function in both immune and epithelial cells.
Influenza viruses express another non-structural protein besides PB1-F2, namely NS1, which has a well-characterized IFN antagonist function.Citation12–Citation14 We thus examined the relationship between PB1-F2 and the NS1 proteins in regards to their IFN antagonism function and observed that PB1-F2 N66S in combination with NS1 inhibits the IFN response more efficiently than NS1 alone.Citation11 In addition, PB1-F2 N66S expressed from a virus with an NS1 protein, which is crippled in its ability to suppress the antiviral IFN response, significantly reduces the induction of IFN compared with PB1-F2 WT.Citation11 Using an IFNβ reporter assay with different stimuli, we found that PB1-F2 affects the induction of IFN at the level of the mitochondrial antiviral signaling (MAVS) protein () and immunofluorescence studies showed a co-localization of PB1-F2 with MAVS at the mitochondria.Citation11 Of note, PB1-F2 does not affect the induction of IFN when triggered by TRIF, an adaptor protein mediating TLR-mediated IFN production.Citation11 Collectively, we identified and characterized a novel function for PB1-F2, which is its IFN antagonism, and demonstrated that the anti-IFN activity of PB1-F2 is exerted at the level of the MAVS adaptor protein.
The Antiviral Innate Immune Response
The innate immune response is the first line of defense against viral infections. It is mediated through the activation of Toll-like receptors (TLRs), Nod-like receptors (NLRs) and the RIG-I-like RNA helicase receptors (RLHs) retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated gene-5 (MDA-5). Upon recognition of characteristic pathogen-associated patterns, the TLRs and RLHs induce the production of type I IFN,Citation15 which binds to specific receptors and thus upregulates antiviral genes through the JAK/STAT pathway in an autocrine or paracrine manner. The mitochondria play an essential role in the antiviral IFN response since MAVS mediates RIG-I and MDA-5 triggered IFN production. Once activated, RIG-I and MDA-5 interact with MAVS via their CARD domains leading to the phosphorylation and activation of IRFs by TBK1 and IKKε. The IRFs then translocate to the nucleus to drive the expression of type I IFN in combination with the NFκB and ATF/c-Jun transcription factors (). It is unclear how MAVS propagates the signal for the IFN production pathway. Some reports indicate that MAVS undergoes a re-organization in the mitochondrial membrane into distinct punctate structuresCitation16 and re-arranges from a higher- to a lower-order complex upon activation.Citation17 It has also been suggested that MAVS can self-oligomerize via its transmembrane domainCitation18 and even form prion-like agglomerates.Citation19 Furthermore, a recent report by Koshiba et al. shows the importance of an intact mitochondrial membrane potential for MAVS-mediated IFN production.Citation20
How does PB1-F2 affect MAVS-dependent IFN production? Preliminary data from our laboratory suggest that PB1-F2 binds to MAVS, with the N66S mutation contributing to an increased interaction affinity. It would be interesting to investigate whether the interaction of PB1-F2 with MAVS affects the ability of MAVS to form complexes with other proteins, or if PB1-F2 interferes with MAVS self-oligomerization and rearrangement. Another possibility would be that PB1-F2 disrupts MAVS-dependent IFN production by disturbing the mitochondrial membrane potential.
The Pro-Apoptotic Function of PB1-F2
Apoptosis can be initiated through an extrinsic or intrinsic pathway. The extrinsic apoptosis cascade is mediated through so-called death receptors while the intrinsic or mitochondrial apoptosis pathway is triggered by cell damage. Initiation of the intrinsic cell death pathway leads to the permeabilization of the mitochondrial membrane. This can occur through Bcl-2 family members or the formation of the permeability-transition pore complex (PTPC). The PTPC mainly consists of the adenine nucleotide translocase 3 (ANT3) in the inner membrane, and the voltage-dependent anion channel 1 (VDAC1) in the outer mitochondrial membrane. The permeabilization of the mitochondrial membrane leads to the efflux of apoptogenic molecules, such as cytochrome c, which activate the caspase cascade. The extrinsic and intrinsic cell death pathways merge into a common effector molecule, caspase 3, which executes the death sentence. Based on previous findings, PB1-F2 can induce apoptosis via two mechanisms: (1) by interacting with VDAC1 and ANT3 to promote the formation of the PTPC () and (2) by forming pores via self-oligomerization ().
The role of apoptosis during influenza virus infection is controversial. Several reports indicate that cell death is a type of host defense mechanism which is antagonized by the NS1 proteinCitation21 through activation of the pro-survival Akt pathway.Citation22 Conversely, other reports show that influenza virus can exploit the apoptosis pathways for efficient replication.Citation23,Citation24 Specifically, it was found that caspase 3 activityCitation23 and mediators of the extrinsic apoptosis pathway, namely TRAIL and Fas/FasL,Citation24 are needed for efficient viral propagation.
MAVS: Molecular Bridge between the Antiviral IFN Response and Mitochondrial Cell Death?
Mitochondria are essential organelles of the cells. They function as “energy factories,” mediate antiviral responses and regulate cell death. Viruses have evolved mechanisms to modulate these mitochondrial functions for their lifecycle. Understanding how viruses can subvert these processes can give insights into the organization of cellular pathways. Viral proteins with multifunctional properties such as the influenza A virus protein PB1-F2 may provide insights into the cross-talks between cellular signaling cascades. It is fascinating that a small protein such as PB1-F2 can exert several functions, i.e., the induction of apoptosis and the inhibition of the antiviral IFN response (among other possible functions). These two activities may be performed independently of each other, but it is equally possible that PB1-F2 unravels a molecular bridge between the mitochondrial cell death pathway and MAVS-dependent IFN production.
Since both the antiviral and the apoptosis pathways are mediated through the mitochondria, the question arises whether there is a crosstalk between these cellular processes at the level of the MAVS adaptor protein. A tight control of the cell death pathway in regards to IFN production would be desirable since it would be unfavorable for an infected cell to induce apoptosis pre-maturely, i.e., before antiviral proteins could be successfully produced and secreted. Interestingly, a recent report indicates a negative regulatory role of MAVS in VDAC1-mediated cell death.Citation25 The results of the study suggest that MAVS interacts with VDAC1 at the outer mitochondrial membrane via its proline-rich domain and thus inhibits the formation of the VDAC1-containing pore complex that would lead to the efflux of cytochrome c to initiate apoptosis. Of note, a pro-apoptotic function for MAVS has also been described which is executed via different mechanisms.Citation26–Citation28 Overall, the function of MAVS in the regulation of apoptosis is more complex than previously thought.
The Multifunctional Influenza A Virus Protein PB1-F2: Are its Functions Linked?
PB1-F2 has been shown to localize to mitochondria via its mitochondrial localization sequence in the C-terminus.Citation29 It has been demonstrated to self-oligomerize to form pores in the mitochondrial membraneCitation30 and it can also interact with VDAC1 and ANT3 to promote apoptosis.Citation1 In addition, PB1-F2 inhibits MAVS-mediated IFN production, possibly by binding to the MAVS adaptor protein.Citation11 We hypothesize that the interaction of PB1-F2 with MAVS has two effects: (1) the decrease in IFN production and (2) the promotion of VDAC1-mediated cell death (). This way, influenza virus can “kill” two undesired cellular processes, i.e., IFN production and inhibition of apoptosis by MAVS, with one protein, PB1-F2. Based on the previously described findings by Xu et al. and preliminary data from our laboratory, we propose that PB1-F2 disrupts the interaction between MAVS and VDAC1, thus lifting the inhibitory effect of MAVS on VDAC1 (). In the absence of the negative regulatory action of MAVS, VDAC1 would then be available for the formation of permeability pores with other proteins such as ANT3 to release apoptogenic molecules into the cytoplasm. In parallel, the interaction of PB1-F2 with VDAC1 and ANT3 would promote the pore formation that can be enhanced in response to exogenous stimuli such as TNFα.
The extent of the pro-apoptotic and anti-IFN effects of PB1-F2 is likely to be cell type-dependent. It is possible that the expression of PB1-F2 varies in immune and epithelial cells and that the expression patterns of the target proteins of PB1-F2 are different so that some cells may be more sensitive to the pro-apoptotic effect than others. To create an optimal environment for viral replication, it is plausible that PB1-F2 decreases the antiviral IFN production in both epithelial and immune cells. Epithelial cells are the primary target cells for replication so it would be undesirable to kill these cell types. Instead, PB1-F2 may specifically promote and accelerate cell death in immune cells to evade the detection of the virus by the immune system. Also, it is possible that the conformation, subcellular localization and post-translational modification of PB1-F2 play a role in the execution of its functions.
It will be important to identify the residues that mediate the different functions of PB1-F2 in order to determine if these functions are linked or executed separately from each other. Also, it has been established that there is great strain variance in PB1-F2 mediated functions, so it would be interesting to examine if the degree of apoptosis induction or IFN suppression can be linked to a viral pathogenicity phenotype. Additionally, a better understanding of the molecular function of PB1-F2 in avian species is needed in view of the high conservation rate of the PB1-F2 open reading frame in avian influenza virus isolates. In recent years, novel hypotheses on the molecular mechanism by which PB1-F2 contributes to pathogenesis have come forward and in the future it will be a challenge to connect all findings into one comprehensive model.
Abbreviations
| ANT3 | = | adenine nucleotide translocator 3 |
| ATF | = | activating transcription factor |
| CARD | = | caspase activation and recruitment domain |
| IFN | = | interferon |
| IKKε | = | IκB kinase ε |
| IRF | = | interferon regulatory transcription factor |
| ISG | = | interferon-stimulated gene |
| JAK | = | Janus kinase |
| MAVS | = | mitochondrial antiviral signaling protein |
| MDA-5 | = | melanoma differentiation-associated gene-5 |
| NDV | = | new-castle disease virus |
| NFκB | = | nuclear factor κB |
| NLR | = | Nod-like receptor |
| PTPC | = | permeability-transition pore complex |
| RIG-I | = | retinoic acid inducible gene-I |
| RLH | = | RIG-I-like RNA helicase receptor |
| STAT | = | signal transduction and activator of transcription |
| TBK1 | = | TANK-binding kinase 1 |
| TNFα | = | tumor-necrosis factor α |
| TLR | = | toll-like receptor |
| TRAIL | = | tumor necrosis factor-related apoptosis-inducing ligand |
| TRIF | = | TIR-domain-containing adapter-inducing interferon-β |
| VDAC1 | = | voltage-dependent anion channel 1 |
Figures and Tables
Figure 1 The anti-interferon activity of PB1-F2. Upon recognition of viral RNA species, RIG-I interacts with and activates MAVS. This leads to the activation of TBK1 and IKKε which phosphorylate IRFS. The IRF transcription factor complex then translocates to the nucleus to mediate the transcription of type I IFN genes (together with NFκB and ATF/c-Jun, not shown here). Our recent study shows that PB1-F2 inhibits the induction of type I IFN at the level of the MAVS adaptor protein. Please note: schematic is not to scale.
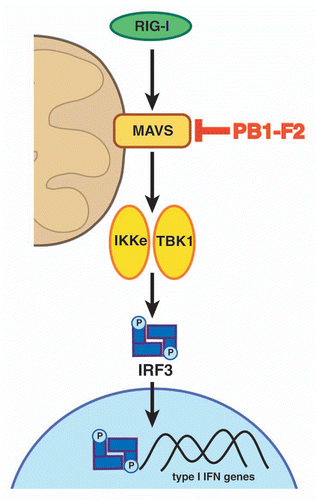
Figure 2 PB1-F2 promotes mitochondrial apoptosis. (A) PB1-F2 can associate with ANT3 and VDAC1 in the inner and outer mitochondrial membrane, respectively, to induce the efflux of cytochrome c (cyto c), which further downstream leads to the activation of caspases. (B) PB1-F2 can also self-oligomerize to form pores in the mitochondrial membrane. Please note: schematic is not to scale.
Figure 3 Model for the molecular bridge of the pro-apoptotic and anti-interferon functions of PB1-F2. MAVS is an important mediator of the type I IFN production pathway. It has also been shown that MAVS has a negative regulatory effect on VDAC1-mediated apoptosis. PB1-F2 affects MAVS-mediated IFN production by possibly binding to MAVS, which may have two effects: (1) the inhibition of the type I IFN response and (2) the lifting of the inhibitory effect of MAVS on VDAC1-mediated cell death. Please see text for further details and discussion. Please note: drawing is not to scale.
Acknowledgments
Work on the PB1-F2 protein in the laboratory of Dr. Peter Palese is supported by the NIAID grant P01AI058113 and the Center of Research in Influenza Pathogenesis (CRIP, NIAID contract HHSN266200700010C). We would like to thank Natalie Pica and Irina Margine for helpful discussions.
Addendum to:
References
- Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog 2005; 1:4; PMID: 16201016; http://dx.doi.org/10.1371/journal.ppat.0010004
- Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 2001; 7:1306 - 1312; PMID: 11726970; http://dx.doi.org/10.1038/nm1201-306
- Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol 2006; 80:7976 - 7983; PMID: 16873254; http://dx.doi.org/10.1128/JVI.00415-06
- McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog 2010; 6:1001014; PMID: 20661425; http://dx.doi.org/10.1371/journal.ppat.1001014
- Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog 2007; 3:1414 - 1421; PMID: 17922571; http://dx.doi.org/10.1371/journal.ppat.0030141
- Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol 2011; 85:652 - 662; PMID: 21084483; http://dx.doi.org/10.1128/JVI.01987-10
- McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007; 2:240 - 249; PMID: 18005742; http://dx.doi.org/10.1016/j.chom.2007.09.001
- Hai R, Schmolke M, Varga ZT, Manicassamy B, Wang TT, Belser JA, et al. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J Virol 2010; 84:4442 - 4450; PMID: 20181699; http://dx.doi.org/10.1128/JVI.02717-09
- Mazur I, Anhlan D, Mitzner D, Wixler L, Schubert U, Ludwig S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol 2008; 10:1140 - 1152; PMID: 18182088; http://dx.doi.org/10.1111/j.1462-5822.2008.01116.x
- McAuley JL, Zhang K, McCullers JA. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol 2010; 84:558 - 564; PMID: 19828614; http://dx.doi.org/10.1128/JVI.01785-09
- Varga ZT, Ramos I, Hai R, Garcia-Sastre A, Fernandez-Sesma A, Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog 2011; 7:1002067; PMID: 21695240; http://dx.doi.org/10.1371/journal.ppat.1002067
- García-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998; 252:324 - 330; PMID: 9878611; http://dx.doi.org/10.1006/viro.1998.9508
- Kochs G, Garcia-Sastre A, Martinez-Sobrido L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 2007; 81:7011 - 7021; PMID: 17442719; http://dx.doi.org/10.1128/JVI.02581-06
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 2008; 89:2359 - 2376; PMID: 18796704; http://dx.doi.org/10.1099/vir.0.2008/004606-0
- Baum A, Garcia-Sastre A. Differential recognition of viral RNA by RIG-I. Virulence 2011; 2:166 - 169; PMID: 21422808; http://dx.doi.org/10.4161/viru.2.2.15481
- Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, et al. Virus-infection or 5′ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog 2010; 6:1001012; PMID: 20661427; http://dx.doi.org/10.1371/journal.ppat.1001012
- Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2009; 2:47; PMID: 19690333; http://dx.doi.org/10.1126/scisignal.2000287
- Tang ED, Wang CY. MAVS self-association mediates antiviral innate immune signaling. J Virol 2009; 83:3420 - 3428; PMID: 19193783; http://dx.doi.org/10.1128/JVI.02623-08
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011; 146:448 - 461; PMID: 21782231; http://dx.doi.org/10.1016/j.cell.2011.06.041
- Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal 2011; 4:7; PMID: 21285412; http://dx.doi.org/10.1126/scisignal.2001147
- Zhirnov OP, Konakova TE, Wolff T, Klenk HD. NS1 protein of influenza A virus downregulates apoptosis. J Virol 2002; 76:1617 - 1625; PMID: 11799156; http://dx.doi.org/10.1128/JVI.76.4.1617-25.2002
- Zhirnov OP, Klenk HD. Control of apoptosis in influenza virus-infected cells by upregulation of Akt and p53 signaling. Apoptosis 2007; 12:1419 - 1432; PMID: 17468837; http://dx.doi.org/10.1007/s10495-007-0071-y
- Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T, Pleschka S, et al. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J 2003; 22:2717 - 2728; PMID: 12773387; http://dx.doi.org/10.1093/emboj/cdg279
- Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H, et al. NFkappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem 2004; 279:30931 - 30937; PMID: 15143063; http://dx.doi.org/10.1074/jbc.M403258200
- Xu Y, Zhong H, Shi W. MAVS protects cells from apoptosis by negatively regulating VDAC1. Mol Cell Biochem 2011; In press PMID: 21110072
- Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J 2010; 29:1762 - 1773; PMID: 20360684; http://dx.doi.org/10.1038/emboj.2010.50
- Lei Y, Moore CB, Liesman RM, O'Connor BP, Bergstralh DT, Chen ZJ, et al. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS ONE 2009; 4:5466; PMID: 19404494; http://dx.doi.org/10.1371/journal.pone.0005466
- Li HM, Fujikura D, Harada T, Uehara J, Kawai T, Akira S, et al. IPS-1 is crucial for DAP3-mediated anoikis induction by caspase-8 activation. Cell Death Differ 2009; 16:1615 - 1621; PMID: 19644511; http://dx.doi.org/10.1038/cdd.2009.97
- Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol 2003; 77:7214 - 7224; PMID: 12805420; http://dx.doi.org/10.1128/JVI.77.13.7214-24.2003
- Bruns K, Studtrucker N, Sharma A, Fossen T, Mitzner D, Eissmann A, et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J Biol Chem 2007; 282:353 - 363; PMID: 17052982; http://dx.doi.org/10.1074/jbc.M606494200