Abstract
Transmissible encephalopathies (TSEs) are believed by many to arise by spontaneous conversion of host prion protein (PrP) into an infectious amyloid (PrP-res, PrPSc) without nucleic acid. Many TSE agents reside in the environment, with infection controlled by public health measures. These include the disappearance of kuru with the cessation of ritual cannibalism, the dramatic reduction of epidemic bovine encephalopathy (BSE) by removal of contaminated feed, and the lack of endemic scrapie in geographically isolated Australian sheep with susceptible PrP genotypes. While prion protein modeling has engendered an intense focus on common types of protein misfolding and amyloid formation in diverse organisms and diseases, the biological characteristics of infectious TSE agents, and their recognition by the host as foreign entities, raises several fundamental new directions for fruitful investigation such as: (1) unrecognized microbial agents in the environmental metagenome that may cause latent neurodegenerative disease, (2) the evolutionary social and protective functions of different amyloid proteins in diverse organisms from bacteria to mammals, and (3) amyloid formation as a beneficial innate immune response to stress (infectious and non-infectious). This innate process however, once initiated, can become unstoppable in accelerated neuronal aging.
If I told him. Would he like it would Napoleon—Gertrude Stein
Introduction
The purpose of this perspective is to communicate several fundamental biologic features of TSE infections that may not be apparent to scientists who do not isolate and characterize infectious particles, or analyze different TSE agent-strains. At high multiplicity, TSE agents can induce pathological misfolding of host PrP into PrP-res amyloid. However, infection is not required for this conversion, and genetic host PrP coding mutations are sufficient to cause PrP amyloid plaques with no infectivity.Citation1-Citation4 “Prion” amyloid states in yeast and bacteria that lack the infectious characteristics of mammalian TSEs illustrate amyloid commonalities, as well as the functional diversity of protein aggregation in evolution. Unity in the “prion” field relates amyloid processes, but not the diverse inciting causes of protein aggregation. Autocatalytic PrP-res accumulation follows a markedly different trajectory than the replication of infectious TSE particles, as detailed below. Because PrP band patterns neither correlate with nor predict agent strain properties that are preserved across species, non-PrP molecules must determine strain characteristics. Mammals interact with trillions of microorganisms on the skin and in the alimentary tract, posing many opportunities for exchange and/or incorporation of foreign nucleic acid. TSE infectious agents can represent the tip of the iceberg of hidden environmental pathogens that cause late onset disease.
For simplicity, composite TSE data are first broadly summarized, with illustrations of well-established facts. TSEs, by definition, are a group of related mammalian infections that are lethal once they invade the brain. Active brain infection separates TSEs from other neurodegenerative diseases, such as Alzheimer disease (AD) and post-encephalitic (post-infectious) Parkinson disease, that share protein misfolding mechanisms and amyloid phenotypes but are not infectious. Amyloid plaques have long been known to form in the brains of normal older animals, particularly primates, and inoculation of AD β-amyloid homogenates in the brain only accelerates aging, but does not transmit a replicating infectious agent.Citation5 The rare genetic cases of human AD and prion pathology, along with transgene (Tg) models for AD β-amyloid and PrP amyloid deposition, underscore non-infectious causes of the same end-stage phenotype or amyloid scar. Cumulative environmental factors, including toxins and past infections that can limit the lifespan of non-dividing neurons, are likely to cause the more common forms non-infectious AD.Citation6 In contrast, a foreign infectious agent is the root cause of TSEs, a fact that has been obscured by the assumption that host-encoded PrP-res is the infectious agent. Or, as more prohibitively stated: “Prions are composed solely of the disease-causing prion protein (PrPSc)”.Citation7 This exclusive view continues to be promulgatedCitation8,Citation9 even though other molecules, including nucleic acids, can be required for replication of TSE infectious agents.Citation10
The structure and integral molecules of the infectious TSE particle remain undefined. To date, PrP-res itself has failed to fulfilled Koch postulates.Citation11,Citation12 Koch postulates remain particularly relevant now because recombinant and synthetic nucleic acids, without the need for culture, are sufficient to recreate infection and its characteristic disease phenotype, as shown for poliovirus. Similarly, many recent TSE investigations have attempted to prove the prion hypothesis by focusing on test tube autocatalytic conversions of normal PrP to a misfolded, presumably infectious PrP-res. In these experiments, infectious material is first used to start or “seed” the amyloidogenic process. These amyloid conversion reactions, sequentially replenished with uninfected material, such as normal brain homogenates, typically generate enormous amounts of PrP-res with little, or no infectivity. Thus the misfolded PrP-res protein alone is not necessarily infectious. PrP-res has also been generated from normal brain. This shows that PrP-res formation and infectious agent replication are two different processes.
Many studies acknowledge that some other “cofactor” is required to make PrP infectious,Citation10,Citation13,Citation14 but only one group has reported that recombinant PrP (recPrP) can be converted into an infectious form without any animal derived additives or “seeds”.Citation15 This result has never been replicated by others over the past 8 y, and serial transmissions from that recPrP in wt mice revealed incubation times and neuropathology of their laboratory scrapie strain (RML); laboratory contamination is an acknowledged problem in test tube PrP-res conversions.Citation16 Another group has also claimed in their abstract that infectivity “can be generated solely from… recPrP without any mammalian or synthetic cofactors”, but the experimental details show that a standard scrapie brain PrP-res preparation, known to contain infectivity (as well as nucleic acid), was added to seed this recPrP reaction.Citation17 In a study that may be more convincing, tissue derived lipids were added to recPrP to produce unusually high levels of lab-like scrapie infectivity.Citation18 During the past 3 y this result apparently has not been reproduced elsewhere by independent investigators. Subsequent conversion studies, with lipid and synthetic poly(rA) RNA also showed only very marginal infectivity in animals, and questionable PrP-res pathology, in contrast to added tissue derived RNA.Citation19 Thus an agent RNA cannot be ruled out as claimed. Notwithstanding the diagnostic potential of these PrP conversions,Citation20 the purpose of this review is to cover substantial biological and analytical evidence that invites a more inclusive appreciation of alternative agent concepts. In particular, the environmental source of infection, the variety and persistence of different agent strains, the species and cell responses to these distinct agent strains, and the presence of nucleic acids in highly infectious particles all lead to consideration of a nucleic acid genome that defines TSE strain virulence and mutability.Citation21
Routes of Infection and Species Determine Prolonged Latency
Many decades of work have shown that even intracerebral (ic) inoculations of species-adapted TSE agents yield very long incubation times to disease. These typically range from 100 d to 500 d in rodents infected with different CJD and scrapie strains.Citation22 The incubation time depends on the TSE agent’s virulence for the species inoculated. Hamsters, rats, mice, guinea pigs and primates have different immune repertoires that affect agent spread and pathology, as has been shown by passage of sporadic CJD (sCJD) agents in these different speciesCitation23 (vide infra). Peripheral infections by intraperitoneal, intravascular and oral routes (the most natural), can take an even longer time to produce disease than the ic route, even with a virulent TSE agent that is “fast” for a given species, such as 263K scrapie that ic kills hamsters in ~65 d. Regardless of route, as later detailed, latency is largely based on host recognition and suppression of a foreign agent that dwells in and derives from the environment.
Oral administration of the 263K scrapie agent in hamsters can yield prolonged asymptomatic incubation times, yet still lead to lethal disease even when administered from old soil samples, the common source of continuous endemic scrapie in UK and Icelandic sheep.Citation24,Citation25 On the other hand, repeated high oral dosing of mice with the virulent Asiatic FU-CJD agent fails to produce disease within the normal mouse lifespan.Citation26 Thus mice can suppress and/or eliminate this agent peripherally. In humans, asymptomatic infections can be as long as 5–30 y from the time of exposure, as established in kuru,Citation27 after peripheral inoculations of sCJD contaminated growth hormone, and in the epidemic spread of the BSE agent to zoo animals and humans (vCJD) via contaminated feed. A current uncontrolled TSE of cervids (CWD) continues to spread geographically across the United States to wild deer and elk. The environmental source of infection is clear in all these major TSE infections. Only in sCJD, a relatively rare and avirulent TSE, is the environmental source not obvious. Hence sCJD is a sporadic, but not necessarily “spontaneous” infection.Citation28
Two related problems further confront an “infectious PrP-res”. First, oral dosing experiments show that the infectious agent, but not PrP-res, survives the digestive juices.Citation29,Citation30 Agent, but not PrP survival is now further substantiated by proteinase K (PK) digestions that destroy all forms of PrP, including the relatively PK resistant “infectious” PrP-res form.Citation31 Moreover, high levels of infectious agent are quantitatively recovered from digested brain, even after ~3 logs of PrP and PrP-res are destroyed. This would logically lead to an examination of non-PrP agent components, especially molecules and viral structures best designed to infect mammals via the oral route. These include viruses, with their protected nucleic acids, that are part of the vast metagenome. (MetagenomeCitation32 refers to previously unknown communities of microbial organisms in the environment that are being discovered using genetic sequencing rather than laboratory cultivation.) The second conundrum raised by prolonged peripheral latency is how any form of PrP, or a spontaneous PrP-res seed, can modulate its own quiescence in a tissue specific way, and then suddenly release itself to unceasingly replicate with a strain-specific doubling time in brain.Citation33 Host controls of a foreign environmental agent would typically be involved in this type of pattern.
Silent Replication of TSE Agents with Late Onset PrP-res
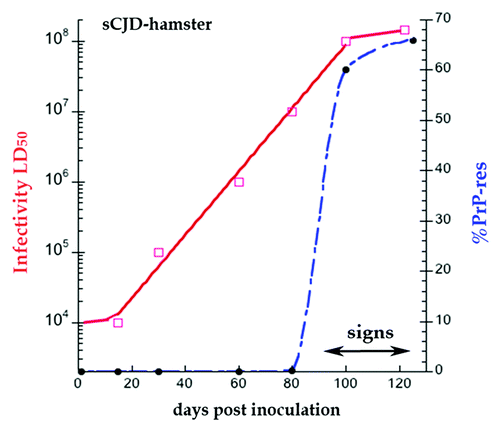
Time course analyses of infectivity titers and PrP conversion after brain inoculation demonstrate a clinically silent phase of exponential agent replication that starts well before late-onset PrP-res pathology. In particular, such studies show why PrP-res is a poor substitute for quantitatively assessing agent replication. shows experimental data for sCJD in hamsters,Citation34 a species that is quite susceptible to this human agent. In hamsters, sCJD causes widespread brain lesions 120 d after ic inoculation, whereas wt mice with sCJD have a 360 d latency with very limited lesions.Citation35,Citation36 In the data of there is a clear disconnect between the time and slopes of accumulating agent (solid red line) vs. those of PrP-res (blue broken line). Note the progressive log accumulation of infectious particles starting at 20 d until a maximal infectivity is reached at 90 d. Agent accumulation is then arrested and remains at a plateau as clinical signs begin. Shortly before agent arrest at 90 d, PrP-res begins to form, and maximal PrP-res (65% of the total brain PrP) rapidly coincides with agent arrest. In the case of PrP-res, there is an observable 2 log rise (from 0.6 at 80 d to 60%). The PrP-res shows an identical sleep slope when plotted on a log scale, or even extrapolated to a level of PrP-res that is not directly detectable. Most simply: (1) PrP-res appears as a late response to high levels of the infectious agent, and (2) high PrP-res can act as part of an innate immune response that limits agent replication and/or survival. Trapping of infectious particles in a PrP-res amyloidCitation37 can be one underlying mechanism for this arrest (see the section “PrP is a Required Cell Binding Site for Infectious TSE Particles”).
Figure 1. Hamster sCJD progression in days post-infection ic. Superimposed data for infectious titer in logs (red solid line) and % PrP-res of the total PrP (blue broken line). PrP is not converted to PrP-res until 80 d, and PrP-res shows a late onset and much steeper trajectory than infectivity.Citation34 The effective doubling time of PrP-res is 65× the rate of agent doubling; this rate includes both replication and destruction (see text). Note clinical signs appear at beginning of the high plateau phase of infectivity and PrP-res.
PrP-res Conversion Progresses at a Rate Very Different from Agent
also demonstrates mathematically important data that allows one to calculate and compare the doubling times of agent and PrP-res conversion. The effective (observed) doubling time (ti) by definition incorporates both replication and elimination,Citation34 and can be applied to a molecule as well as an infectious particle. The sCJD doubling ti in hamsters is 7.6 d. In marked contrast, when calculated by this equation, the ti of PrP-res in is 0.116 d, or >65× greater than the agent rate, a clearly separate process. This observation unifies many diverse bacterial, yeast and TSE protein aggregation phenomena because similar types of protein misfolding can progress rapidly even when initiated by different types of past and cumulative environmental stresses. These include the many toxic chemicals treatments that induce intracellular yeast protein aggregates,Citation38 in addition to the TSE and conventional viral infectious agents that ultimately induce PrP-res and neurofibrillary tangle formation respectively in non-dividing mammalian neurons.Citation39
Discordant rates of TSE agent replication and PrP-res conversion are a consistent feature of TSEs in wt animals. This discordance has not been obvious in the very short incubation 263K hamster scrapie ic model (~65 d to terminal signs). However, additional models with incubation times of >110 d all demonstrate a progressive slow exponential rise in infectivity followed by a sudden increase in PrP-res, as shown in of Asiatic FU-CJD, a strain that is much more virulent for wt mice than sCJD. The FU-CJD agent steadily replicates by >4 logs (from ~20 to 90 d) before provoking a sudden, rapid rise of PrP-res in the brain.Citation40 Note that total PrP (left panel) remains constant as its derivative PrP-res is being misfolded (+PK lanes), i.e., PrP-res misfolding is a process of conversion rather than true replication. Moreover, a new extremely fast CWD agent model in bank voles, which takes only 35 d to terminal disease,Citation41 also shows a marked discrepancy between PrP-res and infectious titer. These diseased brains have high levels of infectivity (8.4 logs/gm) but only low levels of PrP-res. To explain this, it is hypothesized that (1) there are different toxic and infectious PrP-res forms, (2) these PrP forms replicate and mature at different rates, and (3) the infectious PrP-res subset contains additional unique misfolded variants that encode all the fast and slow CWD strains in these voles. Notably, bank voles have relevant attributes beyond PrP. Of particular interest is their susceptibility to many different viral infections, and their ability to act as viral reservoirs in zoonotic infections.Citation42 These viruses may enhance the virulence of some CWD strains, as do retroviruses for CWD infection in other rodent cells (reviewed in ref. Citation21). It is also possible that voles carry some TSE strains asymptomatically and spread TSEs through the environment via excrement, as they do for other viruses. The zoonotic spread of TSEs is not well studied, and TSE concepts and prevention may be radically transformed by metagenomic analyses.
Figure 2. Late onset of PrP-res in FU-CJD wt mice illustrates constant level of PrP in left panel without proteinase K (PK) from 50–130 d. Right panel shows the FU-CJD agent replicates by 5 logs in these mice before PrP-res is first detectable at 90 d (+PK lanes) as shown by the interpolated rise between assay points at ~15 and 100 d. Gel load at 130 d was decreased to 0.5× for detection in the linear range. Log infectivity increase by ic route has been reproduced (n > 3 from different serial passages).

Agent-Induced Neurodegeneration Does Not Require PrP-res
Marked neurodegenerative changes can be initiated by a TSE agent without pathological PrP-res as an accomplice. This is well demonstrated by the very prolonged sCJD rat model. Rats infected ic with sCJD have an incubation time of >350 d vs. an incubation of 120 d in hamster sCJD. Remarkably, in these sCJD infected rats, classical TSE spongiform brain changes and widespread microglial activation become prominent simultaneously as much as 100 d before PrP-res makes its entrance.Citation23 It is often assumed that PrP-res is the cause of neuronal death in TSEs, but these results show that the sCJD agent itself can provoke active spongiform neurodegeneration without appreciable PrP-res, probably mediated, in part, by microglia. This type of data makes it critical to consider early therapeutic interventions against the TSE agent, rather than focusing solely on inhibiting late-stage PrP-res formation.
A Role for PrP In Eliminating Infectious Particles, with Pathologic Sequelae
The prediction that host PrP-res can help limit infection has been verified by recent TSE culture experiments. At very early passages after infection of neuronal GT1 cells, high PrP-res levels coincide with limiting kuru agent accumulation. Conversely, a delayed low level of PrP-res tolerates a 1000-fold higher titer of the FU-CJD agent.Citation33 An even more dramatic demonstration that PrP-res can function to eliminate high levels of agent has now been demonstrated in stationary rat neuronal cell cultures infected with FU-CJD. Four logs of infectious agent were eliminated from cells during a continuing enormous increase in the production of both PrP and PrP-res.Citation39 This finding is contrary to the prion expectation that PrP-res will increase the infectious titer. Furthermore, this non-infectious PrP-res self-perpetuates, underscoring a stress response akin to the stress induced bacterial elaboration of biofilm amyloids,Citation43 as well as to yeast protein aggregates induced by environmental chemicals.Citation38 Self-propagating PrP-res in the cured rat neurons is clearly an amyloidogenic process gone awry, with potential destructive effects on neurons.
Synthetic PrP-res peptides that form fibrils can be toxic for neuronal tumor and cerebellar granule cells.Citation44 On the other hand, abundant intracellular PrP-res amyloid fibrils induced by scrapie and CJD agent infection are insufficient to cause visible degenerative or toxic changes in neuronal GT1 cells.Citation45 Thus (1) infected neuronal cells lack a special toxic form of PrP-resCitation46 or (2) other cellular factors and cell types (such as activated microglia and astrocytes) are required for spongiform change. Because GT1 cells in culture contain very high levels of the infectious agent (≥3 infectious particles per cell), but show no toxic changes, non-neuronal cells in the brain are likely to be part of the progressive TSE neurodegenerative cascade in vivo, one that probably starts as a host defensive mechanism.
The realization that PrP-res is a late response to earlier agent-induced stresses has relevance for AD and other noninfectious degenerative brain diseases. In AD, past infectious insults, in conjunction with cumulative environmental stresses and toxins, may ultimately cause the late stage protein misfolding of β-amyloid and Tau pathology. The progressive accumulation of PrP amyloid, even after the TSE agent has been eliminated from cells,Citation39 gives further credence to self-perpetuating pathological protein misfolding in later progressive stages of neurodegenerative diseases. Indeed, this is when non-infectious seeded amyloid conversions are most relevant, as originally proposed.Citation47
Innate Immune Recognition of Agent Occurs Early in Infection
In TSEs, a role for the immune system has been largely dismissed because no acute lymphocytic infiltrates and neutralizing antibodies are seen. Nevertheless, many classical experiments in scrapie and CJD from 1960–1990 clarified the critical importance of lymphoreticular tissues and myeloid white blood cells in the dissemination and sequestration of TSE agents in peripheral tissues,Citation37 a route that many viruses take within migratory lymphoreticular cells to escape antibody recognition. More critically, Tateishi’s group first used immunodeficient SCID mice to prove that the lymphoreticular system was required for effective spread and persistence of scrapie peripherally.Citation48 Additional Tg models have substantiated this in both scrapie and CJD. However, Tg lymphoreticular ablations only retard disease onset because animals ultimately succumbed to florid infectious disease.Citation49,Citation50 With the acceptance of the infectious PrP or prion hypothesis, it was further assumed that TSE agents would provoke no immune response because PrP is not recognized by its host as foreign. However, the early microglial activation in rat sCJDCitation23 renewed investigations of innate immune responses that might modulate agent replication and latency. For these studies, it was advantageous to use normal wt mice because of their well characterized immune systems.
Many pathogenic microbes, from small viruses to mycobacteria, are sequestered in a latent state in myeloid dendritic cells, and physiologic stresses provoke their recrudescence and dissemination. This pattern fits the 10–30 y dormancy and recrudescence of both iatrogenic sCJD and kuru agents in humans. This process is also apparent in the much shorter FU-CJD mouse model in four respects. First, isolated brain microglia, a relative of peripheral migratory dendritic cells, contain extremely high levels of the FU-CJD agent, but minute amounts of PrP, and no detectable PrP-res (reviewed in ref. Citation11). Second, array studies of relevant molecular markers demonstrated innate immune responses in infected, but not control microglia. Moreover, this specific group of myeloid cell transcripts are substantially elevated in FU-CJD infected microglia, but are not educed by PrP-res.Citation51 Third, some of these innate immune transcripts are provoked during the earliest phase of exponential agent replication, well before PrP-res or neurodegenerative changes begin.Citation40 These innate immune markers clearly signify host-recognition of a foreign infectious particle rather than host PrP-res. Subsequent studies have uncovered additional important early host innate immune responses in scrapie,Citation52 demonstrating these responses are a general feature of TSE infections. Fourth, high scrapie infectious titers have been found in isolated peripheral plasmacytoid dendritic cells that lack any detectable PrP.Citation53 This further extends the centrality of these types of cells in TSE agent latency and tissue spread. It also suggests that PrP may not be an essential or integral component of the minimal infectious particle. It is also unclear how PrP-res, a seed for its own conversion, could arrest itself for years, and then reactivate by seeding itself in specialized dendritic cells that contain no detectable PrP.
Proof That an Animal’s Innate Immune Biochemical Systems Limit Agent Expansion
The above observations strongly implicate complex cellular responses to a foreign agent that are unrelated to either PrP or PrP-res conversion. It has long been known that PrP itself is insufficient to make cells susceptible to infection since an animal’s lung and kidney epithelial cells with PrP do not normally become infected. Thus individual cell and complex animal responses are involved. The critical role of an animal’s cellular defense mechanisms against TSE agents has now been conclusively demonstrated. Basically, when grown in culture, TSE agents are released from multicellular suppressive host controls. Thus TSE agents are freed to replicate rapidly, and some agents, as FU-CJD, achieve even higher titers per cell than in brain. Conversely, when these cultured agents are re-inoculated ic, they can no longer replicate rapidly, i.e., they are again constrained by complex host controls that can target both replication and destructive processes. Five very different scrapie and human TSE agents with widely diverse ti effective doubling times from 4.3 to 32 d in wt mice all suddenly and dramatically reduced their doubling time to a ti of 1 d when grown in GT1 neuronal cells.Citation33 Thus all these different TSE agents are capable of rapid replication when released from multicellular host controls. Rapid agent replication did not depend on PrP-res accumulations because PrP-res in culture spanned from 0% to 50%. When rapidly replicating CJD and scrapie agents in culture were re-inoculated in mice, they recreated their strain-specific regional neuropathology in addition to their original distinctive long latencies.Citation54,Citation55 Thus the replication and survival of these agents are clearly limited by cells and factors not present in monotypic culture. This data shows no evidence for agent mutations as the cause of this change. Moreover, mice clearly can respond to, and differentially retard each of these agents.
At a single cell level, innate immunity also makes cells permissive for particular agents. and even cells in culture that synthesize high levels of homologous PrP can resist infection for unknown reasons. Neuroblastoma (N2a) tumor cells used for many scrapie studies are known to be genetically unstable, and inevitable progressive population variants are differentially receptive to even a single agent strain, such as 22L scrapie.Citation56 Hence, it will be unclear if replication or loss of a given TSE agent is due to altered cell characteristics or mutant strains. The positive infection of genetically stable GT1 cells with very different agents allowed us to demonstrates that these cells ex vivo can differentially recognize and suppress these agents though their innate immune networks. GT1 cells, with their invariant PrP and cell phenotype, clearly show intrinsic preferences for particular TSE agents.Citation22 Since GT1 cells are permissive for infection by all these agents they do not require additional factors. Instead, it is evident than active cellular process can limit infection and even lead to a progressive cure. For example, the murine adapted kuru agent reproducibly and stably replicates for >1 y at a constant level, and continues to elicit large amounts of PrP-res. Yet, as noted, this agent attains an infectious titer that is only 1/1000th of the murine adapted FU-CJD agent.Citation33 Moreover, murine adapted vCJD is able to infect parallel GT1 cells, and induce vCJD-specific PrP-res bands for ~9 passages (3–4 weeks). However, in this case, PrP-res diminishes over the next few weeks and ultimately is undetectable (with no change in the total cell PrP). This suggests these GT1 cells have substantially cured themselves of the vCJD agent. We have repeated this observation >5 times, using different vCJD infected mouse brain samples and different passages of GT1 cells (Liu, Tittman, Kipkorir, Botsios, and Manuelidis, unpublished data). Clearly this agent reduction (also shown by a representative infectious assay) involves an active innate defense process that is agent-specific, rather than a lack of some factor necessary for infection. Some of these differential intrinsic cell controls may be uncovered by high throughput RNA interference screens. Indeed, initiatives that target these non-PrP factors should be of value in arresting and treating CJD.
PrP is a Required Cell Binding Site for Infectious TSE Particles
PrP is a GPI anchored protein and is ultrastructurally abundant on plasma and ER membranes.Citation45 While other membrane molecules and cellular machinery are likely to drive PrP-res accumulation in the living cell, it is clear from PrP knockout studies that host PrP is essential for progressive scrapie agent infection.Citation57 One interpretation of these data posits that some form of PrP must be the infectious agent. Alternatively, host PrP can be a receptor and/or scaffold required for active agent replication and spread. This latter view is based on established viral requirements for a particular host protein, where even a single coding mutation in the host receptor can prevent infection.Citation47 diagrams the PrP null state when there is no scaffold for effective TSE agent replication. Hence agent particles disintegrate and are eliminated. shows infection with two different TSE infectious agents, each inducing a recognizably different PrP-res amyloid aggregate. In , separate processes of agent replication and PrP-res aggregation are diagrammed, including trapping of some agent by PrP amyloid. While there is a clear distinction between the infectious and the purely genetic causes of PrP pathology, both can induce transmembrane forms of PrP that lead to lysosomal processing and amyloid plaque deposition.Citation58
Figure 3. Model of invading ~25 nm TSE viral particle binding its required host PrP receptor to induce PrP-res amyloid. (A) Shows disintegration and elimination of the infectious particle with its nucleic acid core (solid circle) and protective protein capsid cage when there is no PrP on which to dock. (B) Shows two different PrP-res conformers induced by two different TSE viral strains (A and B particles with protective capsid cages). As previously modeled,Citation47 the infectious particle initiates or “seeds” the PrP misfolding. As now shown experimentally, this amyloid can continue to perpetuate itself even after agent elimination.Citation39 (C) Top depicts the manufacture of abundant TSE infectious particles with a residue of limited tightly bound PrP membrane attachment sites. Bottom shows late accumulating host PrP-res amyloid that can trap, protect, and eventually eliminate agent.Citation39
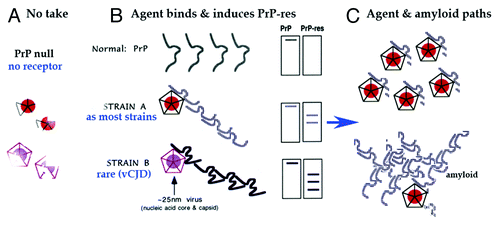
Most strains (as strain A) induce an identical PrP-res band pattern, provided that the cell type and species are not varied. In different species and cell types the PrP band differences are largely due to post-translational glycosylation, and deglycosylation with neuraminidase reveals the underlying homogeneity of PrP, first shown by resolution of a single discrete isoelectric spot in 2D gels.Citation59 Even more extensive deglycosylation that further minimizes PrP variation does not reduce infectivity or alter the strain’s latency and neuropathologic phenotype.Citation37 Nevertheless, we had proposed that a rare or unusual agent (strain B) might bind PrP differently to yield a variant PrP-res band pattern that is maintained in various species and cell types, regardless of glycosylation differences.Citation47 This has now been verified experimentally. The PrP-res doublet induced by the unique BSE/vCJD agent is constant in infected cows, humans, wt mice, and even in monotypic cell cultures.Citation22 In contrast, most other CJD and scrapie strains have now been conclusively shown to induce indistinguishable PrP-res bands in standard wt mouse brains.Citation36 This invites further questions for prion proponents about what observable PrP differences might encode strains.
A PrP receptor model fits many important TSE features, including the species “transmission barrier” linked to host PrP codon mutations. It also accounts for the above retrieval of a biologically identical TSE agent from cells with different PrP-res bands and PrP glycosylation patterns. PrP-res patterns are very different in lymphoid tissues, brain, and cultured cells, but all these cells yield the same agent rather than individually distinct strains. According to the prion concept at least some of these different PrP forms should encode various TSE strains. Despite intense efforts, neither a reproducibly infectious nor a strain-encoding PrP conformer has been identified. In sum, different agent strains give the same PrP-res pattern, and samples with very different PrP-res patterns, even from different species, can propagate the same infectious agent.
The concept that PrP is a required TSE agent receptor, rather than the causal infectious agent, has received little attention. However, recent studies show that PrP is intimately involved in the life cycle of other infectious agents, from viruses to gram-negative bacteria.Citation60,Citation61 Indeed, PrP is the receptor required for a bacterial oral infection, and this finding parallels the PrP requirement by TSE agents for infection. An increase in PrP, even when its sequence does not match the PrP of the infected donor, markedly facilitates infection of human agents to mice. In fact, Tg mice overexpressing wt murine PrP are more susceptible to human vCJD than Tg mice overexpressing human PrP.Citation22 This finding is compatible with PrP as a receptor, even if slightly flawed, but is contrary to the prion dogma that PrP sequence differences will necessarily thwart cross-species infection. A viral induction of pathological protein aggregates, including human paramyxovirus induced neurodegenerative neurofibrillary tangles,Citation39 also gives precedence to TSE agent induced PrP-res, as does the newest demonstration that a viral infection can induce a conformational “prion-like” state in mitochondrial membrane complexes (MAVS).Citation62,Citation63 This last parallel is even more intriguing because MAVS activate and propagate antiviral innate immune responses, a finding remarkably consistent with the innate immune responses in TSEs reviewed above. Such findings further support the model in which shows a TSE viral particle that induces PrP-res aggregation by binding its required host receptor.Citation47
The Function(s) and Location of PrP are Advantageous for TSE Agents
Although it is often stated that the normal function of PrP is unknown, PrP is clearly involved in terminal neural differentiation, cell-to-cell contacts, and synapse formation (reviewed in ref. Citation39). Newly formed nanotubes proliferate as PrP is induced in cultured neurons undergoing terminal differentiation, and nanotubes facilitate the transmission of viruses. Indeed, the proliferation of PrP rich nanotubes result in >120-fold increases in TSE infectious particles per cell.Citation39 Because PrP is not essential for the normal function of an animal, TSE agent binding to host PrP, especially at lower asymptomatic levels, will not result in loss of any critical physiological function. Agent hijacking of PrP thus provides for long-term survival of both the agent and the animal. When PrP binds the infectious particle, it can also help the agent hide from adaptive immune surveillance. Additionally, PrP-res amyloid trapping in myeloid dendritic cellsCitation26 and macrophages can structurally protect the agent from elimination for many years, particularly because PrP-res, as other amyloids, are so impenetrable and insoluble. Finally, apposed plasma membranes between two cells are known to exchange molecular components, and thus agent attachment to PrP will further facilitate cell-to-cell spread, the highly favored route for TSE agents.Citation55 Other PrP functions may confer additional advantages.
TSE Infections with 2 Strains Share Features with Yeast “Prions”
also predicts that two different agent strains, each inducing recognizably unique pathological PrP-res bands, could exist in the same cell. In the prion model, it is claimed that an intermediate PrP-res chimera (of two different PrP conformers) will materialize, and give rise to intermediate infectious properties, as proposed for adaptive cross-species PrP-res conversions.Citation28 In contrast, in the viral model, two different TSE agents would continue to induce independent A and B forms of PrP-res, i.e., two infecting strains would maintain their independent identities, and each PrP-res would continue to seed itself and accumulate in the cell. In fact, two CJD strains have been shown to simultaneously replicate in animals for 400 d, and each maintains its original strain phenotype.Citation11 Moreover, two different agents inducing two different PrP-res conformers can accumulate in a GT1 cell with no evidence for any chimeric PrP-res intermediate.Citation55 This latter observation parallels yeast “prion” amyloid studies that also show two different types of protein aggregates can exist in the same cell. In yeast cells these different cytoplasmic aggregates are mixed by asexual mating, a non-Mendelian genetic processCitation38 very different from infections of mammals with tissue-specific agent targeting and spread. On the other hand, if one ignores the “infectious” designation for yeast prions, both yeast and mammalian amyloids show a striking ability to self-propagate their individual conformations, possibly by using a similar network of cell pathways and mechanisms.
Interestingly, different types of yeast protein aggregates can promote or inhibit each other’s appearance and maintenance.Citation64 In contrast, an inhibiting TSE infectious agent, but not PrP-res aggregates, is required to prevent superinfection by a second agent.Citation55 This is relevant for human infections because vaccination of mice with low levels of the avirulent sCJD agent can completely prevent superinfection by the more virulent FU-CJD strain without the production of any detectable PrP-res (reviewed in ref. Citation11). The presence of different types of diagnostic amyloid in a cell has additional relevance for human disease. Amyloids not linked to a known infectious source can co-exist with those provoked by a TSE agent. Positive serial transmission of sCJD to hamsters was obtained from a brain with many classical AD β-amyloid plaques in addition to spongiform changes and PrP-res typical for sCJD (Manuelidis, unpublished data). Neither amyloid inhibited the other.
Stability, Evolution, and Selection of TSE Agent Strains
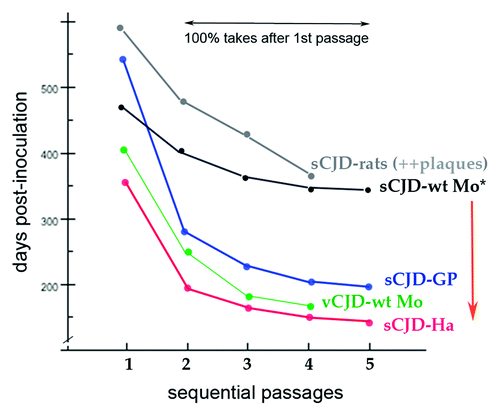
In general, TSE agents can show a remarkable lack of change, even when passaged through different species. Each agent strain, whether human or sheep derived, gives a characteristic incubation time and neuroanatomical lesion profile in a given species.Citation22 is a composite of published experimental data showing relevant incubation time features for human TSE isolates in rodents. First, in sequential ic passages, there is a progressive reduction in incubation time denoting an increased agent virulence for the recipient species. During this evolving agent adaptation, the PrP-res band pattern does not change. These fundamental and progressive agent changes are consistent with known viral adaptation through nucleic acid mutation, or selection from a mixture of related agent strains.Citation23 In contrast, a similar progressive agent change from p1 to p2 is not observed after ic inoculation of recPrP-res amyloid.Citation65 Second, the same agent in a different species changes its incubation (effective doubling) time, e.g., sCJD in rats, wt mice (wt-Mo), guinea pigs (GP), and hamsters (Ha) as shown in . This indicates species-specific defenses for a given agent, as discussed above. The graph of vCJD in wt mice also demonstrates a very different incubation pattern from sCJD, and additional distinctions are obvious with the Chandler (RML) and 22L scrapie strains in standard wt mice.Citation22 Thus each incubation time pattern is determined by the agent, and not by the invariant murine wt PrP, or indeed, by any the other identical background genes. Remarkably, the vCJD agent appears so dominant that it also gives an identical strong hypothalamic targeting pattern in mice, even after long-term replication in cows (BSE) or in humans (vCJD) with different PrP sequences.Citation22 Third, despite growth in a heterologous species, TSE agents can retain their ability to infect their donor species with their original phenotype, e.g., hamster sCJD transmitted to mice becomes very prolonged, yet even after serial mouse passages this sCJD agent still retains its ability to immediately replicate rapidly in hamsters (red arrow).Citation66 This indicates both the preservation of an invariant strain-specific feature, as well as a mutable feature of retained (hamster) adaptation. Again, PrP-res band patterns do not reflect these features.
Figure 4. Composite published experimental data for transmission of sCJD human brain to different species in sequential passages. Guinea pig (GP), hamster (Ha), murine (Mo), and rat sCJD plots contain original data from the first serial transmission of a human TSE agent to these small animalsCitation23,Citation35,Citation70,Citation71 as well as additional transmissions. All show a significant reduction in incubation time in serial passages. The sCJD agent from human and from hamster brain homogenates give the identical disease with the same very prolonged incubation time.Citation31 Even after serial passages in standard wt mice with a prolonged incubation time, the sCJD agent (*) retains and immediately expresses its virulent rapid incubation mutation acquired during its first hamster adaptation (red arrow) as previously documented.Citation66 The sCJD agent in rats induces many plaques and widespread disease,Citation23 unlike the limited lesions provoked in mice.Citation35,Citation36 Thus plaque deposition is a species-specific reaction to an agent, and not necessarily a consequence of a long incubation time as sometimes assumed. Even after serial rat passages FU-CJD also retains its virulence for murine cells.Citation39 Note that human adapted vCJD (as BSE) does not require a human PrP sequence to infect mice; it rapidly adapts to mice, and here exhibits a very different incubation pattern than sCJD. The vCJD agent (and BSE) also induce the same distinctive regional neuropathology in mice.Citation22
A low level of TSE agent nucleic acid mutation, with retention of only few selected variants seems most plausible, concepts first introduced with the seminal transmission experiments of Pattison, Dickinson and Kimberlin in scrapie, and the realization that these agents have an “independent genome” (reviewed in refs. Citation22, Citation37, and Citation67). Cloning of a single mutant agent by extreme end-point dilution was necessary to obtain a pure agent strain with negligible reversion. Such an invariant agent is exemplified by the cloned 263K hamster scrapie agent that became avirulent for mice. There are also scrapie strains that show more frequent change. Interestingly, one scrapie strain with markedly altered transmission properties has recently been selected in mice by modifying the glycosylation of PrP,Citation67 a finding consistent with agent particles tightly bound to, and influenced by host receptor PrP. Such binding can also induce additional host processes involved in agent selection. While the authors of this paper hypothesize multiple misfolded forms or “quasispecies” of infectious PrP, the resolution of these different PrP forms has not been evident. This new scrapie data instead argues most parsimoniously for a mutable agent genome. Furthermore, the long-established reversible switching between the 79A and 139A scrapie agent phenotypesCitation67 suggests that minor agent nucleic acid variants can persist, with each variant coming to the fore in its more favorable environment or species. In sum, there is both selective stability and mutability of TSE agents that mirrors viral behavior and the transfer of strain-specific information encoded by nucleic acid. Unlike with nucleic acids, no molecular model exists to support a comparable protein-based “information” transfer. Moreover, the multiple conformers of PrP res that are presumed to encode all the different scrapie and CJD agent strains propagated in wt mice remain hypothetical. Nor has any specific or consistent misfolding been identified that generally defines the infectious form of PrP. The multiple designations for all of the different capabilities of proposed PrP conformers can be mind-boggling, e.g., the PrP-res forms that must encode major strain classes such as PrPcjd, PrPsc, PrPcwd, PrPBSE, the PrP-d for the disease form, a TPrP for the neurotoxic form, and most recently, a subpopulation of an infectious protease sensitive PrP-sens form (reviewed in ref. Citation31). Perhaps the capacities of PrP are overstated?
The Structure of Infectious Particles
Whereas PrP-res fibrils and amyloid aggregates distribute over a huge size spectrum, TSE infectious agents have a homogeneous 120S size and behave as viral particles that separate from disaggregated PrP.Citation37 Infectious preparations also contain ultrastructural 25 nm electron dense particles that do not bind PrP antibodies, unlike PrP amyloid fibrils.Citation11 Cells infected with 22L scrapie or FU-CJD agents also produce intracellular arrays of comparable 25 nm virus-like particles that are not seen in uninfected controls, extending the reality of these particles first described many years ago in scrapie brains.Citation45 Moreover, when isolated infectious particles are disrupted, they release nucleic acids and protective nucleic acid binding proteins, and simultaneously lose 3 logs of infectivity (reviewed in refs. Citation11 and Citation37). This additional evidence points to a protected nucleic acid genome rather than the PrP-res that was not disrupted by this treatment. Given such findings, and the irrefutable evidence for mutable TSE strains, it is remarkable that there has been so little support for or interest in pursuing purification of nucleic acids from infectious particles. All of the many infectious preparations we have tested, including standard PrP-res fibrils made to seed test-tube amyloid conversions,Citation17 have yielded nucleic acids of >500 nt, yet no major TSE lab in the past 15 y seems to have tested any of their infectious tissue derived seed or co-factor material in quantities sufficient to detect an agent nucleic acid by the insensitive gel staining methods used.Citation21 These analyses also typically exclude molecules of >200 nt. Nor have preparations been tested by any specific molecular method such as PCR, to detect cytoplasmic nucleic acids such as retroviral sequences and protected mitochondrial DNA regions known to co-purify with infectivity.Citation21 Thus the common belief that TSE agents contain no nucleic acid appears to be based on the prion model, rather than on direct examination of highly infectious subcellular preparations. Because neither an infectious form of PrP, nor a causative agent-specific nucleic acid sequence has yet been defined, the nature of the infectious particle remains unknown.
Nucleic Acids in Infectious Preparations
There have been limited attempts to define a nucleic acid that is essential for infection. One TSE group reported that small RNAs extracted from standard 263K hamster PrP-res scrapie brain preparations was able to infect animals when delivered with (or protected by) recPrP.Citation68 This result deserves follow-up, despite the dismissive appended comments about this interesting contribution by two dominant prion proponents.Citation68 Although our major emphasis has been the transmission and characterization of human TSE strains, we have also looked at potential agent-specific nucleic acids, including circular DNAs that co-purify with infectious 25 nm particles. The relative stability of TSE agents, and biological propensities such as tumor formation, suggested a cytoplasmic circular DNA of 1–4 kb,Citation21 rather than an RNA virus like hepatitis C that is unstable, and rapidly produces millions of mutated and phenotypically distinct quasispecies.
Recent analyses of particle nucleic acids extracted from infectious cytoplasm have been facilitated by the development of new simplified stable TSE culture models, rapid infectivity assays for improved isolation of infectious particles, and application of faithful Phi 29 polymerase amplifications.Citation21 The robustness and fidelity of this last approach was validated, because nuclease protected full-length circular mitochondrial DNAs (mtDNA) of 16 kb were retrieved from infectious cytoplasmic fractions. MtDNA have been found by PCR in every infectious preparation examined, including standard brain derived PrP-res fibrils used to seed test-tube PrP conversions. It is likely that all cytoplasmic tissue extracts used to initiate PrP-res will also show mtDNA by PCR, as well as rodent retroviral sequences. While these are unlikely to define or encode TSE agent strains, they provide a good positive control for efficient nucleic acid extraction.
Circular Sphinx DNAs: Unsuspected Exchanges with the Metagenome?
Using multiple displacement amplification (MDA) strategies, several novel circular “Sphinx” DNA elements, compatible with the predicted TSE viral size, have now been isolated from infectious particles propagated in hamster brain (with 263K scrapie) and murine GT1 cells (with FU-CJD). Unlike mtDNA sequences that were species specific and abundant, the isolated Sphinx sequences were present at the low levels expected for a TSE agent in sucrose gradient purified particles. They were homologous by PCR regardless of species or agent strain. These two DNAs were not detectable in any of the enzymes, chemicals, or solutions used, verifying their origin in the particle preparations. Because the ORFs in these two Sphinx elements were also present by PCR, albeit at a much lower level, in uninfected parallel preparations, their role in TSE infection is indeterminate. However, their role can be evaluated using antisense strategies. It may also be informative to examine potential mutations in the uninfected homologs. In infectious particle preparations, a few additional protected circular DNAs have also been identified, and these deserve further study given the independent strain, replication dynamics, and environmental origin of TSEs. These types of sequences alternatively could be involved in the recruitment of non-infectious amyloids, not only in TSEs, but in other neurodegenerative diseases.
Remarkably, both the circular 1.8 kb and a 2.4 kb Sphinx elements contain open reading frame (ORF) homologies with portions of the large circular ~12 kb plasmids of Acinetobacter, but the parent bacterium was not apparent in the infectious extracts. These two Sphinx elements also contain non-coding intervening sequences that are not in the database, implicating an evolutionary difference from the standard bacterial plasmids. Acinetobacter are widespread in the environment, including soil and water, are antibiotic-resistant, and are commensal infections that are not typically pathogenic. Thus they raise two fundamentally new concepts. First, that low pathogenicity environmental DNAs may reside in a quiescent or latent form in mammals until the organism is stressed. Second, Sphinx elements may represent the tip of the iceberg of unexpected metagenomic exchanges with mammals: a rather exciting new direction with broad ramifications.
Summary
The structure and strain-determining molecular components of infectious TSE particles remains unknown. An enormous variety of evidence contradicts the conclusion that TSE infectious agents are made of host PrP without nucleic acid or an environmental component. First, different agent strains give the same PrP-res pattern, and conversely, samples with very different PrP-res patterns can propagate the identical infectious agent. Thus observed PrP conformers do not encode or predict strains. Second, all forms of detectable PrP can be destroyed with no loss of infectivity. PrP has also been undetectable in cells with high infectious titers. Third, the host recognizes and mounts early innate immune defenses against individual TSE agents and this determines latency and incubation time. Fourth, PrP-res amyloid appears as part of a late defense mechanism against a foreign agent, and TSE agent replication demonstrates a vastly different trajectory than the conversion kinetics of PrP-res (the “infectious” form). Fifth, PrP acts as a required host receptor for agent survival. In this capacity it can protect the agent while allowing the host to survive for years. Sixth, as other GPI proteins, PrP facilitates cell-to-cell transfer, and can be used by several infectious agents, not just TSEs. Seventh, as in TSE infections, other viruses can induce misfolded protein “prion” aggregates as part of an antiviral innate immune response. Eighth, the ability to fold into an amyloid state is a property shared by many proteins and peptides.
Amyloid aggregation is a process that is highly conserved in evolution and it does not require infection. In yeast, such conversions are involved in a wide range of functions, including transcriptional and translational regulation.Citation38 Dense resistant bacterial biofilms function in socialization and protectionCitation69 and can be considered together with mammalian amyloids, including PrP and AD plaques that contain many other elements.Citation23 Infection poses additional unexplored opportunities. The discovery of bacterial plasmid-like elements associated with TSE infectious particles raises the issue of unsuspected exchanges between mammals and bacterial viral elements in disease. Some of these viral elements may be the root cause of TSEs as well as a diversity of chronic conditions. Alternatively, such elements may promote the aggregation of fibrillar proteins in neurodegenerations of unknown etiology. Metagenomic elements, along with environmental toxins, surely deserve broader attention as causal factors in AD and other late-onset diseases.
Acknowledgments
This work was supported by NIH grant NINDS grant R01 012674
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998; 279:827 - 34; http://dx.doi.org/10.1126/science.279.5352.827; PMID: 9452375
- Jeffrey M, McGovern G, Chambers EV, King D, González L, Manson JC, et al. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathol 2012; 22:58 - 66; http://dx.doi.org/10.1111/j.1750-3639.2011.00508.x; PMID: 21645162
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, et al. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci U S A 2000; 97:5574 - 9; http://dx.doi.org/10.1073/pnas.97.10.5574; PMID: 10805813
- Cortes CJ, Qin K, Cook J, Solanki A, Mastrianni JA. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Sträussler-Scheinker disease. J Neurosci 2012; 32:12396 - 405; http://dx.doi.org/10.1523/JNEUROSCI.6189-11.2012; PMID: 22956830
- Ridley RM, Baker HF, Windle CP, Cummings RM. Very long term studies of the seeding of beta-amyloidosis in primates. J Neural Transm 2006; 113:1243 - 51; http://dx.doi.org/10.1007/s00702-005-0385-2; PMID: 16362635
- Neve RL, Robakis NK. Alzheimer’s disease: a re-examination of the amyloid hypothesis. Trends Neurosci 1998; 21:15 - 9; http://dx.doi.org/10.1016/S0166-2236(97)01168-5; PMID: 9464679
- Karpuj MV, Giles K, Gelibter-Niv S, Scott MR, Lingappa VR, Szoka FC, et al. Phosphorothioate oligonucleotides reduce PrP levels and prion infectivity in cultured cells. Mol Med 2007; 13:190 - 8; http://dx.doi.org/10.2119/2006-00073.Karpuj; PMID: 17592554
- Saunders SE, Bartz JC, Shikiya RA. Protein misfolding cyclic amplification of prions. J Vis Exp 2012; http://dx.doi.org/10.3791/4075; PMID: 23168797
- Makarava N, Savtchenko R, Baskakov IV. Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J Biol Chem 2013; 288:33 - 41; http://dx.doi.org/10.1074/jbc.M112.419531; PMID: 23168413
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 2012; 109:E1938 - 46; http://dx.doi.org/10.1073/pnas.1206999109; PMID: 22711839
- Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem 2007; 100:897 - 915; http://dx.doi.org/10.1002/jcb.21090; PMID: 17044041
- González L, Thorne L, Jeffrey M, Martin S, Spiropoulos J, Beck KE, et al. Infectious titres of sheep scrapie and bovine spongiform encephalopathy agents cannot be accurately predicted from quantitative laboratory test results. J Gen Virol 2012; 93:2518 - 27; http://dx.doi.org/10.1099/vir.0.045849-0; PMID: 22915693
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90; http://dx.doi.org/10.1016/0092-8674(95)90236-8; PMID: 7553876
- Saá P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmézas CI. Strain-specific role of RNAs in prion replication. J Virol 2012; 86:10494 - 504; http://dx.doi.org/10.1128/JVI.01286-12; PMID: 22811520
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, et al. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog 2011; 7:e1002370; http://dx.doi.org/10.1371/journal.ppat.1002370; PMID: 22114554
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 7; http://dx.doi.org/10.1074/jbc.C110.113464; PMID: 20304915
- Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/10.1126/science.1183748; PMID: 20110469
- Wang F, Zhang Z, Wang X, Li J, Zha L, Yuan CG, et al. Genetic informational RNA is not required for recombinant prion infectivity. J Virol 2012; 86:1874 - 6; http://dx.doi.org/10.1128/JVI.06216-11; PMID: 22090130
- Jones M, Peden AH, Head MW, Ironside JW. The application of in vitro cell-free conversion systems to human prion diseases. Acta Neuropathol 2011; 121:135 - 43; http://dx.doi.org/10.1007/s00401-010-0708-8; PMID: 20535485
- Manuelidis L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 2011; 17:131 - 45; http://dx.doi.org/10.1007/s13365-010-0007-0; PMID: 21165784
- Manuelidis L, Liu Y, Mullins B. Strain-specific viral properties of variant Creutzfeldt-Jakob disease (vCJD) are encoded by the agent and not by host prion protein. J Cell Biochem 2009; 106:220 - 31; http://dx.doi.org/10.1002/jcb.21988; PMID: 19097123
- Manuelidis L, Fritch W, Xi YG. Evolution of a strain of CJD that induces BSE-like plaques. Science 1997; 277:94 - 8; http://dx.doi.org/10.1126/science.277.5322.94; PMID: 9204907
- Seidel B, Thomzig A, Buschmann A, Groschup MH, Peters R, Beekes M, et al. Scrapie Agent (Strain 263K) can transmit disease via the oral route after persistence in soil over years. PLoS One 2007; 2:e435; http://dx.doi.org/10.1371/journal.pone.0000435; PMID: 17502917
- Georgsson G, Sigurdarson S, Brown P. Infectious agent of sheep scrapie may persist in the environment for at least 16 years. J Gen Virol 2006; 87:3737 - 40; http://dx.doi.org/10.1099/vir.0.82011-0; PMID: 17098992
- Shlomchik MJ, Radebold K, Duclos N, Manuelidis L. Neuroinvasion by a Creutzfeldt-Jakob disease agent in the absence of B cells and follicular dendritic cells. Proc Natl Acad Sci U S A 2001; 98:9289 - 94; http://dx.doi.org/10.1073/pnas.161055198; PMID: 11470899
- Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science 1977; 197:943 - 60; http://dx.doi.org/10.1126/science.142303; PMID: 142303
- Prusiner S. The Nobel Lecture: Prions. Nobelprizeorg 1997.
- Jeffrey M, González L, Espenes A, Press CM, Martin S, Chaplin M, et al. Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 2006; 209:4 - 14; http://dx.doi.org/10.1002/path.1962; PMID: 16575799
- Scherbel C, Pichner R, Groschup MH, Mueller-Hellwig S, Scherer S, Dietrich R, et al. Infectivity of scrapie prion protein (PrPSc) following in vitro digestion with bovine gastrointestinal microbiota. Zoonoses Public Health 2007; 54:185 - 90; http://dx.doi.org/10.1111/j.1863-2378.2007.01040.x; PMID: 17542960
- Miyazawa K, Emmerling K, Manuelidis L. High CJD infectivity remains after prion protein is destroyed. J Cell Biochem 2011; 112:3630 - 7; http://dx.doi.org/10.1002/jcb.23286; PMID: 21793041
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 1998; 5:R245 - 9; http://dx.doi.org/10.1016/S1074-5521(98)90108-9; PMID: 9818143
- Miyazawa K, Emmerling K, Manuelidis L. Replication and spread of CJD, kuru and scrapie agents in vivo and in cell culture. Virulence 2011; 2:188 - 99; http://dx.doi.org/10.4161/viru.2.3.15880; PMID: 21527829
- Manuelidis L, Fritch W. Infectivity and host responses in Creutzfeldt-Jakob disease. Virology 1996; 216:46 - 59; http://dx.doi.org/10.1006/viro.1996.0033; PMID: 8615006
- Manuelidis EE, Gorgacz EJ, Manuelidis L. Transmission of Creutzfeldt-Jakob disease with scrapie-like syndromes to mice. Nature 1978; 271:778 - 9; http://dx.doi.org/10.1038/271778a0; PMID: 342977
- Manuelidis L, Chakrabarty T, Miyazawa K, Nduom NA, Emmerling K. The kuru infectious agent is a unique geographic isolate distinct from Creutzfeldt-Jakob disease and scrapie agents. Proc Natl Acad Sci U S A 2009; 106:13529 - 34; http://dx.doi.org/10.1073/pnas.0905825106; PMID: 19633190
- Manuelidis L. Transmissible encephalopathies: speculations and realities. Viral Immunol 2003; 16:123 - 39; http://dx.doi.org/10.1089/088282403322017875; PMID: 12828865
- Li L, Kowal AS. Environmental regulation of prions in yeast. PLoS Pathog 2012; 8:e1002973; http://dx.doi.org/10.1371/journal.ppat.1002973; PMID: 23166488
- Miyazawa K, Kipkorir T, Tittman S, Manuelidis L. Continuous production of prions after infectious particles are eliminated: implications for Alzheimer’s disease. PLoS One 2012; 7:e35471; http://dx.doi.org/10.1371/journal.pone.0035471; PMID: 22509412
- Lu ZY, Baker CA, Manuelidis L. New molecular markers of early and progressive CJD brain infection. J Cell Biochem 2004; 93:644 - 52; http://dx.doi.org/10.1002/jcb.20220; PMID: 15660413
- Di Bari MA, Nonno R, Castilla J, D’Agostino C, Pirisinu L, Riccardi G, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 2013; 9:e1003219; http://dx.doi.org/10.1371/journal.ppat.1003219; PMID: 23505374
- Essbauer SS, Krautkrämer E, Herzog S, Pfeffer M. A new permanent cell line derived from the bank vole (Myodes glareolus) as cell culture model for zoonotic viruses. Virol J 2011; 8:339; http://dx.doi.org/10.1186/1743-422X-8-339; PMID: 21729307
- Schwartz K, Boles BR. Microbial amyloids - functions and interactions within the host. Curr Opin Microbiol 2013; 16:93 - 9; http://dx.doi.org/10.1016/j.mib.2012.12.001; PMID: 23313395
- Thellung S, Florio T, Villa V, Corsaro A, Arena S, Amico C, et al. Apoptotic cell death and impairment of L-type voltage-sensitive calcium channel activity in rat cerebellar granule cells treated with the prion protein fragment 106-126. Neurobiol Dis 2000; 7:299 - 309; http://dx.doi.org/10.1006/nbdi.2000.0301; PMID: 10964602
- Manuelidis L, Yu Z-X, Barquero N, Mullins B. Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles. Proc Natl Acad Sci U S A 2007; 104:1965 - 70; http://dx.doi.org/10.1073/pnas.0610999104; PMID: 17267596
- Zhou M, Ottenberg G, Sferrazza GF, Lasmézas CI. Highly neurotoxic monomeric α-helical prion protein. Proc Natl Acad Sci U S A 2012; 109:3113 - 8; http://dx.doi.org/10.1073/pnas.1118090109; PMID: 22323583
- Manuelidis L. Beneath the emperor's clothes: the body of data in scrapie and CJD. Ann Inst Pasteur (Paris) 1997; 8:311 - 26; http://dx.doi.org/10.1016/S0924-4204(97)86597-3
- Kitamoto T, Muramoto T, Mohri S, Doh-Ura K, Tateishi J. Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. J Virol 1991; 65:6292 - 5; PMID: 1681118
- Klein MA, Frigg R, Flechsig E, Raeber AJ, Kalinke U, Bluethmann H, et al. A crucial role for B cells in neuroinvasive scrapie. Nature 1997; 390:687 - 90; PMID: 9414161
- Manuelidis L, Zaitsev I, Koni P, Lu Z-Y, Flavell RA, Fritch W. Follicular dendritic cells and dissemination of Creutzfeldt-Jakob disease. J Virol 2000; 74:8614 - 22; http://dx.doi.org/10.1128/JVI.74.18.8614-8622.2000; PMID: 10954563
- Baker CA, Manuelidis L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A 2003; 100:675 - 9; http://dx.doi.org/10.1073/pnas.0237313100; PMID: 12525699
- Bradford BM, Mabbott NA. Prion disease and the innate immune system. Viruses 2012; 4:3389 - 419; http://dx.doi.org/10.3390/v4123389; PMID: 23342365
- Castro-Seoane R, Hummerich H, Sweeting T, Tattum MH, Linehan JM, Fernandez de Marco M, et al. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog 2012; 8:e1002538; http://dx.doi.org/10.1371/journal.ppat.1002538; PMID: 22359509
- Arjona A, Simarro L, Islinger F, Nishida N, Manuelidis L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc Natl Acad Sci U S A 2004; 101:8768 - 73; http://dx.doi.org/10.1073/pnas.0400158101; PMID: 15161970
- Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 2005; 310:493 - 6; http://dx.doi.org/10.1126/science.1118155; PMID: 16239476
- Liu Y, Sun R, Chakrabarty T, Manuelidis L. A rapid accurate culture assay for infectivity in Transmissible Encephalopathies. J Neurovirol 2008; 14:352 - 61; http://dx.doi.org/10.1080/13550280802105283; PMID: 18989813
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255 - 64; PMID: 8635458
- Emerman AB, Zhang ZR, Chakrabarti O, Hegde RS. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol Biol Cell 2010; 21:4325 - 37; http://dx.doi.org/10.1091/mbc.E10-09-0742; PMID: 20980618
- Sklaviadis T, Manuelidis L, Manuelidis EE. Characterization of major peptides in Creutzfeldt-Jakob disease and scrapie. Proc Natl Acad Sci U S A 1986; 83:6146 - 50; http://dx.doi.org/10.1073/pnas.83.16.6146; PMID: 3090551
- Caruso P, Burla R, Piersanti S, Cherubini G, Remoli C, Martina Y, et al. Prion expression is activated by Adenovirus 5 infection and affects the adenoviral cycle in human cells. Virology 2009; 385:343 - 50; http://dx.doi.org/10.1016/j.virol.2008.12.005; PMID: 19138779
- Nakato G, Hase K, Suzuki M, Kimura M, Ato M, Hanazato M, et al. Cutting Edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J Immunol 2012; 189:1540 - 4; http://dx.doi.org/10.4049/jimmunol.1103332; PMID: 22772447
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011; 146:448 - 61; http://dx.doi.org/10.1016/j.cell.2011.06.041; PMID: 21782231
- Ye J, Maniatis T. A prion-like trigger of antiviral signaling. Cell 2011; 146:348 - 50; http://dx.doi.org/10.1016/j.cell.2011.07.018; PMID: 21816270
- Derkatch IL, Liebman SW. Prion-prion interactions. Prion 2007; 1:161 - 9; http://dx.doi.org/10.4161/pri.1.3.4837; PMID: 19164893
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 87; http://dx.doi.org/10.1007/s00401-009-0633-x; PMID: 20052481
- Manuelidis L, Murdoch G, Manuelidis EE. Potential involvement of retroviral elements in human dementias. Ciba Found Symp 1988; 135:117 - 34; PMID: 3044706
- Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P, et al. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 2013; 32:756 - 69; http://dx.doi.org/10.1038/emboj.2013.6; PMID: 23395905
- Simoneau S, Ruchoux M, Vignier N, Lebon P, Freire S, Comoy E, et al. Small critical RNAs in the scrapie agent. Nat Precedings 2009; 3344
- Pathak DT, Wei X, Bucuvalas A, Haft DH, Gerloff DL, Wall D. Cell contact-dependent outer membrane exchange in myxobacteria: genetic determinants and mechanism. PLoS Genet 2012; 8:e1002626; http://dx.doi.org/10.1371/journal.pgen.1002626; PMID: 22511878
- Manuelidis EE, Kim J, Angelo JN, Manuelidis L. Serial propagation of Creutzfeldt-Jakob disease in guinea pigs. Proc Natl Acad Sci U S A 1976; 73:223 - 7; http://dx.doi.org/10.1073/pnas.73.1.223; PMID: 1108016
- Manuelidis EE, Gorgacz EJ, Manuelidis L. Interspecies transmission of Creutzfeldt-Jakob disease to Syrian hamsters with reference to clinical syndromes and strains of agent. Proc Natl Acad Sci U S A 1978; 75:3432 - 6; http://dx.doi.org/10.1073/pnas.75.7.3432; PMID: 356055