Abstract
Streptococcus pyogenes (GAS) is a human pathogen that causes millions of infections worldwide. Comparison between GAS strains isolated in different laboratories all over the world is often difficult. Three usually used methods have either low resolution (emm typing), are expensive (MLST) or time- and labor-consuming (PFGE). Our laboratory recently developed new, inexpensive methods of GAS typing—VF (virulence factor profiling) and PP (phage profiling). To improve the typing scheme developed for GAS, we recently proposed a new typing method. Here we present detailed protocol for MLVF analysis.
Introduction
Streptococcus pyogenes (GAS) is a human pathogen that causes millions of infections worldwide. Comparison between GAS strains isolated in different laboratories all over the world is often difficult. Currently three major methods are used to determine relationships between GAS strains: (1) emm typing, which allows for classification of strains into ~150 serotypes, (2) multilocus sequence typing (MLST) and (3) restriction analysis combined with pulse field gel electrophoresis (PFGE). All three methods have either low resolution (emm typing), are expensive (MLST) or time- and labor-consuming (PFGE). Our laboratory recently developed new, inexpensive methods of GAS typing. The first method (VF), allows detection of 20 virulence factors, the second method is based on PCR detection of the mobile genetic elements (MGE) integration sites within S. pyogenes genome (PP, phage profiling).Citation1-Citation3 Nevertheless, both methods are more focused on mobile portion of the genome. To improve and balance the typing scheme developed for GAS and to reflect variability of a core (non-MGE related) genome, we recently proposed a new typing method.Citation4
Multilocus variable tandem repeat analyses (MLVA and MLVF) are methods based on the detection of repeated sequences within bacterial genomes. MLVF (multiple locus variable number tandem repeat fingerprinting) is based on the amplification of several loci of variable size and comparison of generated band patterns with a reference. MLVA (multiple locus variable number tandem repeat analysis) is based on the same principles as MLVF, but instead of pattern analysis, number of repeated sequences within each locus is used to generate unique code that can be stored in the database. Both methods are cheap, fast and do not require specialized equipment, except a thermocycler and electrophoresis tank. Informative results, comparable with PFGE analysis, can be available in less than 10 h.
The method we developed is based on the size variation within seven loci. The number of the size variants varies from several to tens for tested genes. As a result, about 40,000 MLVF patterns can be generatedCitation4. The method is used for routine work and S. pyogenes typing in our laboratory. We analyzed over 700 strains using the procedure described below and observed that homogenous populations, which belong to the same M type and exhibit the same PFGE pattern and MLST profile, can be further differentiated using MLVA typing.Citation4
Reagents
(1) RNase 10 mg/ml (Sigma-Aldrich, R5503)
(2) Lysozyme 20 mg/ml (Sigma-Aldrich, L6876)
(3) Mutanolysin 10 U/µl (Sigma-Aldrich, M9901)
(4) 1 mM stock of four dNTP prepared from 100 mM stock solutions of individual dNTPs (Sigma-Aldrich, DNTP100)
(5) Taq polymerase (Fermentas, EP0402)
(6) 10× Taq buffer with (NH4)2SO4 (Fermentas, B33)
(7) 25 mM MgCl2 stock solution (Fermentas, R0971)
(8) Starters (; Genomed, www.genomed.pl)
Table 1. Starters used for MLVA typing
(9) 2.5% wt/vol NuSieve agarose (Lonza, 50094) (in 1× TBE)
(10) 1× TBE electrophoresis buffer
Equipment
(1) Veriti PCR cycler (Life Technologies).
(2) Gel electrophoresis tank (BioRad, 170-4511) with 26 well combs (BioRad, 170-4525)
Setup
Template
As a template for PCR reaction we use chromosomal DNA isolated using commercially available kits. We have good results with columns Genomic Mini produced by A&A Biotechnology (116-250), but kits available from other manufacturers may be used as well. S. pyogenes cells for DNA isolation are grown on half of the agar plate (Columbia with 5% sheep blood, multiple manufacturers). Prior purification bacteria are scraped from the plate and re-suspended 200 μl of TE buffer. Bacteria are treated with 10 µl of lysozyme, 5 µl of RNase and 2.5 µl of mutanolysin for about 30–45 min at 37°C. Purified DNA used as a template should be diluted 10× to a ~10 ng/μl concentration. We usually run multiple analyses in 96-well plate format, so we dispense diluted DNA to standard 96-well plate and use it in further reaction setup. Diluted template DNA should be kept at 4°C to avoid freezing/thawing cycles.
Starters
Because the amplification efficiency of designed starters varies, we combine various amounts of individual 100 mM primers stocks to prepare primer mastermix that used in the PCR reaction will yield products of comparable intensity. We mix 1 volume of primers 1–6, 0.5 volume of primers 7–10, 2 volumes of primers 11 and 12 and 4 volumes of primers 13 and 14. For example: 30 μl of primers 1 to 6, 15 μl of primers 7 to 10, 60 μl of primers 11 and 12 and 120 μl of primers 13 and 14. Primer mix should be aliquoted into 25–250 μl portions and stored at −20°C until use. Each primer mix portion should never be thawed/frozen more than 2 times. (Problem 1)
Reaction setup
(1) Prepare diluted template DNA (as described above), preferably in 96-well format, or 8-well strips. Template DNA dispensed into wells/strips can be easily transferred to reaction wells with a multichannel pipette.
(2) Mix together all reagents for PCR mastermix according to .
Table 2. Composition of PCR mastermix
(3) Dispense 9 μl of prepared mastermix to each of the reaction wells or PCR tubes.
(4) Transfer 1 μl of DNA template from template DNA plate to the reaction plate with multichannel pipette.
(5) Close tubes or seal the plate.
(6) Mix well and spin for 15 sec (tubes) or 1 min at 500 rpm (plates).
(7) Place the prepared plate/tubes in the thermocycler and run the program.
Equipment setup
No special setup is required. We usually store saved PCR amplification program listed below.
Procedure
(1) PCR amplification
Initial denaturation, 95°C, 5 min
40 cycles of: denaturation, 95°C, 15 sec; annealing, 64°C, 30 sec; elongation, 72°C, 4 min 30 sec
Final elongation, 72°C, 7 min
(2) Gel electrophoresis
The products of multiplex PCR reactions are analyzed by electrophoresis in 2.5% (wt/vol) NuSieve agarose gel with ethidium bromide, in standard TBE buffer. We usually use 50 bp and 100 bp ladder (Fermentas, SM0371, SM0321), which allows precise fragment size determination.
Electrophoresis is performed with constant current 100mA in Sub-Cell Model 192 electrophoresis cell for 3.5 h. Gels are visualized under UV light and photographed.
(3) Gel analysis
Pattern analysis can be performed on multiple levels. (1) When MLVF is simply used to check if two or more strains are identical, visual band matching of two or multiple patterns can be performed. (2) In case of multiple samples that generate variable patterns, we recommend more detailed analysis. Usually, based on the photograph, we estimate size of the amplified products with the imaging software that allows automatic determination of the product size (in our case Molecular Imaging Software by Kodak, supplied with the GelLogic 2000 imaging system). It is much easier to compare product sizes than patterns on the gel photograph. (3) To compare MLVF patterns of samples run on multiple gels we use BioNumerics software. The software allows to normalize patterns between multiple gels/runs, compare them to each other, and cluster similar patterns.
We consider two strains as closely related if they differ by no more than single band within the profile from the reference strain (size or present/absent) (Problem 2).
Timing
(1) DNA isolation ~90 min
(2) Reaction setup ~15 min
(3) PCR ~4 h
(4) Electrophoresis ~3.5 h
Problem Handling
Problem 1
We sometimes observe low yield of PCR products. This is a result of non-efficient PCR amplification that is caused either by low quality of primers or template. Primers and the template used in the reaction are often degraded after multiple freezing/thawing cycles. To maximize PCR efficiency primer mastermixes are kept at −20°C in small portions and are never frozen/thawed more than two times. Also diluted chromosomal DNA is kept in the fridge to avoid multiple freezing cycles.
Problem 2
In some patterns, very strong bands can be observed among weaker ones. It is a result of multiple bands of similar size. Double, more intense bands are often observed for M1 (about 750 bp in size) and M28 (about 630 bp) serotype strains. Depending on the electrophoresis equipment, for better separation of PCR products, time of electrophoresis could be extended.
Anticipated Results
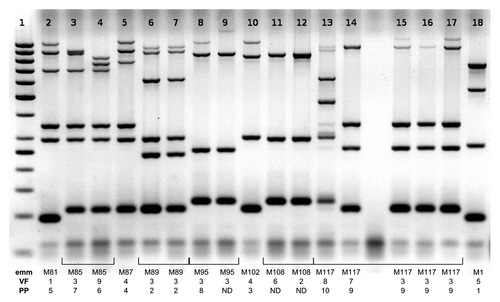
The gel presented in shows typical results of MLVA analysis of multiple strains. MLVA patterns in lanes 6–7 are identical and represent two strains of serotype M89 with identical virulence factor profiles (VF) and phage profiles (PP), patterns 8 and 9 are related and represent two M95 strains with identical virulence factor profile (VF). Patterns 7 and 8 are not related. All bands in these patterns differ in size or are not present.
Figure 1. Results of routine MLVF analysis of 17 GAS strains. Each strain is marked with emm type, VF profileCitation1,Citation2 and PP profile,Citation1,Citation3 each numerical designation denotes unique pattern within the analyzed group of strains, strains with the same designation have identical profiles. Identical patterns 6 and 7 represent two strains of the same serotype (M89), the same VF profile (3) and the same PP profile (2). Patterns 14–17 represent four M117 strains. MLVF patterns of strains15–17, as well as VF and PP profiles, are identical, strain 14 has distinct, but related, MLVF and VF profiles. Lane 1: GeneRuler 50 bp standard (Fermentas).
Acknowledgments
The work was financed by the grant from National Center for Science (NCN) number NN401 536140.
References
- Borek AL, Wilemska J, Izdebski R, Hryniewicz W, Sitkiewicz I. A new rapid and cost-effective method for detection of phages, ICEs and virulence factors encoded by Streptococcus pyogenes. Pol J Microbiol 2011; 60:187 - 201; PMID: 22184925
- Borek AL, Obszańska K, Hryniewicz W, Sitkiewicz I. Detection of Streptococcus pyogenes virulence factors by multiplex PCR. Virulence 2012; 3:529 - 33; http://dx.doi.org/10.4161/viru.21540; PMID: 23076284
- Borek AL, Obszańska K, Hryniewicz W, Sitkiewicz I. Typing of Streptococcus pyogenes strains using the phage profiling method. Virulence 2012; 3:534 - 38; http://dx.doi.org/10.4161/viru.21887; PMID: 23076280
- Obszańska K, Borek AL, Izdebski R, Hryniewicz W, Sitkiewicz I. Multilocus variable number tandem repeat analysis (MLVA) of Streptococcus pyogenes. J Microbiol Methods 2011; 87:143 - 9; http://dx.doi.org/10.1016/j.mimet.2011.08.017; PMID: 21920391