Abstract
The encephalomyocarditis virus (EMCV) is a small non-enveloped single-strand RNA virus, the causative agent of not only myocarditis and encephalitis, but also neurological diseases, reproductive disorders and diabetes in many mammalian species. EMCV pathogenesis appears to be viral strain- and host-specific, and a better understanding of EMCV virulence factors is increasingly required. Indeed, EMCV is often used as a model for diabetes and viral myocarditis, and is also widely used in immunology as a double-stranded RNA stimulus in the study of Toll-like as well as cytosolic receptors. However, EMCV virulence and properties have often been neglected. Moreover, EMCV is able to infect humans albeit with a low morbidity. Progress on xenografts, such as pig heart transplantation in humans, has raised safety concerns that need to be explored. In this review we will highlight the biology of EMCV and all known and potential virulence factors.
Description of EMCV
History
The encephalomyocarditis virus (EMCV) was first isolated in 1945, by Helwig and Schmidt in Miami, FL.Citation1 It was from a captive male gibbon that died suddenly of pulmonary edema and myocarditis. Mice inoculated intravenously, intraperitoneally, subcutaneously, intracranially or by intranasal instillation with filtered edema fluid from the gibbon developed paralysis of the posterior members and a myocarditis followed by death in a week. The pathogenic agent was at that time named the encephalomyocarditis virus.
In 1948, a Mengo virus was isolated by Dick et al.Citation2 in the Mengo district of Entebbe in Uganda. It was isolated from a captive rhesus monkey that had developed hind limb paralysis. In 1949, cross sero-neutralization studies showed that Mengo, EMCV were antigenically indistinct from each other, meaning they are part of the same species. However, they were distinct from Theiler’s murine encephalomyelitis virus (TMEV) another cardiovirus (compare classification) that is responsible, depending on the strain, for an acute fatal-poliomyelitis or a chronic demyelinating disease with persistence of the virus in the central nervous system of the mouse (for review, see refs. Citation3 and Citation4).
Classification
The EMCV belongs to the Picornaviridae (pico = small, rna = ribonucleic acid) family, which is currently divided into 12 genera: Aphtovirus, Avihepatovirus, Cardiovirus, Enterovirus, Erbovirus, Hepatovirus, Kobuvirus, Parechovirus, Tescovirus, Tremovirus, Sapelovirus and Senecavirus.Citation5
The genus Cardiovirus is divided into two species: the Encephalomyocarditis virus (EMCV) and Theilovirus. The EMCV only harbors one serotype, Mengovirus, being a strain of EMCV. In contrast, Theilovirus comprises Theiler’s murine encephalomyelitis virus (TMEV), Vilyuisk human encephalomyelitis virus (VHEV), Thera virus (TRV; isolated from rats), Saffold virus 1–8 (SAFV; isolated from humans, for review about SAFV see ref. Citation6).Citation5 TMEV, SAFV and EMCV share about 50% identity.Citation7-Citation9 Depending on the protein considered, the percentage of identity between TMEV and EMCV can vary from 20 to 70%. The L and 2A, two proteins that are considered as viral security proteins or virulence factor, are the most divergent.Citation9
The EMCV characteristics and genome
The EMCV, like other picornaviruses, is a small non-enveloped virus, with an icosahedric capsid of 30 nm diameterCitation10 and a genome consisting of a positive single-stranded RNA of approximately 7.8 kb that allows direct translation of the RNA into a polyprotein.
The viral RNA is infectious in and of itself, meaning that viral proteins are not required to initiate viral gene expression, and thus its introduction by transfection into cells suffices for production of infectious viral particles. Viral RNA acts as a mRNA during translation and as template during genome replication. The genome has a unique coding region flanked by two untranslated regions (UTR). The 5′ UTR is between 800 to 1,200 nucleotides (nt) long, while the 3′ UTR is about 120 ntCitation11 long and composed of short stem-loop structures followed by a poly(A) tail of variable length (20 to 70 nucleotides) ().
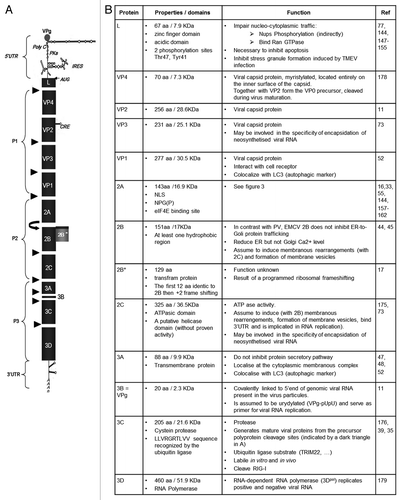
Figure 1. (A) Genomic organization. The EMCV genome is a positive single-strand RNA, covalently linked to the viral protein VPg in 5′. The genomic RNA is composed of two UTR in 5′ and 3′ and an ORF that encodes for a polyprotein (dark gray). The polyproteins is cleaved by the 3C protease, cleavage sites are indicated (black triangles). Ribosome skinping (curved right arrow). A second ORF (clear gray) due to a +2 ribosome frameshifting. cre, Cis replicating element; PKs, pseudoknots; IRES, internal ribosome entry site. (B) Description of EMCV proteins properties and functions known or assumed by similarity with other picornaviruses.
The viral RNA is thus polyadenylated at the 3′ end, but while cellular mRNA are capped with a 7-methyl-guanosine, the 5′ extremity of the viral RNA is covalently linked to a 20 amino acid viral protein called VPg (or 3B).Citation12 Downstream, a poly(C) tract of about 150 nucleotides (depending on the strain) has been found and is specific to aphtoviruses as well as EMCV. In contrast, TMEV is also a cardiovirus but does not possess this poly(C) tract.Citation8 Studies with Mengo virus in murine hosts suggest that the poly(C) length may play an important role in viral pathogenesisCitation13 and will be further discussed in the section “virulence factor.”
Adjacent to the poly(C) tract are some pseudoknots with unknown function. The last unit of the 5′ UTR is the internal ribosome entry site (IRES). The IRES of picornaviruses fall into five categories, based on conserved primary and secondary structureCitation14 (for review see ref. Citation15). EMCV has a type II IRES of about 450 nt with a highly ordered structure made up of hairpin loops. It is subdivided into five structural domains designated H-L, and immediately upstream of the initiation codon AUG a pyrimidine rich tract is found.Citation16 The IRES allows ribosome binding, and thus the initiation of translation of the open reading frame, which encodes a polyprotein of 2,292 amino acids. Recently, the existence of a programmed ribosomal frameshift has been described, leading to an out-of-frame generation of a cryptic 2B* protein.Citation17 This finding indicates that the EMCV genome does not have one, but at least two ORF, encoding for 13 mature proteins. The genome of picornaviruses also contains a higher-order RNA structure, the cis-acting replication element (CRE), used as a template for 3D polymerase-mediated VPg uridylation, necessary for initiation of viral RNA replication. In the case of EMCV, the CRE is located in the VP2 coding region and consists of a stem-loop structure with a conserved AAACA sequence in the loop.Citation18
Viral proteins
The viral RNA encodes a large polyprotein (L-1ABCD-2ABC-3ABCD) that is cleaved to produce approximately 13 mature proteins. The proteins and precursors of EMCV as well as of the other picornaviruses get their names from their position within the polyprotein, that is, the leader protein (L), the precursor P1, comprising capsid proteins 1A, 1B, 1C and 1D, also called VP4, VP2, VP3 and VP1, respectively, once integrated into the virion. The P2 and P3 are precursors of the non-structural proteins: 2A, 2B and 2C, and 3A, 3B (also called VPg), 2B*, the 3C protease and the RNA-dependent RNA polymerase 3D.Citation11
The EMCV genome is represented in and the accompanying table describes EMCV proteins and summarizes, in their respective order within the polyprotein, their known functions. The function of the EMCV proteins have often been assigned by virtue of their similarity to their well-studied counterparts in poliovirus (PV), Theiler’s murine encephalomyelitis virus (TMEV) and foot and mouth disease virus (FMDV). In this review we highlight what has been shown for EMCV and what has been assumed from study of other picornaviruses. (In the next paragraphs we used the term picornaviruses to designate that as far as we know it has not been clearly proven for EMCV but it has been shown for other picornaviruses.)
Three-dimensional structure of the capsid
The Mengovirus has been crystallized and analyzed by X-ray diffraction.Citation10,Citation19 The viral capsid is organized into an icosahedron with exactly 60 copies of each of the structural protein. The basic unit of the capsid is a protomer formed by the association of VP1, VP2, VP3 and VP4. The VP4 protein is located in the internal face of the capsid and interacts with the viral RNA. Five protomers assemble into a pentamer and 12 pentamers are necessary to form the capsid. The capsid is organized around symmetric axes of 2-, 3- and 5-fold symmetry. Five VP1 proteins are grouped around the 5-fold symmetry axis, while VP2 and VP3 proteins alternate around the 3-fold symmetry axis. In the case of Mengo and TMEV the five copies of VP1 form a plateau with a five branches star shape. The extremities of the branches are constituted of prominent structures (loops) that are the most exposed region at the surface of the Mengo capsid. Each branch of the star is separated by a depression called the pit. The pit corresponds to the contact region between VP1 and VP3. The pit is considered as the analog of the “canyon” found on the capsid of PV. By analogy, it has been supposed that the Mengo pit would serve as a receptor binding site. To support this hypothesis, residues lining in the pit are more conserved than other surface residuesCitation20 and computer modeling can fit a sialic acid residue into a pocket near the bottom of the 22-Å-deep pit.Citation21
Viral Cycle
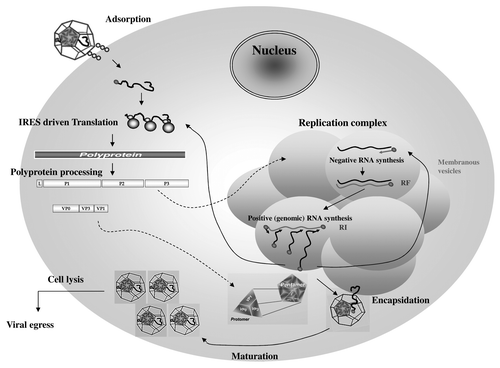
The EMCV viral cycle, represented in , is detailed below. (The time scale given in the figure legend is specific for BHK-21 and might vary upon the cell type and the EMCV strain use for the infection.)
Figure 2. Encephalomyocarditis viral replication cycle. The virion binds to a cellular receptor. After uncoating, the genomic RNA is released by an unknown mechanism. Internalization (~1–2 h). Once in the cytoplasm, the VPg protein is detached to the 5′ end of the genome, the translation is initiated at the IRES that require the PTB cellular protein, the polyprotein is synthesized. Translation (~2.5–3 h). The polyprotein is cleaved during and after the translation, leading to precursors or mature proteins. Some of those proteins allow formation of the membranous vesicles where genome replication will occur. The positive genomic RNA is replicated into negative RNA thanks to the VPg-pUpU that serves as a primer for the 3D polymerase. The negative RNA syntesis leads to the production of a double stranded RNA molecule, the replication form (RF). The newly synthesized negative RNA in turn, serves as templates for synthesis of positive RNAs in the replication intermediate (RI). Replication (~3–4 h). Those new positive genomic RNAs will either serve for translation after removal of VPg, serve as matrix for synthesis of new viral RNAs or will be encapsidated. The viral capsid proteins VP0, VP1 and VP3 auto-assemble into a protomer. Five protomers will assemble into pentamers and 12 of them will form the icosahedric capsid. Encapsidation (~4–6 h). After RNA encapsidation, cleavage of the precursor VP0 into VP2 and VP4 allows maturation of the virion. Virions are then egress by cell lysis. Egress (~6–10 h). (Modified from ref. Citation177).
Adsorption: entry
To initiate the infection, the first step is the attachment of the virus particle to a cell membrane molecule: a receptor or coreceptor.Citation21 This virus-cell interaction and the distribution of the virus receptor are considered to be determinant for the viral host range, tissue tropism and pathogenesis. Vascular cell adhesion molecule 1 (VCAM-1) is a sialoglycoprotein that has been identified as a receptor for EMCV on murine vascular endothelial cells.Citation22
Another 70 KDa sialoglycoprotein found on HeLa and K562 cells and the Sialylated glycophorin A (found on red blood cells) have also been described as being attachment proteins for EMCV.Citation23,Citation24 Sialoglycoproteins are “sticky” proteins that may have non-specific binding properties. Thus, the main receptor for EMCV, as well as its viral binding partner, remains uncertain. A recent study suggests that the capacity of the virus to interact with cell surface sialic acid residues is important for infection of BRL cells.Citation25 However, it is interesting to note that sialic acid binding is not required for all EMCV strains.Citation26
The mechanisms involved in internalization and uncoating of cardioviruses are poorly documented. It is known that infectivity of EMCV and PV does not require diminution in pH. Thus, unlike FMDV whose entry requires receptor-mediated endocytosis and endosomal acidification, it is believed for EMCV that interaction of the viral particle with a cellular receptor suffices to induce the conformational changes of the capsid shell required for viral RNA delivery into the cytoplasm.Citation27,Citation28 These conformational changes has been shown for PV to affect contacts between pentamers and thus initiate the dissociation of the capsid. VP1 N-terminal extremities have an amphipathic helix that inserts into the cell membrane and allows the formation of a pore through which the viral RNA is thought to be injected into the cell.Citation28 However this two-steps dissociation process seems not to fully apply to cardioviruses since contacts between their pentamers are less extensive than for PV.Citation21 Thus it is still unclear how EMCV release its genome into the cytoplasm.
Translation
Once the genomic RNA is in the cytoplasm, it must be translated as it cannot be replicated by cellular RNA polymerases and no viral enzymes are brought along with the viral particle. The 5′ end of the positive single-stranded viral RNA is not capped, but is linked to the VPg viral protein. This VPg does not have any known role in translation and can be detached from the viral RNA by a cellular enzyme.Citation29 The genome is then translated into viral proteins needed for genome replication and the production of new viral particles. For EMCV, initiation of translation is cap-independent due to its IRES. There exist five types of IRES that differ in their primary and secondary structures, the location of the initiation codon and their activity in different cell types. All picornaviruses possess an IRES. EMCV and the other cardioviruses have a type II IRES (like the aphtoviruses, but unlike the enteroviruses that has a type I IRES).Citation30
The EMCV IRES requires binding of cellular factors, including all the eIFs, except eIF4E, the N-terminal region of eIF4G and the polypyrimidine tract binding protein (PTB).Citation31,Citation32 This then allows direct recruitment of the 40S ribosomal subunit together with several translation initiation factors (eIF-1A, eIF-GTP-met-tRNA and eIF3), which permits direct initiation of translation at the start codon. Unlike cap-dependent translation, IRES translation does not require eIF4F complex formation; that is, binding of the eIF4E component to the cap structure at the 5′ mRNA and subsequent recruitment of the 40s ribosomal subunit, which scans the 5′ non-coding region until the authentic start codon is reached. In both cases, the 60S ribosomal subunit joins the complex at the AUG initiation codon, initiation factors are released, and elongation ensues.
The translation of picornaviral RNA leads to synthesis of viral proteins, some of which inhibit cap-dependent translation. Enterovirus 2Apro cleaves eIF4E, a function assumed by the Lpro for aphtoviruses. The cardioviruses that possess only one protease, 3C, seem to have developed different strategies that enable inhibition of host cell translation, even if this inhibition is slower and less efficient.Citation33 The EMCV 2A protein is believed to play an essential role in this process (see and below the description of “EMCV 2A protein” in the section “EMCV Virulence”).
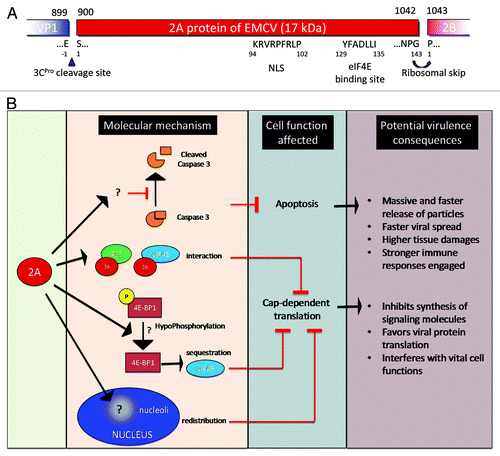
Figure 3. Encephalomyocarditis viral 2A protein. (A) EMCV 2A protein organization and putative sequence. (B) Functionality of EMCV 2A protein and potential consequence on EMCV virulence.
Polyprotein processing
Polyprotein cleavage is mainly performed by the 3C protease (3CPro) of EMCV. However, the first instance of polyprotein processing is not proteolytic, and indeed occurs during translation between the 2A and 2B protein, before EMCV 3Cpro synthesis. The NPG(P) sequence at the junction between these two proteins leads to ribosome skipping; no peptide bond is made between the glutamine (G) of 2A and the proline (P) of 2B, resulting in production of two separate peptides (for details see and description of the 2A protein in the section “EMCV Virulence”).
EMCV 3Cpro, the only protease of EMCV, is an essential protein. It is a cysteine protease that exhibits a high degree of substrate specificity. Comparison of known cleavage sites of the EMCV viral protease has failed to reveal a clear consensus sequence, although some common determinants can be identified. EMCV 3Cpro preferentially cleaves between Q or E and G, S or A residues.Citation34,Citation35 The precursors 3ABC, 3CD and P3 of EMCV are able to cleave as efficiently as the 3C.Citation35 Of note, 3CD of EMCV can cleave P1, whereas 3CD of PV cannot.
EMCV 3Cpro has been described to be extremely labile in vitro and in vivo. An LLVRGRTLVV amino acid motif has been identified as a destruction signal that leads to 3C ubiquitination and degradation by the proteasome.Citation36-Citation38 In contrast to FMDV and PV, only one cellular substrate has been reported for the EMCV 3Cpro. Interestingly, EMCV 3Cpro is able to cleave the RNA recognition sensor RIG-I, at least in vitro.Citation39 This finding could explain why RIG-I is unable to sense EMCV RNA.
During EMCV infection, once the 3C protease is translated, it is immediately active and starts to cleave the polyprotein in cis. When detached from the polyprotein, 3Cpro can also cleave newly synthesized polyproteins in trans. EMCV 3Cpro cleavage sites within the polyprotein are indicated by small black triangles in . The last cleavage occurs during the maturation of the virion, after RNA encapsidation, when VP0 is cleaved into VP4 and VP2 (see paragraph “provirion assembly—encapsidation”).
Replication
Localization-formation of the membranous replication complex
Replication of the picornavirus genome occurs within the cytoplasm. Picornavirus infection leads to intracellular membranous proliferation and rearrangements. The endoplasmic reticulum (ER) and Golgi apparatus are rearranged and the cytoplasm fills up with bilayer membranous vesicles.Citation40 Viral replication takes place at the vesicle surface.Citation41 The 3A and 2BC proteins of PV are known to localize in the ER and are sufficient to induce the formation of these vesicles.
The enterovirus 2B protein is a small hydrophobic protein. It is localized in the ER and the Golgi and reduces Ca2+ levels in these organelles. The 2B protein is a viroporin,Citation42 able to form transmembranous pores, which could explain this calcium flux. In addition to membrane rearrangements, it inhibits protein trafficking through the Golgi complex.Citation43-Citation46 The role of the 2B protein has not been clearly established for EMCV. However, De Jong et al.Citation44 showed that the 2B protein of EMCV (like that of FMDV) shares little similarity (< 20% sequence identity) with the enterovirus 2B proteins, which are much smaller. The 2B protein of EMCV contains one or more hydrophobic regions, but no cationic amphipathic α-helix like that found in all enterovirus 2B proteins. Although EMCV 2B, when lowly expressed in cells, does not show a clear ER localization, it was also found to reduce the level of Ca2+ in the ER but not the Golgi.Citation44 The mechanism underlying this decrease is unknown, and the localization of the 2B protein has yet to be confirmed in the context of an EMCV infection. In the same study, the 2B protein of EMCV and FMDV, unlike the 2B protein of enteroviruses, was shown to be unable, on its own, to interfere with protein trafficking.
In addition, the 3A protein, which is a small transmembraneous hydrophobic protein, has also been found in the membranous complex. While the Enterovirus 3A protein inhibits ER-to-Golgi transport, it has been demonstrated that the 3A protein of FMDV, TMEV and EMCV does not.Citation47,Citation48 Thus, EMCV 3A and EMCV 2B do not block protein transport. It remains to be established whether EMCV has the capacity to block the transport form the ER to the Golgi. Indeed, expression of 2BC or 2B and 2C together are required to block ER-to-Golgi apparatus transport in FMDV infected cells.Citation49,Citation50 However, it has been described that 2BC of TMEV, when express from a plasmid, does not show the same pattern of localization as 2BC of FMDV.Citation51 In conclusion, it is yet to be determined whether protein trafficking is inhibited during EMCV infection. To our knowledge, there is no evidence regarding inhibition of ER-to-Golgi protein trafficking in EMCV-infected cells. Of note, even if EMCV does not inhibit ER-to-Golgi trafficking, it induces intracellular membranous proliferation and rearrangements.Citation52,Citation53
It is assumed that the lipids that make up the membranous vesicles are derived from the ER.Citation46 However, those vesicles might also originate from the autophagic cellular pathway. Indeed, during PV infection, it has been shown that the vesicles of the replication complex resemble those of the autophagic pathway, in that they possess double membrane structures, cytoplasmic components within the vesicles, and typical autophagic markers such as LC3 and LAMP1 (latency-associated membrane protein 1).Citation40,Citation54 Very recently, similar observations have also been described for both EMCV and FMDV. During EMCV infection, colocalization of the 3A and VP1 proteins with LC3 at the surface of double-membrane vesicles has been described.Citation52 Colocalization of 2B, 2C and 3A viral proteins of FMDV with LC3 and LAMP1, as well as colocalization of VP1 with Atg5 (cellular protein required for autophagosome formation), has been reported.Citation53 Some studies showed that inhibition of autophagy led to a decrease in EMCV production whereas its induction with rapamycin led to an increase in viral titer.Citation52,Citation55 This suggests that the autophagic pathway may be hijacked by EMCV, as other picornavirus, to furnish a membranous support for viral replication.Citation54
Thus, viral replication of EMCV is believed to occur in the cytosol within the replication complex localized at the surface of clustered membranous vesicles. The replication complex is essentially composed of the viral RNA-dependent RNA polymerase 3Dpol in association with the 3Cpro, the 2C and the 3AB proteins.
Again, very few studies have addressed the replication of EMCV, and most of our assumptions are based on extrapolation from studies performed with enteroviruses. The model developed for poliovirus replication is believed to hold for other picornaviruses, but this awaits confirmation.
Negative strand RNA synthesis
It is commonly accepted that translation of picornaviruses must stop to allow transcription of the negative strand viral RNA,Citation56 negative RNA synthesis being the first step of viral RNA replication. The mechanism governing the shift from translation to replication is not fully determined, although a model has recently been proposed for PV.Citation57 This model implicates cleavage of the host protein PCBP2 by 3CDpro; however, as this cleavage has never been described for EMCV, further studies will be required to decipher how the transition from translation to replication may occur during EMCV infection.
The initiation of negative strand RNA synthesis of picornaviruses is still controversial. A model for PV proposes that such initiation starts with the circularisation of the genome through the interaction of the cellular protein PCBP [poly r(C) binding protein] with the 3CD protein, which itself binds to the cloverleaf structure of the 5′UTR viral genome.Citation58 For EMCV these interactions might take place at the S fragment or the poly(C). Thus 3′ and 5′ viral genome extremities may temporarily interact via a protein bridge that forms the so-called ribonucleoproteic complex. The formation of this initiation complex permits the uridylylation of the VPg (3B) by 3Dpol using the poly(A) tract or the AAACA motif of cre as a template.
The cre is an RNA stem-loop structure of the viral genome, found in the VP2 coding sequence of EMCV.Citation18 The loop bears an AAACA, the two first A residues of which acting as a template for the synthesis of VPg-pU and VPg-pUpU (uridylylation) and therefore for the initiation of replication.Citation59 Mutation within this conserved motif causes a marked decrease in viral replication. Interestingly, the position of the cre within the genome seems not to be critical and can even be provided in trans.Citation18,Citation60 It is believed that VPg-pUpU matches with the poly(A) 3′ extremity and therefore acts as a primer for negative RNA strand synthesis by 3Dpol. The elongation of the negative strand leads to the formation of a double-stranded RNA structure called the replication form (RF).Citation61 After RF formation, synthesis of new positive RNA can start.
Positive strand RNA synthesis
Picornavirus positive strand synthesis initiation from the RF has not been elucidated. Two main hypotheses have been proposed for VPg-pUpU generation. The first hypothesis suggests that initiation of positive strand synthesis may use VPg proteins that have already been uridylylated, in excess, during negative strand synthesis.Citation62 The second hypothesis proposes that VPg-pUpU is generated at the poly(A) tail of the positive RNA for the synthesis of the negative strand, while the uridylylation at the cre is essentially used for positive strand synthesis.Citation63
The RF structure is presumed to be unwound for positive strand synthesis. The 2C viral protein possesses an ATPase activity and a putative helicase domain (even if helicase activity has never been reported), and seems to be important for positive RNA synthesis.Citation64 It has been reported that the 2C protein binds to the 3′ extremity of the negative strand RNA along with the cellular p38 protein. This interaction probably destabilizes the RF. However, cellular helicases and nuclear proteins might also be implicated, as the RF structure is infectious when used to transfect non-enucleated cells.Citation61,Citation65
The negative strand RNA newly synthetized is then used as a template for positive strand RNAs. Indeed, there is formation of partially double-stranded RNA, corresponding to the replication intermediates (RI) that allows simultaneous synthesis of several positive RNA strands from a single negative RNA template.
Provirion assembly: encapsidation, viral release or egress
The last steps in the EMCV viral cycle are encapsidation of newly synthesized viral RNA, maturation of the provirions and virus release from the cell. The mechanisms involved in encapsidation and maturation are still unresolved and are probably the least studied steps of the viral cycle.
The formation of viral particles occurs in the cytosol through the 3Cpro cleavage of the P1 precursor, releasing VP0, VP1 and VP3 that auto-assemble to form a protomer containing a single copy of each protein. Five protomers form a pentamer and 12 pentamers auto-assemble to form the icosahedric capsid. There are two models for picornavirus RNA encapsidation. A first model proposes that the protomers assemble into empty capsid and that the viral RNA is then inserted into the capsid. This is supported by the observation that empty capsids are found during picornavirus infection, but has never been formally proven. A second model proposes that the pentamers assemble directly around the neo-synthesized viral RNA. Picornaviruses encapsidate only positive single-stranded RNA linked to the VPg protein.Citation66 In addition, only newly synthesized RNAs are encapsidated, suggesting that a link exists between active replication and encapsidation.Citation67
Substitution of P1 by a reporter gene in several picornaviruses does not perturb viral RNA encapsidation when capsid proteins are provided in trans.Citation68-Citation70 This indicates that the putative encapsidation signal is not found within the P1 region of viral RNA. In fact, numerous attempts to identify an RNA encapsidation signal have failed. A single study reports that the encapsidation signal could lie within the 5′UTR of the Aichi virus together with the L protein.Citation71,Citation72 However, a recent paper, which tends to favor the second model of encapsidation, also proposes a mechanism by which the triple interaction vRNA-2C-VP3 would be sufficient to explain the specificity of picornavirus encapsidation.Citation73 The novel mechanism described by Liu et al. suggests that encapsidation takes place at the site where newly synthesized genomes emerge from the replication complex, since morphogenesis is linked to genome replication, 2C ATPase being a component of the replication complex and possessing specific affinity for the VP3 capsid protein. They showed that for chimeric viruses composed of PV and Cox that are unable to support encapsidation, the encapsidation process could be restored if the P1 and 2C proteins originate from the same virus. Previous studies on heterologous encapsidationCitation68 have shown that the PV replicon could be encapsidated into coxsackie B3 virus (CBV3), human rhinovirus 14 (RHV14) and mengovirus capsids. It should be noted that the capsid proteins were provided by the expression of the full viral cDNA under control of a T7 promoter, indicating that their 2C proteins were certainly present.
The presence of the viral RNA in the capsid is required for the maturation step in which VP0 is cleaved into VP2-VP4. This cleavage is considered to be autocatalytic and would result from a local activation of water molecules by a His residue in the VP2. This would lead to a nucleophilic attack on the scissile bond and cleavage.Citation74,Citation75 The maturation is necessary for the generation of infectious viral particles. Indeed, it is possible that VP0 cleavage is required for the subsequent release of viral RNA into the cytoplasm of newly infected cells.Citation76
EMCV is a rapidly lytic virus that causes necrotic cell death within 7 to 10 h.Citation77,Citation78 However, just what may lead to membrane permeabilization and what the signals for cell lysis and viral egress may be remain to be characterized. Several hypotheses have been advanced, such as cell burst due to accumulation of viral particules or proteins in the cell or permeabilization of cell membranes due to viroporins (like the 2C of PV). Irrespective of which process is involved, the efficiency of viral release is likely to influence EMCV virulence (further developed in the section on EMCV Virulence).
Host Spectra, Epidemiology and Zoonotic Potential
EMCV was first isolated from a gibbon in 1945 in Florida,Citation1 and then from swine following an epizootic in Panama in 1958.Citation79 From 1945 to the present, EMCV has been detected in many wild and domestic animals, in many different areas around the world, such as Europe,Citation80,Citation81 Canada,Citation82 South America,Citation79,Citation83,Citation84 Australia,Citation85 KoreaCitation86 and China.Citation87
A non-exhaustive list of animals from which EMCV has been isolated includes voles, squirrels, elephants, swine, wild boar, racoons, antelope, lions, birds and several species of non-human primates.Citation1,Citation81,Citation85,Citation88,Citation89 Human infections have also been reported (see paragraph on zoonotic potential).
All together these studies indicate that EMCV has a worldwide distribution and can infect a wide range of animal species. Its natural reservoir is thought to be rodents (mouse or rats). Indeed rat infections are usually asymptomatic and the virus can replicate and be excreted up to 29 d post-infection.Citation90 The presence of infected rodents in proximity to farms with infected swine also suggests they play an important role in virus spread,Citation91,Citation92 even more so because horizontal transmission between pigs seems limited.Citation93 However, a study performed in Greece points out that the possibility of a reservoir existing in wild boar should not be neglected.Citation88
EMCV infection occurs by ingestion of EMCV-contaminated food, water and diseased carcasses.Citation89,Citation94 EMCV is a small non-enveloped virus that is very resistant and may remain infectious for days, even in a hostile environment. EMCV is quite stable in a large spectrum of pH and resistant to ether treatment. Disinfection methods recommended by veterinary are multiples: the virus can be inactivated by treatment at 60°C for 30 min, H2O containing 0.5 ppm of chlorine, iodine-based disinfectants or mercuric chloride.Citation95 Recently, a non-invasive and environment-friendly method using a femtosecond laser has been proposed to inactivate the virus.Citation96 A large scale usage and its extension to other pathogens could be an interesting development for this method.
Zoonotic potential
The EMCV has often been described as a potential zoonotic agent. Nevertheless, an association between human infection and disease has still not been clearly established. Some experimental infections on human explants or human primary cell cultures have been described and clearly point out that human cells are sensitive to EMCV.Citation97,Citation98
Between 1940 and 1950 childhood infections associated with fever and encephalitis but no myocarditis were described in Germany and Holland. Several EMCV strains (MM, AK, Li32, Ortilb and SVM) were isolated after inoculation of rodents.Citation99 However, the assignment of these strains to the EMCV species was determined on the basis of serological tests, without virological confirmation. Unfortunately, these strains are no longer available for further characterization. In an earlier study, neutralizing antibodies against EMCV were found in 17 soldiers based in Manila, Philippines. These soldiers presented febrile illness for 3 d. Three patients out of the four presented an increase in neutralizing antibody titer.Citation100 In 1948, after isolation of a Mengovirus from a Rhesus monkey, a researcher who was studying the virus and taking care of the animal had developed encephalitis, from which he later recovered. Meanwhile, the virus was isolated from his blood.Citation101
From 1950 to 2009, no EMCV infections associated with clinical signs were reported. However, serological studies performed on healthy persons revealed a prevalence of 2.3% to 15%.Citation102-Citation104 In Austria, a more recent study revealed that 5% of persons with occupational exposure to animals were EMCV seropositive. This percentage reached 15% for hunters.Citation105,Citation106
Two recent studies, performed in Peru, described human cases. Patients presented febrile illness, likely due to EMCV infection. Indeed, virus was isolated during the acute phase for two patients with nausea, headache and dyspnea. Upon molecular diagnosis, no virus other than EMCV was detected.Citation84 Isolation of viruses from sera of acute phase patients supports the idea that EMCV can infect humans and cause disease. A wider study has been performed and indicates that EMCV could be responsible for almost 1% of the febrile illness analyzed during this study in Peru.Citation83 Despite the low morbidity, human exposure to the virus is quite common. Indeed, 6% to 17% of Peruvian town inhabitants are seropositive for EMCV.Citation83 These recent data confirm what R. Tesh said in 1978: “EMC infection in man is fairly common but most human cases are probably asymptomatic and/or unrecognized.”Citation102
EMCV Pathogenesis
EMCV infection occurs by ingestion of EMCV-contaminated food, water and diseased carcasses.Citation89,Citation94 EMCV is a small non-enveloped virus that is very resistant and may remain infectious for days, even in a hostile environment. There is considerable variation in the severity and location of lesions caused by EMCV in the broad range of susceptible hosts. However, EMCV pathogenesis has mostly been studied in rodents, swine and monkeys.
General symptoms in non-human primates, such as baboons, gibbons, chimpanzees, green and rhesus monkeys, are mainly death and labored respiration associated with acute heart failure. The salient necropsy findings are pulmonary congestion and edema, hydropericardium, hydrothorax, ascites, lymph node and splenic hypertrophy, and pale white-to-tan mottled hearts. Placental infection with fetal loss can also occur.Citation107 Sub-cutaneous inoculation of baboons or African green monkeys induces myocardial and nervous disorders leading to death within a week. Infection of monkeys can lead to high mortality in primatology centers, as occurred in a baboon colony in San Antonio in 1992,Citation107 and more recently in a group of rhesus macaques.Citation108 Histologically, affected myocardiums present multifocal infiltration by lymphoplasmacytic cells admixed with necrotic and degenerate myofibers and infrequent mineralization.Citation108
In pigs
EMCV usually induces acute focal myocarditis with sudden death in pigs. Myocarditis is characterized by cardiac inflammation and cardiomyocyte necrosis. Other symptoms have been observed, such as anorexia, apathy, palsy, paralysis or dyspnea.Citation109 Susceptible pigs develop severe myocarditis followed by sudden death, while more resistant pigs develop mild myocarditis and can remain asymptomatic. Experimentally infected piglets show high fever, followed by death within 2 to 11 d, but can sometimes recover with chronic myocarditis. Mortality in piglets before weaning can rise to 100%. Mortality decreases with age.Citation91
After oro-nasal infection of piglets, viruses are thought to spread from tonsils to target organs by means of infected circulating monocytes.Citation110 In swine, the heart is considered to be the major target organ.Citation110 A few days after infection, however, viruses can be isolated from many other organs, such as brain, spleen, intestine, pancreas, liver, kidneys, heart, lungs and lymph nodes.Citation91,Citation109,Citation111 Upon autopsy of experimentally infected piglets, myocardial lesions, hydropericardium, pulmonary edema, ascites and hydrothorax are observed.Citation109,Citation110 Hearts are often dilated and show focal areas of necrosis (with uneven greyish white discoloration). The virus can be detected in the myocardium, even when myocardial lesions are small or absent.Citation109,Citation110
Histological analysis of piglet hearts reveals myocarditis associated with scattered or localized infiltration and accumulation of mononuclear cells, vascular congestion, edema and myocardial fiber degeneration, with necrosis. In the brain, congestion is accompanied by meningitis, perivascular infiltration of mononuclear cells and neuronal degeneration.Citation112,Citation113
Reproductive disorders including abortion, fetal death or mummification have been described in infected females.Citation114
In rodents
EMCV infection can be asymptomatic,Citation90 but in mice generally induces encephalitis,Citation115 member paralysis,Citation78,Citation116-Citation118 myocarditisCitation119 or type 1 diabetes.Citation120 EMCV infection can also lead to reproductive disorders in pregnant mice,Citation121 testicular lesions (orchitis) in miceCitation122 and also to lesions in salivary and lachrymal glands (sialodacryoadenitis) (for review see ref. Citation123).
Susceptibility to EMCV infection differs according to the strain and age of mice, and the viral strain and dose of inoculum. Infection of mice with different EMCV strains results in a wide spectrum of clinical manifestations, ranging from inapparent infection to severe disease and death.Citation119 For instance, the D strain of EMCV induces diabetes, whereas the M strain triggers myocarditis.
Myocarditis in mice
EMCV-induced myocarditis is characterized by myocardial necrosis and cellular infiltration. It has been widely used as a model to study viral-induced myocarditis. Many studies seem to indicate that the inflammatory response is deleterious,Citation124-Citation128 but discordant data do exist.Citation129 In recent years studies have shown that mast cells also play a critical role in the progression of heart failure induced by EMCV in mice.Citation124,Citation125 (This will be further discussed in the section dealing with EMCV virulence).
Hind limb paralysis and encephalitis
Hind limb paralysis and encephalitis have been documented during EMCV infection. This indicates that the murine central nervous system (CNS) is attained and damaged during EMCV infection. Upon administration of a high dose of inoculum, mice show encephalitis and paralysis within 4 d of infection and no recovery.Citation78 Cerebral lesions are mainly perivascular mononuclear infiltrate in the form of perivascular cuffs, multifocal neuronal necrosis and diffuse gliosisCitation78,Citation117,Citation130 and are predominantly located in the hippocampus.Citation78,Citation117 EMCV also induces spinal cord lesions comprising focal degeneration, lymphocytic infiltration, mild neuronal necrosis and demyelinisation.Citation78,Citation118,Citation131,Citation132 Inoculation of a low dose of virus leads to a biphasic hind limb paralysis with possible recovery.Citation131 EMCV, unlike TMEV and JHL, does not lead to persistent infection of the brain. Hind limb paralysis and demyelinisation occurring during EMCV infection have been proven to be partly due to infiltration of macrophages and lymphocytesCitation131,Citation132: treatment of mice with antibodies raised against macrophages, CD4+ and CD8+ T lymphocytes was sufficient to limit the development of the paralytic syndrome and reduced lesions in spinal cords, indicating that these cell types may participate in CNS injury.Citation133,Citation134 In addition, a study demonstrated that after intracranial inoculation of EMCV, oxidative damage to neurons was due to the induction of NADPH oxidase in microglia (brain macrophages).Citation135 Thus it seems that inflammatory and immune responses are deleterious for the host CNS tissues.
Diabetes
Infection of genetically susceptible mouse strains with a high dose of EMCV (D) leads to diabetes within 3 to 4 d. This diabetes seems to be mainly due to acute destruction of pancreatic β cells by viral replication without involvement of the immune system. By contrast, during infection with a low dose of EMCV, it was found that the immune system, especially macrophages, plays a central role in the destruction of pancreatic β cells.Citation136 Inactivation of macrophages prior to viral infection completely prevents EMCV-induced diabetes.Citation137 It appears that upon low-dose infection of mice the virus initially replicates in pancreatic β cells, leading to the recruitment of macrophages that are activated at the pancreatic islets. These activated macrophages produce soluble mediators like interleukin-1 β (IL-1 β) and produce TNFα and nitric oxide (NO), which induce apoptosis in β cells (for review see ref. Citation120).
EMCV Virulence
As described in the previous section, EMCV is known to induce myocarditis, diabetes and reproductive and nervous disorders. It is believed that symptoms and disease are viral strain-specific. Animal species, sex and age are also known to be important factors for EMCV virulence. Few molecular determinants of EMCV virulence have been defined so far. This chapter will describe them and highlight what should be considered to be potential factors in EMCV virulence.
Adsorption and entry
The first primordial steps that can modulate EMCV virulence is the ability of the virus to adsorb to the cell and gain entry. The viral capsid proteins, in their capacity to interact with cellular receptors, are crucial for this entry step and may be considered to be factors that can modulate EMCV virulence. Indeed, mutation of a single amino acid (alanine 776) of the EMCV polyprotein (in the VP1 capsid protein) of a diabetogenic EMCV renders this virus non-diabetogenic. This mutation modifies the native protein conformation of VP1, and thus inhibits virus attachment to the pancreatic β-cell receptor. Indeed, the Ala776 plays a crucial role in promoting viral infection and destruction of β cells, thereby inducing development of diabetes. As regards diabetes induced by EMCV, the genetic background of the mouse is also an important factor.Citation138 An autosomal recessive gene has been described to modulate the level of expression of the viral receptor at the surface of the pancreatic β cells, and seems to be responsible for mouse susceptibility.Citation139 In this regard, it has been established that SJL/J mice are susceptible to EMCV-induced diabetes while C57BL/6 mice are not. This discrepancy has most commonly been attributed to a lower level of expression of the viral receptor at the surface of β cells of C57BL/6 mice.Citation138 Indeed, only certain strains of mice such as SJL/J, SWR/J, DBA/2J and NIH/Swiss develop diabetes, while strains such as C57BL/6J, AKR/J, CBA/J, LP/J and CE/J do not.Citation138
Zhu and colleagues recently observed that mutation of the threonine at position 100 of the VP1 protein decreases the neurovirulence of EMCV.Citation140 This is the first report indicating that a mutation in the viral capsid protein could attenuate its neurovirulence. Substitution of the Thr100 with an isoleucine or a proline reduced the ability of EMCV to grow in the brain of infected mice. The mutants induced only moderate inflammatory responses and less brain damage, but still caused mortality when inoculated at high doses.
The VP1 Thr100 mutation is not as deleterious for EMCV neurovirulence as the mutation in the Ala776, which is determinant for EMCV infection of pancreatic β-cells and gain or loss of diabetogenicity. The role played by the VP1 Thr100 amino acid is unclear and other factors may determine EMCV neurovirulence.
Thus, the VP1 protein of EMCV influences virulent properties, because it is structure by which the virus attaches to its cell receptor and is essential for virus adsorption and entry. However, it should be noted that mutations within viral capsids may also be deleterious for viral assembly as well as release. Indeed, delay in viral release could probably be sufficient to attenuate virulence.Citation78
The length of the EMCV poly(C) tract
The length of the EMCV poly(C) tract has been suspected to be important for Cardiovirus virulence. It has been clearly demonstrated that diminution of the length of the Mengovirus poly(C) strikingly attenuated its virulence, rendering it non-pathogenic in mice.Citation13,Citation141 Of note, a Mengovirus harboring a short poly(C) tract has been investigated as a potentially safe vector for live recombinant vaccines.Citation141 Studies of different Mengo and EMCV viruses have indicated that virulence of these viruses in mice seems to be positively correlated with their poly(C) tract length, even if short-poly(C) EMCV strains were attenuated only slightly if at all compared with Mengovirus.Citation142 Thus, a short poly(C) is less deleterious for EMCV strains.Citation142 Indeed, virulent EMCV strains with very short poly(C) tracts have been described. LaRue et al.Citation117 isolated an EMCV strain exhibiting a short poly(C) tract (7 to 10 nt) that was pathogenic in mice, pigs and cynomolgus macaques. The fact that a short poly(C) tract is less deleterious for EMCV strains than for the Mengovirus strain indicates that the viral genomic context might influence the degree to which virulence is dependent upon the length of the poly(C) tract.
Viral strain sensitivity to the poly(C) tract length is suspected to be due to a defect in viral replication in certain cell types.Citation143 How exactly the length of the poly(C) tract may interfere with viral replication remains to be clarified.
2B*
Very recently, Loughran et al.,Citation17 demonstrated the existence of a programmed ribosomal frameshift that leads to the translation of a cryptic protein of 129 amino acids, called 2B* (represented in ), which had been previously unknown. These findings indicate that the EMCV genome does not have a single ORF but two and encodes not 12 but rather 13 mature proteins. The 12 N-terminal amino acids of the 2B* protein are identical to those of 2B, and then the +2 frame shift occurs at a conserved GGUUUUU motif. By introduction of synonymous mutations within the conserved motif, the authors showed that inhibition of ribosomal frame shifting and/or absence of 2B* led to a virus with a reduced plaque size phenotype. Importantly, they also observed that efficient frame shifting was only observed in virus-infected cells, indicating that it might be a process “programmed” by the virus. They did not, however, address whether the wt phenotype is restored if ribosomal frameshifting is prevented but expression of the 2B* maintained. It would be of interest to determine whether synthesis of the 2B* protein is important or whether it is the ribosomal frameshifting itself. This process could be important in regulating virus gene expression and may play some sort of regulatory role in the viral lifecycle. It would be interesting to determine whether 2B* plays a critical role in EMCV virulence.
The L protein
The L protein has been termed a “viral security protein” that is nonessential for virus replication but has evolved to fulfil functions that are conserved among picornavirus. The main function of viral security proteins is not to increase virus virulence but rather to counteract host defenses to promote spread within the infected host.Citation144
Among picornaviruses that possess an L protein, the L presents considerable diversity as regards both sequence and function (for review, see ref. Citation144). For instance, the aphtovirus L protein has catalytic activity,Citation145 while that of cardiovirus does not. Even among cardioviruses there are noticeable differences. The L protein is one of the proteins that displays the greatest divergence between TMEV and EMCV (35 % amino acid identity).Citation146 The EMCV L protein is 67 amino acids long and contains an atypical zinc finger and an acidic domain. It can be phosphorylated on residues Thr47Citation147 and Tyr41,Citation148 two residues that are not conserved in the TMEV L protein. Moreover, EMCV L lacks the Ser/Thr-rich C-terminal domain present in the TMEV L.
Even if EMCV L protein is devoid of any known enzymatic activity, in contrast to that of aphtovirus, it appears to suppress interferon productionCitation149 and impair nucleo-cytoplasmic trafficking through the binding of Ran-GTPase (Ras-related nuclear -GTPase required for the RNA and protein translocation through the nuclear pores) and through phosphorylation and inactivation of nucleoporins.Citation150-Citation154
Substitution of the L protein of TMEV by that of Mengovirus showed that even if these two L proteins display very little similarity (less than 40% identity), they were both able to inhibit transcription of type I interferon, cytokine and chemokine genes, to interfere with nucleocytoplasmic trafficking of host-cell proteinsCitation146 and to inhibit infection-induced stress granule assembly.Citation155 Stress granules are cytosolic aggregates of stalled translation preinitiation complexes that form in cells exposed to various environmental stresses. They are thought to play a role in translation inhibition during stress, through sequestration of cellular mRNA. When cells recover from stress, stress granules disperse and mRNA can be released for effective translation or targeted to processing bodies (P-bodies). These studies indicate that the L protein counteracts host responses through different pathways, a function that appears to be conserved among different cardiovirus strains and species. Both TMEV and PV have been recently described to inhibit stress granule assembly. However, it is yet to be determined whether EMCV infection induces stress granule formation that is then inhibited by the EMCV L protein. Whether the L protein of EMCV assumes all the functions described when expressed by a TMEV recombinant virus is uncertain. Kinetic analyses of the activity of the EMCV L protein within the context of TMEV recombinant viruses indicate that the L protein of EMCV acts faster than that of TMEV, but that this was deleterious for the recombinant. It should be noted that EMCV replicates faster than TMEV.
This raised the hypothesis that L proteins of TMEV and EMCV diverged during evolution to adapt to the different levels of replication fitness displayed by these viruses.Citation146
TMEV strains are divided in two subgroups, GVDII is highly neurovirulent, inducing acute fatal polioencephalomyelitis in mice. The TO subgroup in contrast are strains that induce non-fatal polioencephalomyelitis followed by chronic inflammatory demyelination with virus persistence in the spinal cord (for review, see refs. Citation3 and Citation4). It has been shown that strains from the TO subgroup of TMEV possess an out of frame L* which inhibits apoptosis, while the TMEV L present in all subgroup induces it.Citation156 The L* is believed to play an important role for the viral persistence. In contrast, EMCV that is not known as a persistent virus, has only one L protein, as described above, that has been shown to be required to inhibit apoptosis.
Indeed it has been demonstrated that mutation in the L protein of Mengovirus abrogates its ability to inhibit apoptosis during viral infection.Citation77 (The inhibition of apoptosis during EMCV infection is further discussed below.)
The EMCV L protein is one of the best characterized proteins of EMCV. It is a multifunctional protein that interferes with many host cell functions. Such interference is likely to be deleterious for the infected cell, and probably for the host, as well. In vivo studies to define whether mutation in the L protein of EMCV would lead to attenuation of virulence are lacking at present. Indeed, an in vivo study would be required to determine unequivocally whether the L protein should be considered to be a virulence factor of EMCV.
The 2A protein, represented in , is considered to be a second viral security protein of EMCV.Citation144 It is a small protein of 17kDa, 143 amino acids in length and highly basic, and which, unlike the 2A protein of enterovirus, is not a protease. However, during elongation, prior to the synthesis of the 3C protease, the 2A and 2B proteins are not linked. This interruption in the polyprotein sequence is the consequence of ribosome skipping at the NPG(P) peptide sequence.Citation157 This interruption, also called “StopGo” translation, is thought to be caused by an interaction between the 2A peptide and the exit tunnel of the ribosome, preventing the interaction of the ribosome with Pro-tRNA (proline is the first amino acid of the 2B protein).
Upon infection with most picornaviruses, host protein synthesis is shut off.Citation30 This function has been attributed to the 2A protein of EMCV, since partial deletion of this protein result in maintenance of cellular mRNA translation.Citation55 The shut-off of host protein synthesis that occurs during EMCV infection is not as rapid or extensive as during PV infection.Citation33 Indeed, in the case of EMCV, it is thought to stem from competition between cap- and IRES-dependent translation, rather than to be a definitive shut-off.Citation16,Citation158 Several non-exclusive mechanisms involving the 2A protein have been described.
The inhibition of cap-dependent translation could be due to the activation of a translational inhibitor. It has been shown that dephosphorylation of 4E-BP1 triggers the sequestration of eIF4E and inhibits cap-dependent translation. EMCV 2A protein expression in BHK-21 cells triggers 4E-BP1 hypophosphorylation,Citation55 and thus could contribute to the inhibition of cap-dependent translation. This mechanism, however, has not been confirmed in HeLa and L cell types,Citation159 where protein synthesis shut-off has also been observed.
Another possibility is that this shut-off is achieved by the interaction of the 2A protein with the translation initiation factor eIF4E. An eIF4E binding site has been located between the amino acids 126–134 of the 2A protein.Citation159 This interaction might impede eIF4E binding to eIF4G (interactions eIF4E-, eIF4G- and eIF4A are required for initiation of cap-dependent translation). However, alteration of the 2A-eIF4E interaction by introduction of point mutations in the 2A protein is not able to restore normal protein synthesis levels.Citation159
Early after EMCV infection, the 2A protein is localized in the nucleoli, owing to its nuclear localization signal (NLS),Citation159 and associates with the nascent ribosomal subunit.Citation160,Citation161 Mutations introduced in the NLS have been shown to decrease inhibition of cap-dependent translation. In the cytosol, the 2A protein interacts with the 40S ribosomal subunit.Citation158,Citation162 This association is believed to induce preferential use of IRES-dependent templates (rather than capped mRNA). Of note, it has been reported that the 2A protein of EMCV binds RNAs but the functional significance of this interaction remain to be established.
Recently, we have shown that BHK-21 cells infected with EMCV bearing a large deletion in the 2A protein undergo apoptosis through caspase 3 activation, and that induction of apoptosis is independent of the ability of 2A to shut off cap-dependent translation.Citation78 Thus the 2A protein of EMCV is important in inhibiting apoptosis.
In addition, deletion in the 2A protein profoundly affects EMCV virulence in vitro and in vivo.Citation55,Citation78 Our study also showed that, despite a large deletion within the 2A protein of EMCV, this virus was still able to infect and replicate in mice. Nevertheless, the virus no longer induced any clinical symptoms, while the wild type virus led to death in all mice within a week. These results were obtained for two different EMCV strains, one that originally induced myocarditis in pigs and another that caused abortion. Thus the EMCV 2A protein is required for viral pathogenesis in mice.
In conclusion, the EMCV 2A protein is important for the virus in counteracting the host defense, as Δ2A viruses do not inhibit cap-dependent translation,Citation55,Citation159 are unable to inhibit apoptosis in vitro and are no longer pathogenic in vivo.Citation78 The 2A protein thus well deserves to be considered a virulence factor or a “viral security protein.”Citation144
Inhibition of apoptosis
The EMCV has mainly been described as a lytic virus that undergoes “necrotic” cell death. Interestingly, observation of viruses bearing modified 2A and L protein (EMCVCitation78 and Mengo,Citation77 respectively) has shown that the infection was responsible for apoptotic cell death. These studies suggest that EMCV has evolved to be able to counteract cell host defense through the inhibition of programmed cell death. The exact mechanisms by which the L and 2A protein may interfere with apoptosis remain to be characterized.
Apoptosis is considered to be a common cellular defense mechanism in many viral infections. This process can prevent generation and spread of viral progeny.
Of note, apoptotic cells have been detected during wild-type EMCV infection in vivo.Citation121,Citation163,Citation164 However, it remains unclear whether apoptosis is a direct or indirect effect of viral infection. As described by Buenz et al.Citation165 for TMEV-infected mice, the possibility that the majority of cells undergoing apoptosis are uninfected cannot be ruled out. Yamada et al.Citation164 reported that the main source of apoptotic cells in the heart of EMCV-infected mice appeared to be the infiltrating mononuclear cells and not the cardiac myocytes.
However, more experiments and quantification will be required to decipher whether induction of apoptosis during EMCV infection in vivo results from the virus itself or through the host response to viral infection. In favor to the hypothesis that apoptosis is inhibited during EMCV infection in vivo, it has been shown that inhibition of apoptosis by NFκB is required for EMCV virulence in mice.Citation166
Indeed, NFκB, a transcription factor known to induce inflammatory responses and to inhibit apoptosis, is activated during EMCV infection.Citation167 Matsumory et al.Citation128 have shown that NFκB activation, by induction of the inflammatory response, is important for EMCV virulence. Thus, inhibition of apoptosis as well as the induction of an inflammatory response might be important factors for EMCV virulence.
Inflammatory response
The inflammatory response induced upon infection plays a critical role in EMCV pathogenesis. As described in the previous chapter, several studies indicate that EMCV neurovirulence might be partly due to CD4+ and CD8+ T lymphocytes and macrophages.Citation133-Citation135 Thus, inflammatory and immune responses actually seem to exacerbate EMCV neurovirulence. This may not be peculiar to development of neurovirulence, as it has also been described for diabetes and myocarditis. For diabetes, inoculation of high doses of EMCV induces β cell destruction through direct infection and replication of the virus in these cells. However, using lower doses and despite little EMCV-induced β cell destruction, most damage is actually due to the action of soluble IL-1β and TNFα secretion as well as the production of iNOS from activated macrophages. Indeed, inactivation of macrophages prior to viral infection almost completely prevents EMC virus-induced diabetes.Citation137
In the early stages of infection, inflammatory responses are also detected in the heart, where high levels of circulating proinflammatory cytokines have been measured.Citation126,Citation128,Citation168 In the heart of EMCV-infected mice expression of cytokine genes increases, and their degree of expression correlates with the severity of disease.Citation168 Thus, proinflammatory cytokines may serve to accelerate the pathology of myocarditis, and may have a negative influence on cardiac function.
In recent years, it has been shown that mast cells also play a critical role in the pathogenesis of EMCV induced myocarditis.Citation124,Citation125,Citation128 Mast cells are powerful producers of multiple cytokines and chemical mediators. Mast cells are considered to be part of the immune system; they present numerous similarities with basophils, but are located in tissues, including the heart. Upon infection of mast cell-deficient mice with EMCV, along with a decrease in production of intracardiac IL-1β, IL-6, TNFα and NO, mice present attenuated myocardial necrosis, diminished cellular infiltration and improved survival rate.Citation124-Citation128 Although EMCV virulence is attenuated when immune and inflammatory responses are downregulated; no significant effect on viral replication has been noted.Citation126,Citation128
Indeed, NFκB is activated during EMCV infections and leads to the expression of many cytokines. Moreover, inhibition of NFκB upon EMCV infection in mice lowered the mortality of the animals, attenuated necrosis and cellular infiltration, and decreased the production of IL-1β, IL-6, TNFα and NO in mouse heart.Citation126,Citation128 In agreement with these observations, two previous studies indicated that mice lacking the p50 subunit of NFκB more readily survived EMCV infection.Citation166,Citation169 In addition, it has been reported that the circulating TNFα level is increased in mice infected with EMCV, and that pre-treatment with an anti-TNFα antibody attenuated myocardial injury and decreased mortality in the acute stage.Citation170 Another group, however, using TNFα knockout mice, reported that TNFα plays a protective role in acute viral myocarditis in mice.Citation129 These conflicting results might arise from the fact that indeed, an efficient immune system is primordial for elimination of the virus, but an excessive cytokine storm might be deleterious for the host, as is well-documented in sepsis, for instance.
Altogether, the literature suggests that a strong inflammatory response is of benefit to EMCV virulence. Therefore, one could assume that blocking intracellular protein trafficking would not be advantageous for the virus, since it could decrease cytokine secretion. Indeed, 2B and 3A proteins of PV block ER-to-Golgi trafficking, although the 2B and 3A proteins of EMCV do not.Citation44,Citation47,Citation48 It has not as yet been determined whether EMCV 2BC, like FMDV 2BC, possesses this ability.Citation50 It is tempting to suggest that EMCV does not inhibit cell protein trafficking because inflammatory and immune responses are advantageous to its virulence.
The RNA-sensing receptors MDA-5 and RIG-I can be specifically activated by distinct viruses and induce an antiviral response. EMCV exclusively activates MDA-5. Both the EMCV 3Cpro and caspases are supposed to be responsible for the degradation of RIG-I upon EMCV infection.Citation39 Of note, RIG-I is also cleaved in Poly(I:C) treated cells.Citation39
EMCV is able to inhibit regulatory T cell (Treg) function in an antigen presenting cell (APC)-independent manner, but requires that both Treg and effector T cells are simultaneously stimulated through MDA-5.Citation171 The major function of Treg is to downregulate the immune response, mainly through the secretion of IL-10, a known anti-inflammatory cytokine. Indeed, it has also been reported that IL-10 expression in EMCV infected mice suppresses the induction of IL-1β and iNOS genes, attenuates myocardial injury and prolongs the survival of mice.Citation172
In addition, it has recently been described that the EMCV attachment to the CCR5 receptor suffices to induce production of inflammatory cytokines in macrophages within 30 min. In this study, they made the intriguing observation that the virus does not need to enter the macrophage to promote inflammation.Citation173 However, they showed that EMCV-infected CCR5-deficient mice produce less iNOS and COX-2, more TNFα and have a 7-fold higher viral RNA load. They propose that the CCR5-induced inflammatory response favors the control of virus spread. No information, however, was provided regarding mouse survival and disease evolution.
All together these data suggest that proinflammatory cytokine production and inflammatory and immune responses are important factors in EMCV virulence in mice. There might be a delicate balance between the beneficial antiviral effect of cytokines and the deleterious effects associated with the cytokine-induced inflammatory responses, which may determine disease development following EMCV infection.
As previously discussed, it has been shown that EMCV induces lytic cell death and inhibits apoptosis, at least in vitro. Yet, it is known that in vivo, lytic cell death gives rise to a greater inflammatory response than apoptosis.Citation174 The inflammatory response seems to exacerbate EMCV virulence instead of shut it down (discussed above). In addition, inhibition of apoptosis by NFκB has been shown to be required for pathogenesis during EMCV infection in mice.Citation128,Citation166 It has also been reported that a virus deleted in 2A, which was no longer able to inhibit apoptosis in vitro, was no longer pathogenic for mice in vivo.Citation78 Thus, both inhibition of apoptosis and induction of the inflammatory response might be important factors for EMCV virulence. In this study, however, it cannot be ruled out that delay in virus release may be important for virus attenuation. This delay could actually correlate with apoptotic cell death. Indeed, necrotic cell death leads to a massive release of virus that is able to spread and invade the host CNS. In contrast, during apoptosis the cell membranes are not permeabilized, which may constrain virus to confined apoptotic bodies. Apoptotic cells are known to attract phagocytic cells. Thus virus might be retained in these cells and be phagocytosed, without induction of a strong inflammatory response. Thus there might be a link between the defect in virus release, the non-inhibition of apoptosis and the fact that the deleted 2A virus was not able to spread throughout the CNS.
Conclusion
The EMCV is a rapidly lytic virus that does not persist in hosts in most cases. It induces sudden death, myocarditis, encephalitis, nervous disorders and diabetes. Its pathogenicity and the diseases induced depend on viral and host factors. The L and 2A proteins are the two major known virulence factors of EMCV, but understanding of some of their functions at the molecular or host levels is limited. However, EMCV pathogenesis seems to correlate with a strong and uncontrolled inflammatory response that may contribute to EMCV virulence in vivo. Many studies have described an improvement in mouse survival when different components of the immune response are selectively down-modulated. Nevertheless, in other studies the inflammatory response was shown to be protective. This apparent contradiction might reflect a delicate balance between the beneficial effect of the antiviral response and the deleterious effect of a strong inflammatory response. In this balance, inhibition of apoptosis could be one of the major viral activities to tip the scale toward the deleterious side. Indeed, studies using 2A deleted virus suggest a link between viral release, anti-apoptotic activity, inflammatory response and the pathogenicity of EMCV. Further investigations are needed to elucidate how EMCV may manipulate cell death processes to improve viral dissemination, knowledge that could make a major contribution to the understanding of the biology of EMCV.
Although EMCV rarely induces severe clinical symptoms in humans, its worldwide distribution and its wide host range, as well as the biological characteristics of RNA viruses and the use of heart pigs for xenografts, all call for on-going vigilance and improvement of our knowledge of this virus.
| Abbreviations: | ||
| EMCV | = | encephalomyocarditis virus |
| FMDV | = | foot and mouth disease virus |
| TMEV | = | Theiler’s murine encephalitis virus |
| SAFV | = | Saffold virus |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| Cre | = | cis-acting replication element |
| IL-1β | = | interleukin-1β |
| TNFα | = | tumor necrosis factor α |
| NO | = | nitric oxide |
| iNOS | = | inducible nitric oxide synthase |
| RIG-I | = | retinoic acid-inducible gene 1 protein |
| MDA-5 | = | melanoma differentiation-associated gene-5 |
Acknowledgments
The author thanks Raphaël Gaudin for his precious help and contribution, and Jennifer Richardson for editing the English version of the manuscript.
References
- Helwig FC, Schmidt CH. A filter-passing agent producing interstitial myocarditis in anthropoid apes and small animals. Science 1945; 102:31 - 3; http://dx.doi.org/10.1126/science.102.2637.31; PMID: 17787415
- Dick GWA, Smithburn KC, Haddow AJ. Mengo Encephalomyelitis Virus. Isolation and Immunological Properties. Br J Exp Pathol 1948; 29:547
- Brahic M, Bureau JF, Michiels T. The genetics of the persistent infection and demyelinating disease caused by Theiler’s virus. Annu Rev Microbiol 2005; 59:279 - 98; http://dx.doi.org/10.1146/annurev.micro.59.030804.121242; PMID: 16153171
- Roos RP. Pathogenesis of Theiler’s murine encephalomyelitis virus‐induced disease. Clinical and Experimental Neuroimmunology 2010; 1:70 - 78; http://dx.doi.org/10.1111/j.1759-1961.2010.00008.x
- Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, et al. Picornaviridae. In: Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Ed: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. pp 855-880. San Diego: Elsevier. 2011.
- Himeda T, Ohara Y. Saffold virus, a novel human Cardiovirus with unknown pathogenicity. J Virol 2012; 86:1292 - 6; http://dx.doi.org/10.1128/JVI.06087-11; PMID: 22114344
- Drexler JF, Luna LK de S, Stöcker A, Almeida PS, Ribeiro TC, Petersen N, et al. Circulation of 3 lineages of a novel Saffold cardiovirus in humans. Emerg Infect Dis 2008; 14:1398 - 405; http://dx.doi.org/10.3201/eid1409.080570; PMID: 18760006
- Pevear DC, Calenoff M, Rozhon E, Lipton HL. Analysis of the complete nucleotide sequence of the picornavirus Theiler’s murine encephalomyelitis virus indicates that it is closely related to cardioviruses. J Virol 1987; 61:1507 - 16; PMID: 3033278
- Michiels T, Dejong V, Rodrigus R, Shaw-Jackson C. Protein 2A is not required for Theiler’s virus replication. J Virol 1997; 71:9549 - 56; PMID: 9371618
- Luo M, Vriend G, Kamer G, Minor I, Arnold E, Rossmann MG, et al. The atomic structure of Mengo virus at 3.0 A resolution. Science 1987; 235:182 - 91; http://dx.doi.org/10.1126/science.3026048; PMID: 3026048
- Palmenberg AC, Kirby EM, Janda MR, Drake NL, Duke GM, Potratz KF, et al. The nucleotide and deduced amino acid sequences of the encephalomyocarditis viral polyprotein coding region. Nucleic Acids Res 1984; 12:2969 - 85; http://dx.doi.org/10.1093/nar/12.6.2969; PMID: 6324136
- Hruby DE, Roberts WK. Encephalomyocarditis virus RNA. III. Presence of a genome-associated protein. J Virol 1978; 25:413 - 5; PMID: 202751
- Duke GM, Osorio JE, Palmenberg AC. Attenuation of Mengo virus through genetic engineering of the 5′ noncoding poly(C) tract. Nature 1990; 343:474 - 6; http://dx.doi.org/10.1038/343474a0; PMID: 2153940
- Pan M, Yang X, Zhou L, Ge X, Guo X, Liu J, et al. Duck Hepatitis A virus possesses a distinct type IV internal ribosome entry site element of picornavirus. J Virol 2012; 86:1129 - 44; http://dx.doi.org/10.1128/JVI.00306-11; PMID: 22090106
- Belsham GJ. Divergent picornavirus IRES elements. Virus Res 2009; 139:183 - 92; http://dx.doi.org/10.1016/j.virusres.2008.07.001; PMID: 18675861
- Duke GM, Hoffman MA, Palmenberg AC. Sequence and structural elements that contribute to efficient encephalomyocarditis virus RNA translation. J Virol 1992; 66:1602 - 9; PMID: 1310768
- Loughran G, Firth AE, Atkins JF. Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proceedings of the National Academy of Sciences. 2011.
- Lobert PE, Escriou N, Ruelle J, Michiels T. A coding RNA sequence acts as a replication signal in cardioviruses. Proc Natl Acad Sci U S A 1999; 96:11560 - 5; http://dx.doi.org/10.1073/pnas.96.20.11560; PMID: 10500216
- Krishnaswamy S, Rossmann MG. Structural refinement and analysis of Mengo virus. J Mol Biol 1990; 211:803 - 44; http://dx.doi.org/10.1016/0022-2836(90)90077-Y; PMID: 2156078
- Rossmann MG, Palmenberg AC. Conservation of the putative receptor attachment site in picornaviruses. Virology 1988; 164:373 - 82; http://dx.doi.org/10.1016/0042-6822(88)90550-8; PMID: 2835857
- Scraba DG. Functional aspects of the capsid structure of Mengo virus. J Struct Biol 1990; 104:52 - 62; http://dx.doi.org/10.1016/1047-8477(90)90057-J; PMID: 1965133
- Huber SA. VCAM-1 is a receptor for encephalomyocarditis virus on murine vascular endothelial cells. J Virol 1994; 68:3453 - 8; PMID: 7514674
- Burness AT, Pardoe IU. A sialoglycopeptide from human erythrocytes with receptor-like properties for encephalomyocarditis and influenza viruses. J Gen Virol 1983; 64:1137 - 48; http://dx.doi.org/10.1099/0022-1317-64-5-1137; PMID: 6302211
- Jin YM, Pardoe IU, Burness AT, Michalak TI. Identification and characterization of the cell surface 70-kilodalton sialoglycoprotein(s) as a candidate receptor for encephalomyocarditis virus on human nucleated cells. J Virol 1994; 68:7308 - 19; PMID: 7933115
- Guy M, Chilmonczyk S, Crucière C, Eloit M, Bakkali-Kassimi L. Efficient infection of buffalo rat liver-resistant cells by encephalomyocarditis virus requires binding to cell surface sialic acids. J Gen Virol 2009; 90:187 - 96; http://dx.doi.org/10.1099/vir.0.004655-0; PMID: 19088288
- Hammoumi S, Guy M, Eloit M, Bakkali-Kassimi L. Encephalomyocarditis virus may use different pathways to initiate infection of primary human cardiomyocytes. Arch Virol 2012; 157:43 - 52; http://dx.doi.org/10.1007/s00705-011-1133-6; PMID: 21989795
- Madshus IH, Olsnes S, Sandvig K. Different pH requirements for entry of the two picornaviruses, human rhinovirus 2 and murine encephalomyocarditis virus. Virology 1984; 139:346 - 57; http://dx.doi.org/10.1016/0042-6822(84)90380-5; PMID: 6097029
- Hogle JM. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 2002; 56:677 - 702; http://dx.doi.org/10.1146/annurev.micro.56.012302.160757; PMID: 12142481
- Ambros V, Baltimore D. Purification and properties of a HeLa cell enzyme able to remove the 5′-terminal protein from poliovirus RNA. J Biol Chem 1980; 255:6739 - 44; PMID: 6248532
- Bedard KM, Semler BL. Regulation of picornavirus gene expression. Microbes Infect 2004; 6:702 - 13; http://dx.doi.org/10.1016/j.micinf.2004.03.001; PMID: 15158778
- Kaminski A, Hunt SL, Patton JG, Jackson RJ. Direct evidence that polypyrimidine tract binding protein (PTB) is essential for internal initiation of translation of encephalomyocarditis virus RNA. RNA 1995; 1:924 - 38; PMID: 8548657
- Balvay L, Soto Rifo R, Ricci EP, Decimo D, Ohlmann T. Structural and functional diversity of viral IRESes. Biochim Biophys Acta 2009; 1789:542 - 57; PMID: 19632368
- Jen G, Detjen BM, Thach RE. Shutoff of HeLa cell protein synthesis by encephalomyocarditis virus and poliovirus: a comparative study. J Virol 1980; 35:150 - 6; PMID: 6251263
- Blom N, Hansen J, Blaas D, Brunak S. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci 1996; 5:2203 - 16; http://dx.doi.org/10.1002/pro.5560051107; PMID: 8931139
- Parks GD, Baker JC, Palmenberg AC. Proteolytic cleavage of encephalomyocarditis virus capsid region substrates by precursors to the 3C enzyme. J Virol 1989; 63:1054 - 8; PMID: 2536819
- Lawson TG, Gronros DL, Werner JA, Wey AC, DiGeorge AM, Lockhart JL, et al. The encephalomyocarditis virus 3C protease is a substrate for the ubiquitin-mediated proteolytic system. J Biol Chem 1994; 269:28429 - 35; PMID: 7961784
- Lawson TG, Gronros DL, Evans PE, Bastien MC, Michalewich KM, Clark JK, et al. Identification and characterization of a protein destruction signal in the encephalomyocarditis virus 3C protease. J Biol Chem 1999; 274:9871 - 80; http://dx.doi.org/10.1074/jbc.274.14.9871; PMID: 10092679
- Schlax PE, Zhang J, Lewis E, Planchart A, Lawson TG. Degradation of the encephalomyocarditis virus and hepatitis A virus 3C proteases by the ubiquitin/26S proteasome system in vivo. Virology 2007; 360:350 - 63; http://dx.doi.org/10.1016/j.virol.2006.10.043; PMID: 17150238
- Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, et al. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 2009; 393:311 - 8; http://dx.doi.org/10.1016/j.virol.2009.08.009; PMID: 19733381
- Schlegel A, Giddings TH Jr., Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol 1996; 70:6576 - 88; PMID: 8794292
- Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 1994; 202:129 - 45; http://dx.doi.org/10.1006/viro.1994.1329; PMID: 8009827
- Agirre A, Barco A, Carrasco L, Nieva JL. Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J Biol Chem 2002; 277:40434 - 41; http://dx.doi.org/10.1074/jbc.M205393200; PMID: 12183456
- de Jong AS, Schrama IWJ, Willems PHGM, Galama JM, Melchers WJ, van Kuppeveld FJ. Multimerization reactions of coxsackievirus proteins 2B, 2C and 2BC: a mammalian two-hybrid analysis. J Gen Virol 2002; 83:783 - 93; PMID: 11907327
- de Jong AS, de Mattia F, Van Dommelen MM, Lanke K, Melchers WJ, Willems PH, et al. Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J Virol 2008; 82:3782 - 90; http://dx.doi.org/10.1128/JVI.02076-07; PMID: 18216106
- de Jong AS, Wessels E, Dijkman HBPM, Galama JM, Melchers WJ, Willems PH, et al. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J Biol Chem 2003; 278:1012 - 21; http://dx.doi.org/10.1074/jbc.M207745200; PMID: 12244057
- Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J 1995; 14:894 - 907; PMID: 7889939
- Choe SS, Dodd DA, Kirkegaard K. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology 2005; 337:18 - 29; http://dx.doi.org/10.1016/j.virol.2005.03.036; PMID: 15914217
- Wessels E, Duijsings D, Lanke KHW, van Dooren SH, Jackson CL, Melchers WJ, et al. Effects of picornavirus 3A Proteins on Protein Transport and GBF1-dependent COP-I recruitment. J Virol 2006; 80:11852 - 60; http://dx.doi.org/10.1128/JVI.01225-06; PMID: 17005635
- Moffat K, Howell G, Knox C, Belsham GJ, Monaghan P, Ryan MD, et al. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J Virol 2005; 79:4382 - 95; http://dx.doi.org/10.1128/JVI.79.7.4382-4395.2005; PMID: 15767438
- Moffat K, Knox C, Howell G, Clark SJ, Yang H, Belsham GJ, et al. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J Virol 2007; 81:1129 - 39; http://dx.doi.org/10.1128/JVI.00393-06; PMID: 17121791
- Murray L, Luke GA, Ryan MD, Wileman T, Knox C. Amino acid substitutions within the 2C coding sequence of Theiler’s Murine Encephalomyelitis virus alter virus growth and affect protein distribution. Virus Res 2009; 144:74 - 82; http://dx.doi.org/10.1016/j.virusres.2009.04.001; PMID: 19728403
- Zhang Y, Li Z, Xinna G, Xin G, Yang H. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 2011; 7:613 - 28; http://dx.doi.org/10.4161/auto.7.6.15267; PMID: 21460631
- O’Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011; 410:142 - 50; http://dx.doi.org/10.1016/j.virol.2010.10.042; PMID: 21112602
- Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 2009; 335:323 - 33; http://dx.doi.org/10.1007/978-3-642-00302-8_16; PMID: 19802573
- Svitkin YV, Hahn H, Gingras AC, Palmenberg AC, Sonenberg N. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. J Virol 1998; 72:5811 - 9; PMID: 9621041
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev 1998; 12:2293 - 304; http://dx.doi.org/10.1101/gad.12.15.2293; PMID: 9694795
- Daijogo S, Semler BL. Mechanistic intersections between picornavirus translation and RNA replication. Adv Virus Res 2011; 80:1 - 24; http://dx.doi.org/10.1016/B978-0-12-385987-7.00001-4; PMID: 21762819
- Herold J, Andino R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J Virol 2000; 74:6394 - 400; http://dx.doi.org/10.1128/JVI.74.14.6394-6400.2000; PMID: 10864650
- Gerber K, Wimmer E, Paul AV. Biochemical and genetic studies of the initiation of human rhinovirus 2 RNA replication: identification of a cis-replicating element in the coding sequence of 2A(pro). J Virol 2001; 75:10979 - 90; http://dx.doi.org/10.1128/JVI.75.22.10979-10990.2001; PMID: 11602738
- Tiley L, King AMQ, Belsham GJ. The foot-and-mouth disease virus cis-acting replication element (cre) can be complemented in trans within infected cells. J Virol 2003; 77:2243 - 6; http://dx.doi.org/10.1128/JVI.77.3.2243-2246.2003; PMID: 12525659
- Montagnier L, Sanders FK. Replicative form of Encephalomyocarditis virus ribonucleic acid. Nature 1963; 199:664 - 7; http://dx.doi.org/10.1038/199664a0; PMID: 14074552
- Paul A. Possible unifying mechanism of picornavirus genome replication. In: Semler BL, Wimmer E, Eds. Molecular biology of picornaviruses. ASM Press: Washington DC. 2002:227-246
- Murray KE, Barton DJ. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol 2003; 77:4739 - 50; http://dx.doi.org/10.1128/JVI.77.8.4739-4750.2003; PMID: 12663781
- Pfister T, Jones KW, Wimmer E. A cysteine-rich motif in poliovirus protein 2C(ATPase) is involved in RNA replication and binds zinc in vitro. J Virol 2000; 74:334 - 43; http://dx.doi.org/10.1128/JVI.74.1.334-343.2000; PMID: 10590122
- Detjen BM, Lucas J, Wimmer E. Poliovirus single-stranded RNA and double-stranded RNA: differential infectivity in enucleate cells. J Virol 1978; 27:582 - 6; PMID: 212582
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol 1991; 65:3384 - 7; PMID: 1851886
- Nugent CI, Johnson KL, Sarnow P, Kirkegaard K. Functional coupling between replication and packaging of poliovirus replicon RNA. J Virol 1999; 73:427 - 35; PMID: 9847348
- Jia XY, Van Eden M, Busch MG, Ehrenfeld E, Summers DF. trans-encapsidation of a poliovirus replicon by different picornavirus capsid proteins. J Virol 1998; 72:7972 - 7; PMID: 9733835
- McInerney GM, King AM, Ross-Smith N, Belsham GJ. Replication-competent foot-and-mouth disease virus RNAs lacking capsid coding sequences. J Gen Virol 2000; 81:1699 - 702; PMID: 10859374
- Porter DC, Ansardi DC, Morrow CD. Encapsidation of poliovirus replicons encoding the complete human immunodeficiency virus type 1 gag gene by using a complementation system which provides the P1 capsid protein in trans. J Virol 1995; 69:1548 - 55; PMID: 7853488
- Sasaki J, Nagashima S, Taniguchi K. Aichi virus leader protein is involved in viral RNA replication and encapsidation. J Virol 2003; 77:10799 - 807; http://dx.doi.org/10.1128/JVI.77.20.10799-10807.2003; PMID: 14512530
- Sasaki J, Taniguchi K. The 5′-end sequence of the genome of Aichi virus, a picornavirus, contains an element critical for viral RNA encapsidation. J Virol 2003; 77:3542 - 8; http://dx.doi.org/10.1128/JVI.77.6.3542-3548.2003; PMID: 12610129
- Liu Y, Wang C, Mueller S, Paul AV, Wimmer E, Jiang P. Direct interaction between two viral proteins, the nonstructural protein 2C and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog 2010; 6:e1001066; http://dx.doi.org/10.1371/journal.ppat.1001066; PMID: 20865167
- Curry S, Fry E, Blakemore W, Abu-Ghazaleh R, Jackson T, King A, et al. Dissecting the roles of VP0 cleavage and RNA packaging in picornavirus capsid stabilization: the structure of empty capsids of foot-and-mouth disease virus. J Virol 1997; 71:9743 - 52; PMID: 9371640
- Hindiyeh M, Li QH, Basavappa R, Hogle JM, Chow M. Poliovirus mutants at histidine 195 of VP2 do not cleave VP0 into VP2 and VP4. J Virol 1999; 73:9072 - 9; PMID: 10516013
- Knipe T, Rieder E, Baxt B, Ward G, Mason PW. Characterization of synthetic foot-and-mouth disease virus provirions separates acid-mediated disassembly from infectivity. J Virol 1997; 71:2851 - 6; PMID: 9060641
- Romanova LI, Lidsky PV, Kolesnikova MS, Fominykh KV, Gmyl AP, Sheval EV, et al. Antiapoptotic activity of the cardiovirus leader protein, a viral “security” protein. J Virol 2009; 83:7273 - 84; http://dx.doi.org/10.1128/JVI.00467-09; PMID: 19420082
- Carocci M, Cordonnier N, Huet H, Romey A, Relmy A, Gorna K, et al. Encephalomyocarditis virus 2A protein is required for viral pathogenesis and inhibition of apoptosis. J Virol 2011; 85:10741 - 54; http://dx.doi.org/10.1128/JVI.00394-11; PMID: 21849462
- Murnane TG, Craighead JE, Mondragon H, Shelokov A. Fatal disease of swine due to encephalomyocarditis virus. Science 1960; 131:498 - 9; http://dx.doi.org/10.1126/science.131.3399.498; PMID: 14425363
- Maurice H, Nielen M, Brocchi E, Nowotny N, Kassimi LB, Billinis C, et al. The occurrence of encephalomyocarditis virus (EMCV) in European pigs from 1990 to 2001. Epidemiol Infect 2005; 133:547 - 57; http://dx.doi.org/10.1017/S0950268804003668; PMID: 15962562
- Canelli E, Luppi A, Lavazza A, Lelli D, Sozzi E, Martin AM, et al. Encephalomyocarditis virus infection in an Italian zoo. Virol J 2010; 7:64; PMID: 20298561
- Dea S, Bilodeau R, Sauvageau R, Martineau GP. Outbreaks in Quebec pig farms of respiratory and reproductive problems associated with encephalomyocarditis virus. J Vet Diagn Invest 1991; 3:275 - 82; http://dx.doi.org/10.1177/104063879100300401; PMID: 1662074
- Czechowicz J, Huaman JL, Forshey BM, Morrison AC, Castillo R, Huaman A, et al. Prevalence and risk factors for encephalomyocarditis virus infection in Peru. Vector Borne Zoonotic Dis 2011; 11:367 - 74; http://dx.doi.org/10.1089/vbz.2010.0029; PMID: 21395427
- Oberste MS, Gotuzzo E, Blair P, Nix WA, Ksiazek TG, Comer JA, et al. Human febrile illness caused by encephalomyocarditis virus infection, Peru. Emerg Infect Dis 2009; 15:640 - 6; http://dx.doi.org/10.3201/eid1504.081428; PMID: 19331761
- Reddacliff LA, Kirkland PD, Hartley WJ, Reece RL. Encephalomyocarditis virus infections in an Australian zoo. J Zoo Wildl Med 1997; 28:153 - 7; PMID: 9279403
- An D-J, Jeong W, Jeoung H-Y, Yoon SH, Kim HJ, Choi CU, et al. Encephalomyocarditis in Korea: serological survey in pigs and phylogenetic analysis of two historical isolates. Vet Microbiol 2009; 137:37 - 44; http://dx.doi.org/10.1016/j.vetmic.2009.01.005; PMID: 19200668
- Ge X, Zhao D, Liu C, Wang F, Guo X, Yang H. Seroprevalence of encephalomyocarditis virus in intensive pig farms in China. Vet Rec 2010; 166:145 - 6; http://dx.doi.org/10.1136/vr.b4766; PMID: 20118474
- Billinis C. Encephalomyocarditis virus infection in wildlife species in Greece. J Wildl Dis 2009; 45:522 - 6; PMID: 19395765
- Gainer JH. Encephalomyocarditis virus infections in Florida, 1960-1966. J Am Vet Med Assoc 1967; 151:421 - 5; PMID: 4293832
- Psalla D, Psychas V, Spyrou V, Billinis C, Papaioannou N, Vlemmas I. Pathogenesis of experimental encephalomyocarditis: a histopathological, immunohistochemical and virological study in rats. J Comp Pathol 2006; 134:30 - 9; http://dx.doi.org/10.1016/j.jcpa.2005.06.008; PMID: 16330040
- Littlejohns IR, Acland HM. Encephalomyocarditis virus infection of pigs. 2. Experimental disease. Aust Vet J 1975; 51:416 - 22; http://dx.doi.org/10.1111/j.1751-0813.1975.tb15790.x; PMID: 173274
- Spyrou V, Maurice H, Billinis C, Papanastassopoulou M, Psalla D, Nielen M, et al. Transmission and pathogenicity of encephalomyocarditis virus (EMCV) among rats. Vet Res 2004; 35:113 - 22; http://dx.doi.org/10.1051/vetres:2003044; PMID: 15099508
- Kluivers M, Maurice H, Vyt P, Koenen F, Nielen M. Transmission of encephalomyocarditis virus in pigs estimated from field data in Belgium by means of R0. Vet Res 2006; 37:757 - 66; http://dx.doi.org/10.1051/vetres:2006035; PMID: 16973116
- Murnane T. Encephalomyocarditis virus, p 353-357 in HL Foster, JD Small and JG Fox (eds), The mouse in biomedical research, Vil II. Academic Press, New York, NY. 1982.
- Koenen F. Chapter 17: Encephalomyocarditis Virus. In: Straw BE, D’Allaire S, Zimmerman JJ, Taylor DJ, Eds. Disease of the Swine. 9th ed. Boston: Blackwell Science 2006.
- Tsen KT, Tsen S-WD, Fu Q, Lindsay SM, Li Z, Cope S, et al. Studies of inactivation of encephalomyocarditis virus, M13 bacteriophage, and Salmonella typhimurium by using a visible femtosecond laser: insight into the possible inactivation mechanisms. J Biomed Opt 2011; 16:078003; http://dx.doi.org/10.1117/1.3600771; PMID: 21806295
- Adachi M, Amsterdam D, Brooks SE, Volk BW. Ultrastructural alterations of tissue cultures from human fetal brain infected with the E variant of EMC virus. Acta Neuropathol 1975; 32:133 - 42; http://dx.doi.org/10.1007/BF00689567; PMID: 170775
- Wellmann KF, Amsterdam D, Volk BW. EMC virus and cultured human fetal pancreatic cells. Ultrastructural observations. Arch Pathol 1975; 99:424 - 9; PMID: 167704
- Gajdusek DC. Encephalomyocarditis virus infection in childhood. Pediatrics 1955; 16:902 - 6; PMID: 13273129
- Smadel JE, Warren J. The virus of encephalomyocarditis and its apparent causation of disease in man. J Clin Invest 1947; 26:1197; PMID: 20340975
- Dick GW, Best AM, et al. Mengo encephalomyelitis; a hitherto unknown virus affecting man. Lancet 1948; 2:286 - 9; http://dx.doi.org/10.1016/S0140-6736(48)90652-7; PMID: 18877580
- Tesh RB. The prevalence of encephalomyocarditis virus neutralizing antibodies among various human populations. Am J Trop Med Hyg 1978; 27:144 - 9; PMID: 204206
- Kirkland PD, Gleeson AB, Hawkes RA, Naim HM, Boughton CR. Human infection with encephalomyocarditis virus in New South Wales. Med J Aust 1989; 151:176 - 8, 178; PMID: 2547141
- Craighead JE, Peralta PH, Shelokov A. Demonstration of encephalomyocarditis virus antibody in human serums from Panama. Proc Soc Exp Biol Med 1963; 114:500 - 3; PMID: 14104611
- Juncker-Voss M, Prosl H, Lussy H, Enzenberg U, Auer H, Lassnig H, et al. [Screening for antibodies against zoonotic agents among employees of the Zoological Garden of Vienna, Schönbrunn, Austria]. Berl Munch Tierarztl Wochenschr 2004; 117:404 - 9; PMID: 15495931
- Deutz A, Fuchs K, Nowotny N, Auer H, Schuller W, Stünzner D, et al. [Sero-epidemiological studies of zoonotic infections in hunters--comparative analysis with veterinarians, farmers, and abattoir workers]. Wien Klin Wochenschr 2003; 115:Suppl 3 61 - 7; PMID: 15508783
- Hubbard GB, Soike KF, Butler TM, Carey KD, Davis H, Butcher WI, et al. An encephalomyocarditis virus epizootic in a baboon colony. Lab Anim Sci 1992; 42:233 - 9; PMID: 1320151
- Masek-Hammerman K, Miller AD, Lin KC, et al. Epizootic Myocarditis Associated With Encephalomyocarditis Virus in a Group of Rhesus Macaques (Macaca mulatta). Vet Pathol 2012; 49:386 - 92; http://dx.doi.org/10.1177/0300985811409254; PMID: 21653204
- Papaioannou N, Billinis C, Psychas V, Papadopoulos O, Vlemmas I. Pathogenesis of encephalomyocarditis virus (EMCV) infection in piglets during the viraemia phase: a histopathological, immunohistochemical and virological study. J Comp Pathol 2003; 129:161 - 8; http://dx.doi.org/10.1016/S0021-9975(03)00026-4; PMID: 12921722
- Gelmetti D, Meroni A, Brocchi E, Koenen F, Cammarata G. Pathogenesis of encephalomyocarditis experimental infection in young piglets: a potential animal model to study viral myocarditis. Vet Res 2006; 37:15 - 23; http://dx.doi.org/10.1051/vetres:2005041; PMID: 16336922
- Foni E, Barigazzi G, Sidoli L, Marcato PS, Sarli G, Della Salda L, et al. Experimental encephalomyocarditis virus infection in pigs. Zentralbl Veterinarmed B 1993; 40:347 - 52; PMID: 8237207
- Psychas V, Papaioannou N, Billinis C, Paschaleri-Papadopoulou E, Leontides S, Papadopoulos O, et al. Evaluation of ultrastructural changes associated with encephalomyocarditis virus in the myocardium of experimentally infected piglets. Am J Vet Res 2001; 62:1653 - 7; http://dx.doi.org/10.2460/ajvr.2001.62.1653; PMID: 11592335
- Vlemmas J, Billinis C, Psychas V, Papaioannou N, Paschaleri-Papadopoulou E, Leontides S, et al. Immunohistochemical detection of encephalomyocarditis virus (EMCV) antigen in the heart of experimentally infected piglets. J Comp Pathol 2000; 122:235 - 40; http://dx.doi.org/10.1053/jcpa.1999.0365; PMID: 10805976
- Koenen F, Vanderhallen H. Comparative study of the pathogenic properties of a Belgian and a Greek encephalomyocarditis virus (EMCV) isolate for sows in gestation. Zentralbl Veterinarmed B 1997; 44:281 - 6; PMID: 9270350
- Psalla D, Psychas V, Spyrou V, Billinis C, Papaioannou N, Vlemmas I. Pathogenesis of experimental encephalomyocarditis: a histopathological, immunohistochemical and virological study in mice. J Comp Pathol 2006; 135:142 - 5; http://dx.doi.org/10.1016/j.jcpa.2006.04.003; PMID: 16952370
- Kassimi LB, Boutrouille A, Gonzague M, Mbanda AL, Cruciere C. Nucleotide sequence and construction of an infectious cDNA clone of an EMCV strain isolated from aborted swine fetus. Virus Res 2002; 83:71 - 87; http://dx.doi.org/10.1016/S0168-1702(01)00425-7; PMID: 11864742
- LaRue R, Myers S, Brewer L, Shaw DP, Brown C, Seal BS, et al. A wild-type porcine encephalomyocarditis virus containing a short poly(C) tract is pathogenic to mice, pigs, and cynomolgus macaques. J Virol 2003; 77:9136 - 46; http://dx.doi.org/10.1128/JVI.77.17.9136-9146.2003; PMID: 12915530
- Takeda M, Hirasawa K, Doi K. Lesions in the central nervous system of DBA/2 mice infected with the D variant of encephalomyocarditis virus (EMC-D). J Vet Med Sci 1991; 53:1013 - 7; http://dx.doi.org/10.1292/jvms.53.1013; PMID: 1665080
- Cerutis DR, Bruner RH, Thomas DC, Giron DJ. Tropism and histopathology of the D, B, K, and MM variants of encephalomyocarditis virus. J Med Virol 1989; 29:63 - 9; http://dx.doi.org/10.1002/jmv.1890290112; PMID: 2555446
- Yoon J-W, Jun H-S. Viruses cause type 1 diabetes in animals. Ann N Y Acad Sci 2006; 1079:138 - 46; http://dx.doi.org/10.1196/annals.1375.021; PMID: 17130545
- Nakayama Y, Su W, Ohguchi A, Nakayama H, Doi K. Experimental encephalomyocarditis virus infection in pregnant mice. Exp Mol Pathol 2004; 77:133 - 7; http://dx.doi.org/10.1016/j.yexmp.2004.02.003; PMID: 15351236
- Yamanouchi-Ueno A, Nakayama Y, Doi K. Characteristics of testicular lesions in mice infected with a low dose of encephalomyocarditis (EMC) virus. Exp Mol Pathol 2004; 77:72 - 6; http://dx.doi.org/10.1016/j.yexmp.2003.12.008; PMID: 15215053
- Doi K. Experimental encephalomyocarditis virus infection in small laboratory rodents. J Comp Pathol 2011; 144:25 - 40; http://dx.doi.org/10.1016/j.jcpa.2010.05.001; PMID: 20594559
- Higuchi H, Hara M, Yamamoto K, Miyamoto T, Kinoshita M, Yamada T, et al. Mast cells play a critical role in the pathogenesis of viral myocarditis. Circulation 2008; 118:363 - 72; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.741595; PMID: 18606918
- Matsumori A, Yamamoto K, Shimada M. Cetirizine a histamine H1 receptor antagonist improves viral myocarditis. J Inflamm (Lond) 2010; 7:39; http://dx.doi.org/10.1186/1476-9255-7-39; PMID: 20682082
- Iwasaki A, Matsumori A, Yamada T, Shioi T, Wang W, Ono K, et al. Pimobendan inhibits the production of proinflammatory cytokines and gene expression of inducible nitric oxide synthase in a murine model of viral myocarditis. J Am Coll Cardiol 1999; 33:1400 - 7; http://dx.doi.org/10.1016/S0735-1097(98)00692-5; PMID: 10193745
- Wang J-F, Meissner A, Malek S, Chen Y, Ke Q, Zhang J, et al. Propranolol ameliorates and epinephrine exacerbates progression of acute and chronic viral myocarditis. Am J Physiol Heart Circ Physiol 2005; 289:H1577 - 83; http://dx.doi.org/10.1152/ajpheart.00258.2005; PMID: 15923319
- Matsumori A, Nunokawa Y, Yamaki A, Yamamoto K, Hwang MW, Miyamoto T, et al. Suppression of cytokines and nitric oxide production, and protection against lethal endotoxemia and viral myocarditis by a new NF-kappaB inhibitor. Eur J Heart Fail 2004; 6:137 - 44; http://dx.doi.org/10.1016/j.ejheart.2003.10.007; PMID: 14984720
- Wada H, Saito K, Kanda T, Kobayashi I, Fujii H, Fujigaki S, et al. Tumor necrosis factor-alpha (TNF-alpha) plays a protective role in acute viralmyocarditis in mice: A study using mice lacking TNF-alpha. Circulation 2001; 103:743 - 9; PMID: 11156888
- Nasu-Nishimura Y, Taniuchi Y, Nishimura T, Sakudo A, Nakajima K, Ano Y, et al. Cellular prion protein prevents brain damage after encephalomyocarditis virus infection in mice. Arch Virol 2008; 153:1007 - 12; http://dx.doi.org/10.1007/s00705-008-0086-x; PMID: 18408883
- Takeda M, Itagaki S, Doi K. Biphasic disease of central nervous system induced in DBA/2 mice by the D variant of encephalomyocarditis virus (EMC-D). Int J Exp Pathol 1993; 74:493 - 9; PMID: 8217784
- Takeda M, Miura R, Shiota K, Hirasawa K, Lee MJ, Itagaki SI, et al. Distribution of viral RNA in the spinal cord of DBA/2 mice developing biphasic paralysis following infection with the D variant of encephalomyocarditis virus (EMC-D). Int J Exp Pathol 1995; 76:441 - 7; PMID: 8652364
- Sriram S, Topham DJ, Huang SK, Rodriguez M. Treatment of encephalomyocarditis virus-induced central nervous system demyelination with monoclonal anti-T-cell antibodies. J Virol 1989; 63:4242 - 8; PMID: 2550666
- Takeda M, Ohtsuka R, Nakayama Y, Doi K. The role of CD4(+) T cells in biphasic hind limb paralysis induced by the D variant of encephalomyocarditis virus (EMC-D) in DBA/2 mice. Exp Anim 2004; 53:31 - 5; http://dx.doi.org/10.1538/expanim.53.31; PMID: 14993738
- Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett 2010; 469:39 - 43; http://dx.doi.org/10.1016/j.neulet.2009.11.040; PMID: 19945511
- Hirasawa K, Jun HS, Maeda K, Kawaguchi Y, Itagaki S, Mikami T, et al. Possible role of macrophage-derived soluble mediators in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J Virol 1997; 71:4024 - 31; PMID: 9094680
- Hirasawa K, Jun HS, Han HS, Zhang ML, Hollenberg MD, Yoon JW. Prevention of encephalomyocarditis virus-induced diabetes in mice by inhibition of the tyrosine kinase signalling pathway and subsequent suppression of nitric oxide production in macrophages. J Virol 1999; 73:8541 - 8; PMID: 10482607
- Kang Y, Yoon JW. A genetically determined host factor controlling susceptibility to encephalomyocarditis virus-induced diabetes in mice. J Gen Virol 1993; 74:1207 - 13; http://dx.doi.org/10.1099/0022-1317-74-6-1207; PMID: 8389806
- Onodera T, Yoon JW, Brown KS, Notkina AL. Evidence for a single locus controlling susceptibility to virus-induced diabetes mellitus. Nature 1978; 274:693 - 6; http://dx.doi.org/10.1038/274693a0; PMID: 209341
- Zhu S, Ge X, Gong X, Guo X, Chen Y, Yang H. Alteration of encephalomyocarditis virus pathogenicity due to a mutation at position 100 of VP1. Sci China Life Sci 2011; 54:535 - 43; http://dx.doi.org/10.1007/s11427-011-4172-z; PMID: 21706414
- Altmeyer R, Girard M, van der Werf S, Mimic V, Seigneur L, Saron MF. Attenuated Mengo virus: a new vector for live recombinant vaccines. J Virol 1995; 69:3193 - 6; PMID: 7707549
- Hahn H, Palmenberg AC. Encephalomyocarditis viruses with short poly(C) tracts are more virulent than their mengovirus counterparts. J Virol 1995; 69:2697 - 9; PMID: 7884926
- Martin LR, Neal ZC, McBride MS, Palmenberg AC. Mengovirus and encephalomyocarditis virus poly(C) tract lengths can affect virus growth in murine cell culture. J Virol 2000; 74:3074 - 81; http://dx.doi.org/10.1128/JVI.74.7.3074-3081.2000; PMID: 10708422
- Agol VI, Gmyl AP. Viral security proteins: counteracting host defences. Nat Rev Microbiol 2010; 8:867 - 78; http://dx.doi.org/10.1038/nrmicro2452; PMID: 21060318
- Devaney MA, Vakharia VN, Lloyd RE, Ehrenfeld E, Grubman MJ. Leader protein of foot-and-mouth disease virus is required for cleavage of the p220 component of the cap-binding protein complex. J Virol 1988; 62:4407 - 9; PMID: 2845152
- Paul S, Michiels T. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J Gen Virol 2006; 87:1237 - 46; http://dx.doi.org/10.1099/vir.0.81642-0; PMID: 16603526
- Zoll J, Melchers WJG, Galama JMD, van Kuppeveld FJM. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-kappa B. J Virol 2002; 76:9664 - 72; http://dx.doi.org/10.1128/JVI.76.19.9664-9672.2002; PMID: 12208945
- Dvorak CM, Hall DJ, Hill M, Riddle M, Pranter A, Dillman J, et al. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology 2001; 290:261 - 71; http://dx.doi.org/10.1006/viro.2001.1193; PMID: 11883190
- Hato SV, Ricour C, Schulte BM, Lanke KH, de Bruijni M, Zoll J, et al. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol 2007; 9:2921 - 30; http://dx.doi.org/10.1111/j.1462-5822.2007.01006.x; PMID: 17991048
- Lidsky PV, Hato S, Bardina MV, Aminev AG, Palmenberg AC, Sheval EV, et al. Nucleocytoplasmic traffic disorder induced by cardioviruses. J Virol 2006; 80:2705 - 17; http://dx.doi.org/10.1128/JVI.80.6.2705-2717.2006; PMID: 16501080
- Bardina MV, Lidsky PV, Sheval EV, Fominykh KV, van Kuppeveld FJ, Polyakov VY, et al. Mengovirus-induced rearrangement of the nuclear pore complex: hijacking cellular phosphorylation machinery. J Virol 2009; 83:3150 - 61; http://dx.doi.org/10.1128/JVI.01456-08; PMID: 19144712
- Porter FW, Brown B, Palmenberg AC. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J Virol 2010; 84:12538 - 48; http://dx.doi.org/10.1128/JVI.01484-09; PMID: 20881039
- Porter FW, Palmenberg AC. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition by cardioviruses. J Virol 2009; 83:1941 - 51; http://dx.doi.org/10.1128/JVI.01752-08; PMID: 19073724
- Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc Natl Acad Sci U S A 2006; 103:12417 - 22; http://dx.doi.org/10.1073/pnas.0605375103; PMID: 16888036
- Borghese F, Michiels T. The leader protein of cardioviruses inhibits stress granule assembly. J Virol 2011; 85:9614 - 22; http://dx.doi.org/10.1128/JVI.00480-11; PMID: 21752908
- Okuwa T, Taniura N, Saito M, Himeda T, Ohara Y. Opposite effects of two nonstructural proteins of Theiler’s murine encephalomyelitis virus regulates apoptotic cell death in BHK-21 cells. Microbiol Immunol 2010; 54:639 - 43; PMID: 21140599
- Hahn H, Palmenberg AC. Deletion mapping of the encephalomyocarditis virus primary cleavage site. J Virol 2001; 75:7215 - 8; http://dx.doi.org/10.1128/JVI.75.15.7215-7218.2001; PMID: 11435606
- Groppo R, Palmenberg AC. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J Virol 2007; 81:13067 - 74; http://dx.doi.org/10.1128/JVI.00185-07; PMID: 17728235
- Groppo R, Brown BA, Palmenberg AC. Mutational analysis of the EMCV 2A protein identifies a nuclear localization signal and an eIF4E binding site. Virology 2011; 410:257 - 67; http://dx.doi.org/10.1016/j.virol.2010.11.002; PMID: 21145089
- Aminev AG, Amineva SP, Palmenberg AC. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res 2003; 95:59 - 73; http://dx.doi.org/10.1016/S0168-1702(03)00163-1; PMID: 12921996
- Aminev AG, Amineva SP, Palmenberg AC. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res 2003; 95:45 - 57; http://dx.doi.org/10.1016/S0168-1702(03)00162-X; PMID: 12921995
- Medvedkina OA, Scarlat IV, Kalinina NO, Agol VI. Virus-specific proteins associated with ribosomes of Krebs-II cells infected with encephalomyocarditis virus. FEBS Lett 1974; 39:4 - 8; http://dx.doi.org/10.1016/0014-5793(74)80003-7; PMID: 4369440
- Ohguchi A, Nakayama Y, Yasoshima A, Doi C, Mikami T, Uetsuka K, et al. Encepalomyocarditis virus-induced apoptosis and ultrastructural changes in the lacrimal and parotid glands of mice. Exp Mol Pathol 2006; 80:201 - 7; http://dx.doi.org/10.1016/j.yexmp.2005.06.003; PMID: 16038897
- Yamada T, Matsumori A, Wang WZ, Ohashi N, Shiota K, Sasayama S. Apoptosis in congestive heart failure induced by viral myocarditis in mice. Heart Vessels 1999; 14:29 - 37; http://dx.doi.org/10.1007/BF02481740; PMID: 10543311
- Buenz EJ, Sauer BM, Lafrance-Corey RG, Deb C, Denic A, German CL, et al. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute neurovirulent picornavirus infection. Am J Pathol 2009; 175:668 - 84; http://dx.doi.org/10.2353/ajpath.2009.081126; PMID: 19608874
- Schwarz EM, Badorff C, Hiura TS, Wessely R, Badorff A, Verma IM, et al. NF-kappaB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J Virol 1998; 72:5654 - 60; PMID: 9621024
- Roos FC, Roberts AM, Hwang IIL, Moriyama EH, Evans AJ, Sybingco S, et al. Oncolytic targeting of renal cell carcinoma via encephalomyocarditis virus. EMBO Mol Med 2010; 2:275 - 88; http://dx.doi.org/10.1002/emmm.201000081; PMID: 20623734
- Shioi T, Matsumori A, Sasayama S. Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Circulation 1996; 94:2930 - 7; PMID: 8941123
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995; 80:321 - 30; http://dx.doi.org/10.1016/0092-8674(95)90415-8; PMID: 7834752
- Yamada T, Matsumori A, Sasayama S. Therapeutic effect of anti-tumor necrosis factor-alpha antibody on the murine model of viral myocarditis induced by encephalomyocarditis virus. Circulation 1994; 89:846 - 51; PMID: 8313574
- Anz D, Koelzer VH, Moder S, Thaler R, Schwerd T, Lahl K, et al. Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol 2010; 184:939 - 46; http://dx.doi.org/10.4049/jimmunol.0901245; PMID: 19966212
- Nakano A, Matsumori A, Kawamoto S, Tahara H, Yamato E, Sasayama S, et al. Cytokine gene therapy for myocarditis by in vivo electroporation. Hum Gene Ther 2001; 12:1289 - 97; http://dx.doi.org/10.1089/104303401750270940; PMID: 11440622
- Christmann BS, Moran JM, McGraw JA, Buller RML, Corbett JA. Ccr5 Regulates Inflammatory Gene Expression in Response to Encephalomyocarditis Virus Infection. Am J Pathol 2011; 179:2941 - 51; http://dx.doi.org/10.1016/j.ajpath.2011.08.012; PMID: 22001348
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995; 146:3 - 15; PMID: 7856735
- Banerjee R, Tsai W, Kim W, Dasgupta A. Interaction of poliovirus-encoded 2C/2BC polypeptides with the 3′ terminus negative-strand cloverleaf requires an intact stem-loop b. Virology 2001; 280:41 - 51; http://dx.doi.org/10.1006/viro.2000.0770; PMID: 11162817
- Eldin P, Papon L, Oteiza A, Brocchi E, Lawson TG, Mechti N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol 2009; 90:536 - 45; http://dx.doi.org/10.1099/vir.0.006288-0; PMID: 19218198
- Carocci M, Bakkali Kassimi L. Encephalomyocarditis virus, in Molecular Detection of Human Viral Pathogens. 1st Ed. CRC Press; 2010.
- Chow M, Newman JF, Filman D, Hogle JM, Rowlands DJ, Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature 1987; 327:482 - 6; http://dx.doi.org/10.1038/327482a0; PMID: 3035380
- Dmitrieva TM, Alexeevski AV, Shatskaya GS, Tolskaya EA, Gmyl AP, Khitrina EV, et al. Significance of the C-terminal amino acid residue in mengovirus RNA-dependent RNA polymerase. Virology 2007; 365:79 - 91; http://dx.doi.org/10.1016/j.virol.2007.02.038; PMID: 17467026