Keywords: :
In a recent paper appearing in the Proceedings of the National Academy of Sciences of the United States of America, Young and colleagues report the case of a persistent carrier of Staphylococcus aureus who went on to develop a virulent bloodstream infection (Young et al., Proc Natl Acad Sci USA 2012). By sequencing the genomes of bacteria sampled from the nose and bloodstream over an extended period, they pieced together the evolutionary changes that accompanied the transition from asymptomatic carriage to invasive disease. Among an unusual cluster of knockout mutations that coincided with the beginning of a period of declining health in the patient, they identified the truncation of an AraC family transcriptional regulator as a likely modifier of virulence, demonstrating that genetic changes that occur in bacterial genomes during prolonged carriage could play a role in pathogenesis. Here we discuss the alternative evolutionary hypotheses for the observed excess of knockout mutations and review the evidence in favor of a causal role for regulatory gene dysfunction in bacterial virulence. We consider the prospects for systematic studies into the evolution of virulence in bacterial carriage populations.
Introduction
Many bacterial pathogens are, for the most part, commensal organisms that cause disease infrequently compared with their high rates of asymptomatic carriage in the population. Staphylococcus aureus is one such example. S. aureus is a major healthcare-associated pathogen, responsible for an estimated 17,000 deaths in the United States between 1999 and 2007, making it one of the two most frequently identified bacterial causes of mortality, second only to Clostridium difficile (WHO Mortality Update 2011). Yet the frequency of asymptomatic carriage of S. aureus is high. In studies of nasal carriage, 27% of healthy adults have been found to carry S. aureus (Wertheim et al., Lancet Inf Dis 2005). Fatal disease is therefore a rare occurrence in the lifecycle of the bacterium. Epidemiological studies have investigated the risk factors that predispose to invasive disease, but the role of novel genetic changes caused by bacterial evolution within the host has been understudied. However, that is about to change because advances in DNA sequencing make it increasingly feasible to characterize the genomes of large numbers of bacteria rapidly and inexpensively.
In an early example of the potential that high-throughput whole-genome sequencing has for unraveling the genetic basis of bacterial virulence, Young and colleagues charted the evolutionary changes that accompanied the transition from nasal carriage to invasive bloodstream infection in one persistent carrier. The subject was enrolled in a longitudinal study of S. aureus carriage. S. aureus was cultured from six of eight nasal swabs collected over a 12 mo period. Fifteen months after enrolling in the study, the participant was admitted to hospital with a highly virulent S. aureus bloodstream infection.
Whole genome sequencing showed that despite negative swabs in month 10 and at the time of bloodstream infection, all the bacterial colonies isolated from the nasal swabs and from the bloodstream infection were extremely closely related, representing a single cohesive population. A total of 34 genetic variants were observed across 68 colonies, comprising 30 single nucleotide polymorphisms (SNPs) and four insertions or deletions (indels). This represents extremely sparse genetic variation in a genome that is nearly three million nucleotides long. Genome annotation revealed a typical methicillin-sensitive S. aureus (MSSA) profile, with no evidence for the acquisition of additional virulence factors or toxin genes beyond the normal repertoire found in carried strains.
Although carriage is a known risk factor for S. aureus infection (von Eiff et al., N Engl J Med 2001) and invasive disease may stem from ingress of commensal flora into the bloodstream through compromised epithelia, no source for the bloodstream infection was found: there were no endovascular catheters or evidence of endocarditis or surgical-site infection associated with a previously fitted pacemaker. However, genetic analysis revealed that the bacterial population formed three distinct clades: colonies isolated from the early nasal swabs prior to any medical intervention (early nasal cultures, ENC), colonies isolated from the late nasal swab, post-antibiotic treatment (late nasal culture, LNC), and colonies isolated from the bloodstream infection (late blood culture, LBC). These clades were distinguished by multiple mutations, leaving open the possibility that evolution in the bacterial population during carriage had resulted in the spontaneous emergence of a more virulent strain.
Further investigation revealed unusual patterns of evolution between the clades. First, the two branches that separated bacteria isolated early during asymptomatic nasal carriage (ENC clade) from bacteria isolated from the bloodstream infection (LBC clade) were significantly longer than expected, meaning that these groups were surprisingly differentiated. Second, half of the eight mutations that occurred on these two branches resulted in premature stop codons that were predicted to knock out protein function. This was a highly statistically significant excess of premature stop codons given that none were observed among the other 26 mutations. Further evidence that these patterns were highly unusual was provided by an analysis of longitudinal bacterial evolution in two other participants from the same carriage study that did not go on to develop invasive disease.
One of the eight premature stop codons was singled out as a potential modifier of virulence phenotype. It occurred in a gene belonging to the AraC family of transcriptional regulators (AFTRs) that are regulators of carbon metabolism, stress response and virulence, and which respond to environmental conditions such as antibiotic use and oxidative stress (Yang et al., Trends Microbiol 2011). Knockout mutations in genes such as AFTRs could play an important role in bacterial pathogenesis because of their ability to quickly effectuate functional change. In what follows we discuss the possible evolutionary processes underlying the observed patterns of mutation between carriage and invasive bacteria, and the evidence in favor of a role for regulatory gene dysfunction in bacterial pathogenesis.
Evolutionary Explanations for Observed Patterns
The significant excess of mutations separating the early asymptomatically carried nasal bacteria from the late invasive bloodstream bacteria may be consistent with a number of evolutionary scenarios that have different implications for how we view the evolution of virulence within hosts, and we take each of these in turn ().
Figure 1. Evolutionary processes that may underlie observed patterns of mutation between carried and invasive bacteria. (A) Gene flow between bacterial sub-populations may generate genetic heterogeneity. In the example, bacteria normally resident in the nose (gold cocci) and axilla (green cocci) occasionally migrate between sites, leading to mixed populations. (B) Latent bacterial populations may persist in privileged sites only to re-emerge later. For example, the contemporary population (gold) may be re-seeded with ancestral bacteria (green) lying dormant in neutrophil vesicles. (C) During a population bottleneck natural selection is weak, allowing an increase in frequency of deleterious mutants. Prior to the bottleneck the bacterial population contains a fitness gradient ranging from the optimal genotype (solid gold) to deleterious mutants (partially gold). Survival during the bottleneck is random, and deleterious mutants may rise to high frequency by chance. (D) Mutants that have a selective advantage may sweep through the population. In the example, a single mutant (red) out-replicates the original genotype (gold). Over time, the fitter mutant replaces the original genotype.
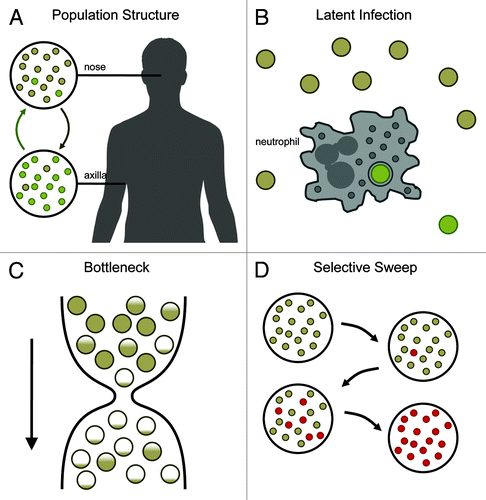
Cryptic populations of differentiated bacteria
Unexpectedly high genetic differentiation within a population that is assumed to be homogeneous may be due to cryptic population structure. For example, S. aureus is routinely isolated from the nose, axilla and groin of asymptomatic individuals, with concurrent colonization of multiple sites common. If multiple sub-populations evolve mostly in isolation but with limited gene flow between body sites, genetically heterogeneous populations may be observed. Other forms of population structure can also lead to genetic heterogeneity. If the sub-populations are ultimately descended from a single founding colonization event, the degree of differentiation is likely to be slight.
Re-seeding by a latent population of ancestral genotypes
It has been suggested that S. aureus can survive intracellularly, possibly within privileged sites such as the phagosomes of neutrophils, and this may contribute to relapsing infections (Thwaites and Gant, Nat Rev Microbiol 2011). This model would allow for the sporadic re-seeding of the S. aureus carriage population with latent bacteria. If rates of mutation are retarded during latency, latent bacteria would more closely resemble ancestral genotypes than the contemporary population. Such a process would lead to an apparent excess of mutations between bacteria because some genotypes would be relics from the past.
Relaxed selection associated with a population bottleneck
Neither population structure nor re-seeding by latent bacteria can, by themselves, explain the excess of premature stop codons observed on the branches separating the early nasal bacteria and late invasive bacteria compared with all other branches of the tree. Premature stop codons are generally much rarer than expected under strictly neutral evolution because they are disfavored by natural selection as a result of their drastically deleterious effect on protein function. An apparent burst of mutations inducing premature stop codons could be explained by a temporary relaxation of selection pressures. Such a phenomenon could be caused by a transient but severe reduction in population size (Otto and Whitlock, Genetics 1997). There is some evidence to support this theory: nasal swabs at months 10 and 15, both of which followed antibiotic treatment, showed no growth although the bacterial population was later shown not to have been completely eradicated.
Adaptive evolution
Intense competition for resources between bacteria within the host may result in selective sweeps if spontaneously occurring mutants can out-replicate non-mutant bacteria. The potential for such mutants to arise might be considerable. For example, any trade-off between the exploitation of the host and transmissibility will lead to a situation in which strains that successfully colonize new hosts are susceptible to out-competition by spontaneously arising mutants that better exploit host resources at the cost of future transmission. Alternatively, mutants that are able to exploit sterile sites in the body such as the bloodstream will quickly multiply in the absence of competition. An important difference between relaxed selection associated with bottlenecks and adaptive evolution is that in the former, spontaneously arising virulent bacteria are less fit than their predecessors, whereas in the latter they are more fit. Since the evolution of highly virulent genotypes is likely to end badly for the pathogen population as well as for the host, in the former they might be viewed as an accelerated form of mutational meltdown while in the latter they could be viewed as a tragedy of the commons.
Evidence for a Role of Regulatory Genes in Pathogenesis
There is increasing evidence that mutations in key regulatory genes play an important role in bacterial pathogenesis. Recent work has provided examples of this phenomenon in S. aureus and other major bacterial pathogens.
The agr locus of S. aureus
The accessory gene regulator (agr) locus is a quorum-sensing system responsible for the downregulation of S. aureus cell surface proteins and the upregulation of toxin genes during the switch from exponential to stationary growth phase (Gilot et al., J Clin Microbiol 2002). An independent association has been demonstrated between dysfunction of the agr locus and 30 d mortality in severely ill patients with S. aureus bacteremia: a cohort study demonstrated a 91% increase in mortality with loss of agr function (Schweizer et al., Antimicrob Agents Chemother 2011). Loss of agr function is believed to result in diminished cell autolysis, and thus impaired killing of S. aureus by bactericidal antibiotics. This association was only seen in the most unwell quartile of patients, suggesting interplay between host vulnerability and pathogen dysregulation. Other studies have shown an association between agr dysfunction and increased duration of bacteraemia as well as decreased vancomycin sensitivity (Fowler et al., J Hosp Infect 2007). While methicillin resistance and toxin production have previously been the focus of much research in S. aureus virulence, these findings highlight the importance of regulatory proteins in the pathogenesis of S. aureus disease and open new directions for investigation.
AFTR-mediated response to environmental conditions
The AraC family of transcriptional regulators (AFTRs) have been implicated in the pathogenesis of bacterial disease (Yang et al., Trends Microbiol 2011; Fantappiè et al., Microbiology 2011). Yang and colleagues review the ways in which AFTRs respond to small molecules in the environment by upregulating gene transcription. For example, the intestinal pathogen Vibrio cholerae responds to bicarbonate secretion in the small intestine by the ToxT mediated transcription of cholera toxin. AFTRs have been demonstrated to play a similar role in numerous Gram-positive and Gram-negative bacterial pathogens of humans, animals and plants. These organisms respond to stimuli specific to the environment in which they are suited to pathogenesis (bicarbonate within the intestine, urea in the urinary tract and cellobiose in plants) by altering gene transcription to enable them to colonize and survive. The Oxfordshire case study represents the first time the role of AFTRs has been associated with the development of virulence in S. aureus infection.
Phylogenetic association between AFTR knockouts and hypervirulence
There is evidence from other species that truncation of AFTRs is associated with altered pathogenicity. One of three AFTRs identified in Neisseria meningitidis—MpeR—has been recently shown to be part of the Fur pathway: a system that is responsive to iron limitation in the environment (a metabolic pathway key to microbial survival within the host), as well as regulating toxin expression and transcriptional activation (Fantappiè et al., Microbiology 2011). A frameshift mutation resulting in loss of the MpeR protein was detected in all sequenced isolates of a hypervirulent clonal complex of N. meningitidis (ST32) which were selected from different outbreaks in disparate countries, but not in any other isolates. Like N. meningitidis, S. aureus is well adapted to a biological niche (in this case, the anterior nares). The loss of a gene known to be subject to regulation by environmental stimuli has in the case of N. meningitidis been associated with increased likelihood of causing meningitis or septicemia. It is possible that the loss of regulation by environmental stimuli can likewise drive a transition to the bloodstream for S. aureus.
Future Studies
The study by Young and colleagues demonstrates the promise that high-throughput whole genome sequencing holds for understanding the evolutionary dynamics of bacteria both during asymptomatic carriage and in disease. These tools might be profitably applied to a number of bacterial pathogens such as N. meningitidis, Streptococcus pneumoniae and Haemophilus influenzae that are generally carried by human hosts without adverse effect, but which can cause life-threatening illnesses. Systematic studies are now needed to yield deeper insights into the genomic underpinnings of pathogenesis.
A major challenge to the design of systematic studies is the unpredictability of the bacterial-host interaction. Of more than 1,100 individuals in a cohort of healthy adults, a single S. aureus carrier developed invasive disease during the period of the study to date (just over three years). Very large numbers of people are therefore required for systematic follow-up in order to capture significant numbers of cases. Evolution over shorter time periods may be easier to capture with large cohort studies in areas of high S. aureus disease prevalence. An alternative but equally important route that we have not explored here are studies that focus on comparing the frequency of gene acquisition or knockout in large numbers of disease-associated cases and non-disease controls. A genome-wide association study, testing for non-random distribution of changes between these groups, faces several analytic challenges: most importantly, it must adequately detect and control for both relatedness and deeper population structure in a clonal organism like S. aureus (Falush and Bowden, Trends Microbiol 2006).
Functional follow-up is required to investigate what phenotypic changes, if any, are induced by the knockout of regulatory genes such as AFTRs in S. aureus. It is critical to establish whether changes to biological correlates of virulence can be observed as a direct effect of mutations that occur in vivo. Animal studies using knockout and knock-in strains would be an obvious route for investigation, but if the repertoire of candidate genes identified by whole genome studies becomes large, alternatives that do not require laboratory animals would be preferable. There is a need for in vitro assays that reliably replicate the host environment, both that of nasal epithelium and the bloodstream, with their variable conditions and host defenses. There is much work yet to do in this area, but the early signs are that evolutionary changes that occur within the host may play an important role in tipping the fine balance between asymptomatic carriage and invasive disease.
Acknowledgments
The authors would like to thank R. Bowden, T.E.A. Peto and D.W. Crook for their comments on the manuscript.