Abstract
We recently developed a method that allows fast differentiation between Streptococcus pyogenes (GAS) strains. The method named phage profiling (PP) is based on a simple assumption that a regular PCR reaction with Taq polymerase and relatively short elongation time is not able to yield long DNA fragment, such as ~40–50 kb integrated prophage. Only fragments without any integrated DNA or short fragments inserted between integration sites can be efficiently amplified. We designed primers that anneal upstream and downstream prophage integration sites, so in simple PCR reaction we can test if any additional DNA is integrated into particular site. Profiling of integrated elements can be used as rapid, high resolution typing method, with the resolution as high as PFGE and is excellent predictor of PFGE type.
Introduction
Streptococcus pyogenes (GAS) is a pathogen that causes infections of human skin and mucosal surfaces that vary in their intensity from mild to severe and life threathening.Citation1 Every year GAS is responsible for over 600 million infections and half a million deaths worldwide, as a result of infections and postinfectional sequelae.Citation2S. pyogenes often causes seasonal outbreaksCitation3,Citation4 and is responsible for infections in hospitalsCitation5,Citation6 and long-term care facilities.Citation7
Epidemiological investigations of GAS infections and outbreaks usually include classification into one of ~150 serotypes (emm typingCitation8) and sequence type (MLST, http://spyogenes.mlst.net). Both methods rely on sequencing of one (emm typing) or seven (MLST) genes. Both methods are relatively fast and uncomplicated, but require specialized equipment and can be too expensive for routine use. In addition, they both have too low resolution to distinguish between closely related strains. PFGE has been for many years the method of choice used to investigate differences between strains at the genome level. But PFGE analysis requires specialized equipment, skilled personnel, and is difficult to analyze and compare.
We recently developed a method that allows fast differentiation between Streptococcus pyogenes (GAS) strains.Citation9 The method named phage profiling (PP) is based on a simple assumption that a regular PCR reaction with Taq polymerase and relatively short elongation time is not able to yield long DNA fragments, such as ~40–50 due to the presence of integrated phage. Only fragments without any integrated DNA, or short fragments inserted between integration sites can be efficiently amplified. Twenty one prophage and ICE (integrative conjugative element) integration sites (named from A to U) are well defined within GAS core chromosome.Citation10 We designed primers that anneal upstream and downstream of known prophage integration sites, so simple PCR reactions can be used to test if DNA is integrated into particular sites. Profiling of integrated elements can be used as rapid, high resolution typing method. The resolution of PP is as high as PFGE and is excellent predictor of PFGE type.Citation9
Reagents
(1) 1 mM stock of four dNTP prepared from 100 mM stock solutions of individual dNTPs (Sigma-Aldrich, DNTP100)
(2) Taq polymerase (Fermentas, EP0402)
(3) 10× Taq buffer with (NH4)2SO4 (Fermentas, B33)
(4) 25 mM MgCl2 stock solution (Fermentas, R0971)
(5) Primers (, Genomed, www.genomed.pl)
Table 1. Primer pairs detecting phage and ICEs integration sites in GAS genomes
(6) 1.5% (wt/vol) SeaKemLE agarose (Lonza, 90004) in 1× TBE buffer
(7) 1× TBE electrophoresis buffer
Equipment
(1) Veriti PCR cycler (Life Technologies)
(2) Gel electrophoresis tank (BioRad, 170-4511) with 26 well combs (BioRad 170-4525)
Reagents Setup
Template
DNA template was isolated using commercially available kits (Qiagen or A&A Biotechnology) or classic methods with phenol extraction and ethanol precipitation.Citation11 A critical step in chromosomal DNA isolation is the treatment of bacteria with mutanolysin to digest cell wall. Purified DNA should be diluted to ~10 ng/ul. Multiple analyses should be run in 96-well plate format, diluted template DNA should be dispensed to standard 96 well plate and used in further reaction setup. Diluted DNA should be kept at 4°C to avoid freezing/thawing cycles.
Primers
Mix equal volumes of individual 10 mM primer stock solutions into appropriate mixes: “Phages Mix 1,” “Phages Mix 2,” “Phages Mix 3” and “Phages Mix 4” according to . Each mix includes primers that amplify regions encompassing phage integration sites and primers that amplify fragment of dnaA gene (positive control). To avoid degradation, primers pre-mixes should be aliquoted into 50–100 μl portions sufficient to run the whole 96-well PCR plate without multiple freezing-thawing cycles. Aliquoted primer mixes solution should be kept at −20°C.
Reaction setup
(1) Prepare diluted template DNA, preferably in 96-well format, or 8-well strips so it can be easily dispensed with a multichannel pipette.
(2) Mix together all reagents for PCR mastermix according to .
Table 2. Composition of mastermixes for the detection of integrated elements per single reaction and per 96 well plate
(3) Dispense 2.2 μl (mastermix Phages Mix 1, 2 and 4); 2.3 μl (mastermix Phages Mix 3) or 4 μl (if you run separate reactions for detecting Phage F, see Anticipated Results section) of prepared mastermix to each of the wells or PCR tubes.
(4) Using multichannel pipette, transfer DNA template prepared in step 1 to wells with dispensed mastermixes from step 3. Volume of DNA depends on the reaction; 2.8 μl of the template DNA should be added to wells with mastermixes 1, 2 and 4; 2.7 μl of the template DNA should be added to wells with mastermix 3; 1 μl of DNA template should be added wells with primers that detect phage F.
(5) Close tubes or seal the plate
(6) Mix well and spin for 15 sec (tubes) or 1 min at 1,000 × g (plates)
(7) Place the prepared plate/tubes in the thermocycler and run appropriate program.
Equipment setup
No special setup is required.
Procedure
(1) PCR amplification
Initial denaturation, 95°C, 4 min
40 cycles of: denaturation, 95°C, 15 sec; annealing, 64°C, 30 sec; elongation, 72°C, 3 min 30 sec
Final elongation, 72°C, 7 min
(2) Gel electrophoresis
We analyze products of multiplex PCR reaction by electrophoresis in 1.5% (wt/vol)
SeaKemLE agarose gel in TBE buffer, with addition ethidium bromide. Separation is performed with current 160 mV for about 100 min. Gels are photographed under UV light.
(3) Gel analysis
As a result of each multiplex reaction a set of bands of various sizes can be observed. Presence of a band on a gel means that no element is integrated into particular site. Lack of a band means that integrated element is longer than ~5 kb and cannot be amplified. We usually score 1 or 0 for the presence/absence of the band on the gel. It allows to generate string of 21 characters that is characteristic for a strain.
Timing
(1) DNA isolation, ~90 min.
(2) Reaction setup, ~15 min for 96-well plate, but only using multichannel pipettes and pre-dispensed template masterplate.
(3) PCR amplification, ~4 h.
(4) Gel electrophoresis, ~100 min.
Problem Handling
Problem 1
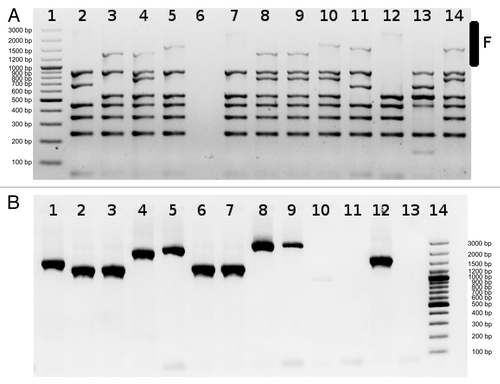
Even in the absence of a whole phage integrated into site F, very often integration of small elements (1–2 kb) has been observed. To avoid the problem of lower amplification efficiency with primers detecting integrated phage in site F () we sometimes use MIX3 without primers detecting integration in F site and run separate reaction with primers F ().
Figure 1. Detection of phages integrated into F site using MIX3 (A) or individual primers (B). Lanes 1 (A) and 14 (B) Size standard O’GeneRuler™ 100 bp Plus DNA Ladder marker (Fermentas SM1153). It is clearly visible that amplification efficiency of large fragments in the multiplex reaction is lower than amplification using individual primers. In (B), lanes 1–9 and 12 various small elements integrated into site F, lanes 10, 11 and 13 large element integrated into F site. PCR products presented in (A and B) were generated using different chromosomal DNAs as templates.
Anticipated Results
Usually interpretation of the results is simple and straightforward. Primers for the detection of the mobile genetic elements were designed based on the comparison of genomic sequences described by Beres and Musser.Citation10 When we designed primers, sequence analysis revealed that PCR product generated for the analysis of F integration site can widely vary in size. We characterized over 1,000 GAS strains using PP method and in some cases we observe small differences in detected product size for other integration sites. Differences are not as frequent as in case of site F, but noticeable. We think that changes in the predicted product sizes are caused by the integration sites acting as hot spots for any integration and changes in product size reflect insertions of small DNA fragments.
We detected changes in product sizes corresponding with M, D and I integration sites. The largest size differences between predicted and observed product sizes were detected for integration site M (over 1 kb, ). The larger product was predominantly detected among M89 strains, and in some cases in M80, M78, M73, M108, M119 and M50. For the integration site D we often detect a band that is slightly (~20–30 bp) smaller than the expected, the change in size is predominantly detected for the serotypes M77 and M75 (). For the integration site I, we observed slightly larger fragments differing in size about 100 bp between strains (). Fragments size different than expected were detected mostly in serotype M75, and to less extent in M89, M4 and M66. All reactions that generated fragments of suspicious size were individually tested to confirm that the variable size M, D or I product is indeed generated with appropriate primers.
Figure 2. Detected changes in size of M (A), D (B) and I (C) PCR products and “ghost bands” in panel (C). Size standard O’GeneRuler™ 100 bp Plus DNA Ladder marker (Fermentas SM1153).
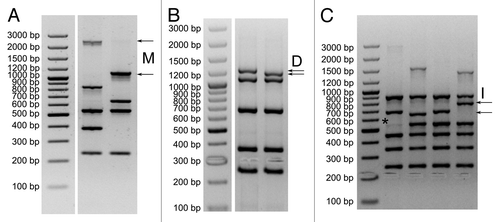
Sometimes we also observed very faint “ghost bands” (*). This is PCR signal derived from the minute fraction of GAS cells carrying prophage/ICE element where it was probably temporarily cleaved out of the genome. The ghost band should be interpreted as a site with integrated element. All size differences were confirmed by PCR reactions separately with individual primer sets for the specific integration site.
The method can be automated and upgraded with the use of primers with fluorescent labels.
Acknowledgments
The work was financed by the grant from National Center for Science (NCN) number NN401 536140.
References
- Sitkiewicz I, Hryniewicz W. Pyogenic streptococci--danger of re-emerging pathogens. Pol J Microbiol 2010; 59:219 - 26; PMID: 21466038
- Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis 2005; 5:685 - 94; http://dx.doi.org/10.1016/S1473-3099(05)70267-X; PMID: 16253886
- Lamagni TL, Darenberg J, Luca-Harari B, Siljander T, Efstratiou A, Henriques-Normark B, et al, Strep-EURO Study Group. Epidemiology of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol 2008; 46:2359 - 67; http://dx.doi.org/10.1128/JCM.00422-08; PMID: 18463210
- Tse H, Bao JY, Davies MR, Maamary P, Tsoi HW, Tong AH, et al. Molecular characterization of the 2011 Hong Kong scarlet Fever outbreak. J Infect Dis 2012; 206:341 - 51; http://dx.doi.org/10.1093/infdis/jis362; PMID: 22615319
- Montes M, Tamayo E, Oñate E, Pérez-Yarza EG, Pérez-Trallero E. Outbreak of Streptococcus pyogenes infection in healthcare workers in a paediatric intensive care unit: transmission from a single patient. Epidemiol Infect 2012; In press http://dx.doi.org/10.1017/S0950268812000751; PMID: 22717031
- Daneman N, Green KA, Low DE, Simor AE, Willey B, Schwartz B, et al, Ontario Group A Streptococcal Study Group. Surveillance for hospital outbreaks of invasive group a streptococcal infections in Ontario, Canada, 1992 to 2000. Ann Intern Med 2007; 147:234 - 41; PMID: 17709757
- Jordan HT, Richards CL Jr., Burton DC, Thigpen MC, Van Beneden CA. Group a streptococcal disease in long-term care facilities: descriptive epidemiology and potential control measures. Clin Infect Dis 2007; 45:742 - 52; http://dx.doi.org/10.1086/520992; PMID: 17712760
- Beall B, Facklam R, Thompson T. Sequencing emm-specific PCR products for routine and accurate typing of group A streptococci. J Clin Microbiol 1996; 34:953 - 8; PMID: 8815115
- Borek AL, Wilemska J, Izdebski R, Hryniewicz W, Sitkiewicz I. A new rapid and cost-effective method for detection of phages, ICEs and virulence factors encoded by Streptococcus pyogenes. Pol J Microbiol 2011; 60:187 - 201; PMID: 22184925
- Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2007; 2:e800; http://dx.doi.org/10.1371/journal.pone.0000800; PMID: 17726530
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, 2nd edition 1989.