Abstract
In the process of host–pathogen interactions, bacterial pathogens always employ some special genes, e.g., virulence factors (VFs) to interact with host and cause damage or diseases to host. A number of VFs have been identified in bacterial pathogens that confer upon bacterial pathogens the ability to cause various types of damage or diseases. However, it has been clarified that some of the identified VFs are also encoded in the genomes of nonpathogenic bacteria, and this finding gives rise to considerable controversy about the definition of virulence factor.
Here 1988 virulence factors of 51 sequenced pathogenic bacterial genomes from the virulence factor database (VFDB) were collected, and an orthologous comparison to a non-pathogenic bacteria protein database was conducted using the reciprocal-best-BLAST-hits approach. Six hundred and twenty pathogen-specific VFs and 1368 common VFs (present in both pathogens and nonpathogens) were identified, which account for 31.19% and 68.81% of the total VFs, respectively. The distribution of pathogen-specific VFs and common VFs in pathogenicity islands (PAIs) was systematically investigated, and pathogen-specific VFs were more likely to be located in PAIs than common VFs. The function of the two classes of VFs were also analyzed and compared in depth. Our results indicated that most but not all T3SS proteins are pathogen-specific. T3SS effector proteins tended to be distributed in pathogen-specific VFs, whereas T3SS translocation proteins, apparatus proteins, and chaperones were inclined to be distributed in common VFs. We also observed that exotoxins were located in both pathogen-specific and common VFs. In addition, the architecture of the two classes of VFs was compared, and the results indicated that common VFs had a higher domain number and lower domain coverage value, revealed that common VFs tend to be more complex and less compact proteins.
Introduction
Bacterial pathogens often cause various epidemic diseases, which threaten human health and lives.Citation1,Citation2 A growing number of researchers have conducted related research and made great achievements in the field of bacterial pathogens. Bacterial pathogens are parasitic organisms with specialized adaptations that allow them to interact with hosts. In the process of host–pathogen interactions, bacterial pathogens utilize a number of mechanisms, including adherence, invasion, antiphagocytosis, protein, or toxin secretion etc.Citation3 That is to say that bacterial pathogens employ a number of special factors or so-called “virulence factors” during these interactions, so they can grow, reproduce, spread, and cause host damage ranging from mild annoyance to death.
In the past two decades, the term “virulence factor” (VF) has been defined as a factor that was produced by a pathogen and causes diseases.Citation4,Citation5 A number of virulence factors (VFs) based on different types of mechanisms employed by bacterial pathogens were identified using molecular biological techniques. However, as the number and diversity of completed bacterial genomes began to increase, some identified VFs were also discovered to be encoded in the genomes of non-pathogenic bacteria or commensal bacteria.Citation6,Citation7 Moreover, some biological experiments also discovered this phenomenon, e.g., with the help of microarray analyses, many of the known virulence factors in pathogenic Escherichia coli and Neisseria spp. were identified as being present in the closely-related commensal Escherichia coli and non-pathogenic Neisseria lactamica.Citation8,Citation9
Now two views about the definition of virulence factor have emerged and their difference concerns whether the virulence factor must be absent in non-pathogenic bacteria. The first definition was defined using comparative genomics and considered that any virulence factors that should not be found in non-pathogens, whereas the second definition was defined using genetic techniques and models of infection.Citation10,Citation11 Confronted with this unresolved debate, some researchers have proposed the hypothesis of host interactions, namely that many so-called “virulence genes” are most likely involved in more general interactions between the microorganism and the host or the environment.Citation7 Generally, the host–bacteria interaction can be classified as symbiotic, commensal, or pathogenic interactions.Citation12,Citation13 These divisions should be viewed as a “homeostasis” and this kind of equilibrium is constantly evolving. According to the ecological and evolutionary view of bacterial pathogenomics, pathogenic bacteria, symbiotic, and commensal bacteria often share their habitats with bacteriophages and other bacteria. In this mixed ecology, almost all of these bacteria also utilize similar strategies and molecular systems to interact with eukaryotic hosts and their maintained homeostasis is usually disrupted by the mechanisms of horizontal gene transfer or gene loss,Citation6,Citation11 for example, pathogenic strains of Enterococcus faecium have evolved from a commensal species via horizontal gene transfer.Citation14 Therefore, it is not surprising that genes encoding “virulence factors” are present in both pathogenic and nonpathogenic bacteria.
Sui et al. performed the first systematic analysis across diverse genera, and found that virulence factors (VFs) are disproportionately associated with genomic islands (GIs, clusters of genes in a prokaryotic genome of probable horizontal origin). Both pathogen-associated VFs and common VFs (having homologs in both pathogens and non-pathogens) were identified by performing a sequence similarity and all were associated with GIs.Citation15 In the current paper, the method of ortholog prediction was adopted between pathogenic and non-pathogenic bacteria.Citation16,Citation17 1988 virulence factors collected from the virulence factor database (VFDB)Citation18 and their orthologous proteins in a nonpathogenic bacteria protein database were identified by carrying out the reciprocal-best-BLAST-hits (RBH) approach.Citation19 Each VF was identified as pathogen-specific (there is no orthologous protein in non-pathogenic bacteria), or common (there is one or more orthologous proteins in non-pathogenic bacteria). The distribution of the identified pathogen-specific and common VFs in pathogenicity islands, functional categories, protein architecture were systemically investigated. We believed that this research would be instructive for us to study the virulence factors in bacterial pathogens and to elucidate the pathogenic mechanism and the evolution of pathogenic bacteria in the future.
Results
Identification of pathogen-specific VFs and common VFs
The 1988 VFs and the proteins from the non-pathogenic bacteria protein database were compared by conducting the reciprocal-best-BLAST-hits. If a VF had one or more orthologous proteins in the non-pathogenic bacteria protein database, the VF was identified as a common VF that was not only found in bacterial pathogens, but also in bacterial non-pathogens. Otherwise, the VF was identified as a pathogen-specific VF that was only present in bacterial pathogen. The cutoff e-value was routinely set to 1e−7, and protein pairs with lower e-values were considered orthologous pairs.
In the 1988 VFs, we identified 620 pathogen-specific VFs and 1368 common VFs, which account for 31.19% and 68.81% of the total VFs, respectively. Moreover, among the 1368 common VFs, there were 1239 VFs (90.57%) that had two or more orthologous proteins in the non-pathogenic bacterial protein database, which ensured the correctness of most of the ortholog predictions.
Pathogenicity islands (PAIs) contained a higher proportion of pathogen-specific VFs
It is well known that pathogenicity islands (PAIs), which are involved in virulence and most likely acquired from horizontal gene transfer (HGT),Citation20,Citation21 belong to a subclass of genomic islands. In the past, many novel virulence factors of pathogenic bacteria, e.g., adhesins, invasins, toxins, secretion systems (especially the type III secretion system [T3SS] and type IV secretion system [T4SS]), iron uptake systems, and others, have been identified in pathogenicity islands,Citation20-Citation22 and these findings suggest that horizontal gene transfer, especially the horizontal gene transfer mediated by pathogenicity islands, has played key roles in the evolution of bacterial pathogens.Citation20,Citation21,Citation23
In order to investigate the distribution of pathogen-specific VFs and common VFs in pathogenicity islands, we tabulated the number of pathogen-specific VFs in PAIs, outside of PAIs, and number of common VFs in PAIs and outside of PAIs in a 2 × 2 contingency table according to the VFDB classification and used a Chi-square test with Yates correction for continuity correction.
The statistical result showed that pathogen-specific VFs were more likely to be located in the PAIs, and common VFs were more likely to be located outside of PAIs (P < 2.20e−16; ), which implied that pathogen-specific VFs might be acquired by horizontal gene transfer, e.g., the horizontal gene transfer mediated by pathogenicity islands. The results also imply that pathogen-specific VFs might be more closely connected to pathogenicity and might play key roles in the evolution of bacterial virulence.
Table 1. Distributions of the VFs from the VFDB inside vs. outside of PAIs
The distribution of pathogen-specific VFs and common VFs in each functional category was different
According to its classification scheme, the VFDB mainly contained 49 different virulence functional categories, e.g., flagella, capsule, toxin, etc. (see ). In order to investigate the distribution of pathogen-specific VFs and common VFs in each functional category, we tabulated the number of pathogen-specific VFs and the number of common VFs in each functional category in a 49 × 2 contingency table according to the VFDB classification and then used a Chi-square test with Yates correction with corrections for multiple testing to acquire the statistical results.
Table 2. The distribution of pathogen-specific and common VFs in each functional category according to the VFDB classification
In our statistical results (see ), proteins belonging to the exotoxin, T4SS, T3SS unclassified protein, T3SS effector protein, and PAI, which were all directly associated with the form of virulence, were more inclined to be distributed in the pathogen-specific VFs, and this suggested that pathogen-specific VFs might be closely connected with the form of virulence. Conversely, flagella, capsule, endotoxins, iron uptake protein, regulation protein, the type VI secretion system (T6SS), and type II secretion system (T2SS) that were apt to be involved in host interactions, were more inclined to be common VFs, and this finding indicated that common VFs might be involved in host interaction. In addition,, there were some functional categories whose distribution in both pathogen-specific VFs and common VFs did not have statistical significance, and we observed that these categories contained some antagonistic proteins, protease, immune evasion, general secretion system, bacterial adherence, cell structure proteins and invasion, etc. We would discuss each functional category included among pathogen-specific VFs and common VFs in more details below.
Exotoxins were included in both among the pathogen-specific and common VFs
Many bacterial pathogens can synthesize exotoxins, which are toxic to host cells and always play a central role in the pathogenesis of microbial diseases.Citation24 In our statistical results, most of the exotoxins were specific to pathogens and had no orthologous proteins in non-pathogenic bacteria (P = 1.49e−14, see ). However, there were still some exotoxins among the common VFs.
According to the VFDB classification, exotoxins can be further classified into three functional categories: membrane-acting toxins, membrane damaging toxins, and intracellular toxins (see ). Membrane-acting toxins bind to a receptor on the cell surface and stimulate intracellular signaling pathways, membrane damaging toxins exhibit hemolysin or cytolysin activity in vitro, and intracellular toxins possess enzymatic activity and affect internal cellular bio-mechanisms or inhibit protein synthesis.Citation25 From , we found pathogen-specific exotoxins mainly including membrane-acting superantigen and enterotoxin, membrane-damaging pore-forming toxin except for the RTX toxin (repeat in structural toxin), and most of intracellular toxin (e.g., N-glycosidase, neurotoxin, adenylate cyclase, ADP-ribosyltransferase toxins, etc.).
Table 3. Proportions of pathogen-specific exotoxins from the VFDB according to the VFDB classification
However, some membrane-damaging toxin, for example, pore-forming RTX toxin and membrane-damaging phospholipases C were also included in the common VFs (see ).
The prototype of RTX toxins was the Escherichia coli α-hemolysin(HlyA), which was the best-characterized RTX protein secreted by a type I secretion system.Citation26 The synthesis, activation and secretion of E. coli HlyA were controlled by the hlyCABD operon, which included hlyC, hlyA, hlyB, and hlyD genes. In hlyCABD operon, the hlyC was a fatty acid acyltransferase, which was responsible for acylation of Pro-HylA and was independent of the secretion of HlyA, the hlyA encoded a structural toxin, whereas the hlyB and hlyD encoded a type I secretion system and they were components of the HlyA secretion apparatus.Citation26 In our research, 14 RTX toxin genes that were contained in our data were all identified as common VFs, and this was consistent with the studies of the RTX toxin in nonpathogens.Citation27,Citation28 Among the 14 genes, there were four structural toxin genes that had same function as hlyA and the rest had the same functions as hlyC, hlyB, or hlyD. Previous work had shown that the hlyA was an α-hemolysin and characterized by a domain consisting of tandemly arranged glycine-rich nonameric repeats near the protein C terminus, which was responsible for Ca2+ binding.Citation29 However, the membrane insertion of α-hemolysin was independent from membrane lysis and the calcium binding was essential for toxin activity.Citation30 So the RTX toxin was not directly involved in virulence and their roles in bacterial non-pathogens need to be studied further.
As for the membrane-damaging phospholipase C toxins, they were synthesized by many widespread bacteria and possessed an enzymatic activity. So far, it had not been substantiated that all phospholipases C would have lethal properties.Citation31 In addition, more and more research indicated that the measurement of the cytolytic potential or lethality of phospholipases C could not accurately indicate their roles in the pathogenesis of disease. Through the investigation of the genetic diversity of the four Mycobacterium tuberculosis phospholipase C-encoding genes (plcA, plcB, plcC, and plcD), it was suggested that the plcD region was significantly associated with the pathogenesis of the tuberculosis and that the plcD gene might play a more important role in the pathogenesis of thoracic tuberculosis.Citation32
Through the above analysis, we found most of exotoxins that were directly involved in virulence were pathogen-specific, and this indicated that the pathogen-specific exotoxins were essential to bacterial virulence and played a central role in pathogenicity. Whereas, as for the roles of the common “exotoxins” in pathogenicity (e.g., RTX toxin, phospholipases C toxin, etc.), some were associated with the secretion of exotoxins and some were still in dispute on whether directly involved in virulence.
Type III secretion system proteins belonged to different classes between pathogen-specific and common VFs
Many gram-negative bacteria use type III secretion systems (T3SS) to secrete virulence factors into the cytosol of host cells. The gene clusters encoding T3SS are often located on virulence plasmids or in pathogenicity islands.Citation21,Citation23 Sui et al.Citation15 analyzed VFDB functional classes and demonstrated that type III and type IV secretion systems were pathogen-associated VFs and associated with GIs.
T3SS proteins are grouped into four classes: bacterial membrane apparatus proteins, translocon proteins, effector proteins, and type III chaperones. We noticed that not all classes of T3SS proteins were pathogen-specific. In fact, T3SSs have also been discovered in commensal and symbiotic bacteria.Citation6,Citation33
In our further result, in T3SS proteins, only effector proteins and T3SS unclassified proteins were included among the pathogen-specific VFs, and most of the T3SS translocation proteins, type III chaperone proteins and secretion apparatus proteins were not pathogen-specific and had many orthologous proteins in nonpathogenic bacteria in our ortholog’s prediction (see ). For the T3SS proteins, effector molecules are injected into eukaryotic host cells, and specifically interfere with the eukaryotic cells functions, resulting in the unbalance of host cells functions.Citation34
Some previous studies have indicated that the type III secretion systems were assembled from core components of the flagellar machine,Citation35 and this had resulted that T3SS translocation proteins and secretion apparatus proteins were not pathogen-specific. As for the chaperones in T3SSs, they had functions that are focused on protein folding and stress repair, and possessed nearly no virulence properties.
From the above analysis, T3SS effector proteins that were directly involved in virulence, were inclined to be distributed in pathogen-specific VFs, whereas the translocation proteins, secretion apparatus proteins and T3SS chaperones that assisted the secretion of effector proteins were inclined to be distributed in common VFs.
Most type IV secretion system effector proteins were pathogen-specific
Type IV secretion systems (T4SS) were versatile systems, which could mediate conjugal transfer between bacteria by a variety of bacteria. Like T3SS, T4SS are also used by bacterial pathogens to secrete “effectors” into host plant or animal cells,Citation36 and are found in pathogenicity islands mediated by horizontal gene transfer. For example, the T4SS in H. pylori is encoded by the cag PAI.Citation37 Up to now, T4SSs has been discovered and identified in some bacterial pathogens, such as Bordetella pertussis, Bartonella spp., Legionella pneumophila, Brucella spp., and Helicobacter pylori.Citation36 However, the components of T4SS are not as well identified as the components of T3SS and the identified components are mainly effector proteins.
In our result (see ), most of the T4SS-encoding genes tended to be distributed among the pathogen-specific VFs (P = 4.01e−10). However, many T4SS-encoding genes were included in the common VFs.
As we know, effector proteins often played a critical role in bacterial pathogenicity. To examine whether effector proteins secreted by T4SSs were included in our identified pathogen-specific VFs, we collected the identified effector proteins of four well-studied bacterial pathogens from related literature (see ). Some of the identified effector proteins were not listed as definitive effector proteins in VFDB database. As observed in , we noted that 83.87% of the effector proteins from four related bacterial pathogens were pathogen-specific. Because the archetypal T4SSs are bacterial conjugation machines, which are widespread in bacteria, it was not surprising to find that some components of T4SSs were not pathogen-specific.
Table 4. T4SSs of four well-studied pathogenic bacteria and their proportions of pathogen-specific effectors
Common VFs tended to be involved in general host interaction
During the process of bacterial evolution, vertical descent and duplication might be considered the primary events of genome evolution.Citation38 Orthologs and paralogs are two types of homologous sequences in genetics. Orthologs are commonly defined as genes that have evolved by vertical descent from a common ancestor and tend to perform the same function.
In our research, the VFs that had one or more orthologous proteins among non-pathogenic bacteria were identified as common VFs. Orthologs in different species were commonly defined as genes that had evolved by vertical descent from a common ancestor and tend to perform the same function.Citation38 In our results, the VFs that were in flagella, capsule, endotoxin, iron uptake, regulation, T6SS, and T2SS categories tended to be found among the common VFs (). These findings indicated that these VFs were universal in both pathogenic and non-pathogenic bacteria and tended to carry the same functions. In , we list the characteristics and functions of those functional categories that were more inclined to be found in common VFs, and found that common VFs were more likely to be involved in general host interaction.
Table 5. The characteristics and functions of each functional category that was more inclined to be found in common VFs
In order to verify the relationship between common VFs and general host interaction further, we collected 278 non-pathogenic bacterial strains and information on their habitats status. In the end, we obtained 65 host-associated and 213 non-host-associated nonpathogenic bacterial strains. According to our orthologs prediction, 1169 (85.45%) of 1368 common VFs had orthologous proteins in the “host-associated” non-pathogenic bacteria strains, and this finding suggested that most of common VFs were more inclined to be involved in general host interaction.
Domain architecture of VFs’ proteins
Domains are considered as the basic units of protein folding, evolution, and function.Citation39 Decomposing each protein into modular domains is a basic prerequisite for the accurate functional classification of biological molecules. The function of a protein is determined by its structure, which is mostly embodied in its domain architecture. In order to investigate the protein domain characteristics of pathogen-specific VFs and common VFs, we inspected the DN and calculated the DC for each VF.
Our results showed that common VFs had a larger DN than pathogen-specific VFs on average (see ), and the proportions of multi-domain proteins in common VFs was higher than that of in pathogen-specific VFs (χ-squared = 38.1187, df = 1, P = 6.657e−10, see ). This indicated that common VFs were more likely to be multi domain protein than pathogen-specific VFs, and vice versa. Both the DC of common VFs and that of pathogen-specific VFs varied in a large range but with slightly different medians. The average DC value for the two types of VFs was 0.66 and 0.70, respectively (see ).
Figure 1. The proportion of multi-domain proteins among the VF proteins.
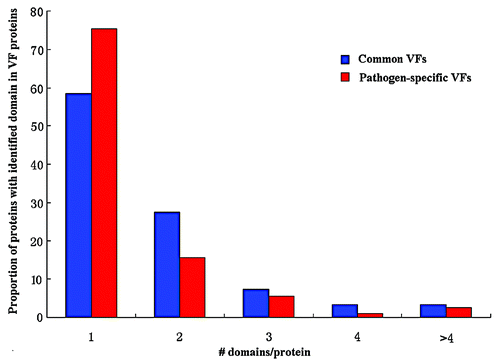
Table 6. Distributions of the VFs from the VFDB in single-domain vs. in multi-domain proteins
Figure 2. The DC of common and pathogen-specific VF proteins.
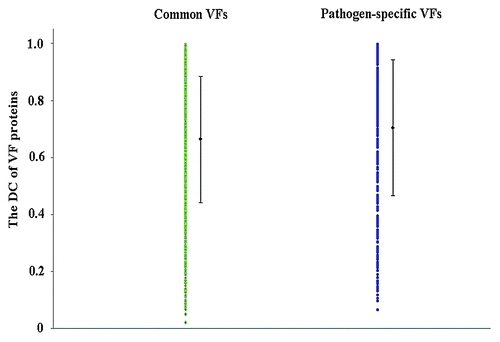
DN can be regarded as an indicator of the complexity of a protein structure. The larger the DN, the more complex the protein is. DC can be used as a parameter representing the structural compactness of a protein. The smaller the DC, the looser the protein is. From our results, it can be inferred that pathogen-specific VF proteins tend to be simpler structures than common VF proteins. The fact that the common VFs had a higher DN than pathogen-specific VFs but with a slightly lower DC values indicates that common VFs tend to be more complex and less compact proteins than pathogen-specific VFs.
Discussion
Pathogen-specific VFs are the divergence that distinguished pathogens from non-pathogens and are exclusively found in pathogens. Conversely, common VFs are shared by the pathogenic and non-pathogenic bacteria and are involved in general host interaction, and survival or maintenance of basic functions in the host. For example, the surface organelles of bacteria, flagella, and fimbriae are primarily the structural components of the organisms.Citation38 Within the host, using the motility and chemotaxis provided by flagella, bacteria can move to their destinations or their target tissues.
At the same time, in order to avoid phagocytosis, bacteria have evolved surface components that prevent the attachment and engulfment of macrophages and other host cellular immune responses. Gram-positive bacteria are naturally surrounded by a thick cell wall (capsule), whereas gram-negative bacterial lipopolysaccharide (LPS) or “endotoxin” can protect against complement-mediated lysis. However, bacterial capsule and lipopolysaccharide are primarily cell wall structural components. In addition to the common capsule and lipopolysaccharides, there were several antigens that are pathogen-specific among bacterial pathogens and can inhibit adsorption, such as streptococcal protein M and staphylococcal protein A.Citation40,Citation41
In addition, bacteria usually use adhesins to adhere to the specific tissues or host cells. Bacterial adhesins can be divided into two major types: pili (fimbriae) and nonpilus adhesins (afimbrial adhesins). Fimbriae are mainly structural components. In order to colonize the human gut mucosa, EHEC O157:H7 and the commensal E. coli K12 use the common pilus adherence factor: E. coli common pilus (ECP) for epithelial cell colonization, as proven in previous experiments.Citation42 With respect to afimbrial adhesins, many pathogenic bacteria, e.g., Staphylococcus aureus and Streptococcus pyogenes., share the same ability to adhere to distinct components of the extracellular matrix (ECM). However, the same specific binding occurs between lactobacilli and components of the extracellular matrix (ECM), including collagen and fibronectin.Citation43
After entering a host cell, the host environment is continuously changing and may not always be ideal for bacterial survival. In order to adapt to the host surroundings, bacteria have to evolve some strategies. For example: the iron uptake factors, e.g., transferrins, hemoglobin protease, hemolysins, and siderophores, are used for iron uptake and heme utilization by bacteria and are indispensable for survival in the host. At the same time, along with the changing of the host environment, bacteria have to use regulatory factors to regulate the expression of various genes and, ultimately, to adapt to the new niche.
Bacterial secretion systems are mainly used to deliver toxins or effector proteins into the eukaryotic host cells and modulate the interactions of bacteria with their environments. Currently, seven different types of secretion systems (referred to as Type I–VII or T1SS–T7SS) have been identified, most of which have been carefully investigated. The toxins or effector proteins secreted by secretion systems play a central role in pathogenesis. However, the presence of secretion systems in nonpathogenic bacteria suggests that the involvement of secretion systems is not limited to virulence, such as T6SS and T2SS in nonpathogenic bacteria, and that such systems may also be implicated in functions such as host/symbiont communication, exchange, and cell–cell communication.
Conclusion
In this paper, pathogenic-specific VFs and common VFs were systematically identified in bacterial pathogens by ortholog predictions between the VFs from VFDB and non-pathogenic bacteria. In VFDB, most VFs (more than 68%) were common to both bacterial pathogens and non-pathogens, whereas only approximately 31% of VFs were pathogen-specific. The VFs that were directly involved in virulence, such as exotoxins, T3SS effector proteins, T4SS effector proteins, and PAIs, tended to be distributed among pathogen-specific VFs. Conversely, the VFs that were associated with the pathogenicity closely and did not directly to cause damage to host cells, such as T3SS translocation proteins, T3SS apparatus proteins and chaperones, flagella, capsule, endotoxins, iron uptake proteins, regulatory proteins, T6SS, and T2SS, were inclined to be located among the common VFs and might be associated with the general interaction between bacteria and host. In addition, the common VFs had a higher DN and a lower DC, which indicated that common VFs tend to be complex and less compact proteins.
Materials and Methods
Data
There are some available public virulence factor databases, including PRINTS,Citation44 VFDB, MvirDB,Citation45 etc. Of these databases, the virulence factor database (VFDB) is the most high-quality data set of bacterial VFs. The VFDB contains experimentally demonstrated bacterial VFs that were collected first based on the original research papers appearing in PubMed. Now, the VFDB contains 24 PAIs and over 2294 virulence-related genes from 24 different pathogen genera, including the most well-known medically important pathogens. For each genus, those experimentally demonstrated VFs were first collected based on the original research papers appearing in PubMed to form the primary database. Each VF entry is grouped into the functional categories, and is accompanied by relevant original literature articles or important reviews accessible through direct links to PubMed, as well as detailed information about related genes, keywords, structural features, functions, and pathogenic mechanisms. One thousand, nine hundred and eighty-eight of those VFs were in bacteria whose genome sequences had been completed, and they were from 51 medically significant bacterial pathogens.
We chose 278 well-defined non-pathogenic bacterial genome sequences from the non-pathogenic bacteria protein database to identify VF orthologous proteins in nonpathogenic bacteria. All 278 strains were not pathogenic to the human, animals, or plants, and the information on their habitats status is known. Owning to the complexity of the evolution of bacterial pathogens, the opportunistic bacterial pathogens were excluded from our non-pathogenic bacteria protein database. The Genomic Standards Consortium (GSC) introduced the minimum information about a genome sequence (MIGS) specification, and the “habitat” was a key metadata descriptor in the proposed MIGS specification.Citation46,Citation47 They defined habitat as the place or environment where an organism naturally or normally lives and grows.Citation47 Currently, GenBank and Genomes Online Database (GOLD) were the two major data sources about the MIGS specification.Citation24
The phenotypes of the 278 non-pathogenic bacterial strains and their habitats status were obtained from the GenBank (http://www.ncbi.nlm.nih.gov/Genbank/): prokaryotic attributes table (e.g., pathogenic in, disease and environment: habitat fields) and Genomes Online Database (GOLD) (http://www.genomesonline.org/): organism information (e.g., phenotype, disease, and habitat fields).Citation47,Citation48 We classified the habitat status into two categories: host-associated and non-host-associated (including aquatic, terrestrial, specialized, and multiple). We obtained 65 host-associated and 213 non-host-associated nonpathogenic bacterial strains from the 278 non-pathogenic bacterial strains.
Identification of the VFs' orthologous proteins
Ortholog prediction is paramount when conducting whole genome comparisons.Citation16,Citation17,Citation38 Generally, orthologous genes are identified by phylogenetic analysis. However, sophisticated phylogenetic analysis is not easily automated and not high throughout.Citation49 Therefore, ortholog prediction for large genome-scale data sets is typically performed using a reciprocal-best-BLAST-hits (RBH) approach and there are numerous orthologous resources that use this method, including the Clusters of Orthologous Groups (COG) database,Citation50 the Institute for Genomic Research (TIGR)’s EGO database,Citation51 and INPARANOID.Citation52,Citation53 In this paper, we mainly adopted the reciprocal-best-BLAST-hits (RBH) approach to identify the VF orthologous proteins. With the RBH method, genes from species A and species B are predicted to be orthologs if they are both the “best BLAST hit” of the other, when all genes from species A are compared with all genes from species B by BLAST analysis. The cutoff e-value was set to 1e−7, which was used to exclude distant homologs.
Identification of domain composition and calculation of domain coverage
The protein domain definition used in this study came from Pfam.Citation54 The domain assignments were made by scanning libraries of HMMs against the protein sequences using HMMER-2.0s. A domain was assigned to a region of a query protein if a match to a domain HMM with an e-value lower than 0.001 was observed.
In order to obtain the non-overlapping domain architecture of multi-domain proteins, we resolved overlapping domains according to some rules. We defined two domains as overlapping if more than 10% of the predicted domain locations were overlapping (based on the relative length of the domains). If, in the case of overlapping domains, the e-value difference was larger than 5 (on a –log10 scale), we kept the domain with the highest e-value. In cases where the difference was smaller, we kept the longest model. If both overlapping models had the same length, we considered differences in e-value.
Based on the non-overlapping domain architecture, the domain number (DN) and domain coverage (DC) for the VFs were calculated. The DN in one protein is the number of annotated domains in the sequence of this protein, whereas the DC of a protein is the percentage of the amino acid sequence that defines the identified domains over the whole protein sequence. DN was calculated by including all non-overlapping domains in one protein. DC refers to the percentage of the entire length of all identified domains in a protein to its whole sequence length.
The procedure for the DN and DC analyses employed in this study has been previously described.Citation55
Statistical analysis
In the tables of this paper, for those categories with small values (<5), the Fisher Exact Test was used instead. When multiple categories were examined in parallel, the Benjamini and Hochberg False Discovery Rate correction for multiple testing was performed for all functional category analyses. We considered P values smaller than 0.05 to be significant. All statistical analyses were performed using the R statistics package.
Acknowledgments
This work was supported by the National Key Program for Infectious Diseases of China (2011ZX10004-001) and the Tianjin Science and technology support program (12ZCZDSF00900).
Submitted
03/22/13
Revised
06/28/13
Accepted
07/11/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Binder S, Levitt AM, Sacks JJ, Hughes JM. Emerging infectious diseases: public health issues for the 21st century. Science 1999; 284:1311 - 3; http://dx.doi.org/10.1126/science.284.5418.1311; PMID: 10334978
- Daszak P, Cunningham AA, Hyatt AD. Emerging infectious diseases of wildlife--threats to biodiversity and human health. Science 2000; 287:443 - 9; http://dx.doi.org/10.1126/science.287.5452.443; PMID: 10642539
- Wilson JW, Schurr MJ, LeBlanc CL, Ramamurthy R, Buchanan KL, Nickerson CA. Mechanisms of bacterial pathogenicity. Postgrad Med J 2002; 78:216 - 24; http://dx.doi.org/10.1136/pmj.78.918.216; PMID: 11930024
- Falkow S. Molecular Koch’s postulates applied to bacterial pathogenicity--a personal recollection 15 years later. Nat Rev Microbiol 2004; 2:67 - 72; http://dx.doi.org/10.1038/nrmicro799; PMID: 15035010
- Wu HJ, Wang AH, Jennings MP. Discovery of virulence factors of pathogenic bacteria. Curr Opin Chem Biol 2008; 12:93 - 101; http://dx.doi.org/10.1016/j.cbpa.2008.01.023; PMID: 18284925
- Pallen MJ, Wren BW. Bacterial pathogenomics. Nature 2007; 449:835 - 42; http://dx.doi.org/10.1038/nature06248; PMID: 17943120
- Holden M, Crossman L, Cerdeño-Tárraga A, Parkhill J. Pathogenomics of non-pathogens. Nat Rev Microbiol 2004; 2:91; http://dx.doi.org/10.1038/nrmicro825; PMID: 15040257
- Dobrindt U, Agerer F, Michaelis K, Janka A, Buchrieser C, Samuelson M, et al. Analysis of genome plasticity in pathogenic and commensal Escherichia coli isolates by use of DNA arrays. J Bacteriol 2003; 185:1831 - 40; http://dx.doi.org/10.1128/JB.185.6.1831-1840.2003; PMID: 12618447
- Snyder LA, Saunders NJ. The majority of genes in the pathogenic Neisseria species are present in non-pathogenic Neisseria lactamica, including those designated as ‘virulence genes’. BMC Genomics 2006; 7:128; http://dx.doi.org/10.1186/1471-2164-7-128; PMID: 16734888
- Wassenaar TM, Gaastra W. Bacterial virulence: can we draw the line?. FEMS Microbiol Lett 2001; 201:1 - 7; http://dx.doi.org/10.1111/j.1574-6968.2001.tb10724.x; PMID: 11445159
- Brown NF, Wickham ME, Coombes BK, Finlay BB. Crossing the line: selection and evolution of virulence traits. PLoS Pathog 2006; 2:e42; http://dx.doi.org/10.1371/journal.ppat.0020042; PMID: 16733541
- Steinert M, Hentschel U, Hacker J. Symbiosis and pathogenesis: evolution of the microbe-host interaction. Naturwissenschaften 2000; 87:1 - 11; http://dx.doi.org/10.1007/s001140050001; PMID: 10663126
- Ehrlich GD, Hiller NL, Hu FZ. What makes pathogens pathogenic. Genome Biol 2008; 9:225; http://dx.doi.org/10.1186/gb-2008-9-6-225; PMID: 18598378
- Leavis HL, Willems RJ, van Wamel WJ, Schuren FH, Caspers MP, Bonten MJ. Insertion sequence-driven diversification creates a globally dispersed emerging multiresistant subspecies of E. faecium. PLoS Pathog 2007; 3:e7; http://dx.doi.org/10.1371/journal.ppat.0030007; PMID: 17257059
- Ho Sui SJ, Fedynak A, Hsiao WWL, Langille MGI, Brinkman FSL. The association of virulence factors with genomic islands. PLoS One 2009; 4:e8094; http://dx.doi.org/10.1371/journal.pone.0008094; PMID: 19956607
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science 1998; 282:2022 - 8; http://dx.doi.org/10.1126/science.282.5396.2022; PMID: 9851918
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, et al. Comparative genomics of the eukaryotes. Science 2000; 287:2204 - 15; http://dx.doi.org/10.1126/science.287.5461.2204; PMID: 10731134
- Yang J, Chen L, Sun L, Yu J, Jin Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res 2008; 36:Database issue D539 - 42; http://dx.doi.org/10.1093/nar/gkm951; PMID: 17984080
- Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2008; 24:319 - 24; http://dx.doi.org/10.1093/bioinformatics/btm585; PMID: 18042555
- Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 2000; 54:641 - 79; http://dx.doi.org/10.1146/annurev.micro.54.1.641; PMID: 11018140
- Hentschel U, Hacker J. Pathogenicity islands: the tip of the iceberg. Microbes Infect 2001; 3:545 - 8; http://dx.doi.org/10.1016/S1286-4579(01)01410-1; PMID: 11418328
- Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol 2006; 8:1707 - 19; http://dx.doi.org/10.1111/j.1462-5822.2006.00794.x; PMID: 16939533
- Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev 2004; 17:14 - 56; http://dx.doi.org/10.1128/CMR.17.1.14-56.2004; PMID: 14726454
- Fabbri A, Travaglione S, Falzano L, Fiorentini C. Bacterial protein toxins: current and potential clinical use. Curr Med Chem 2008; 15:1116 - 25; http://dx.doi.org/10.2174/092986708784221430; PMID: 18473807
- Schmitt CK, Meysick KC, O’Brien AD. Bacterial toxins: friends or foes?. Emerg Infect Dis 1999; 5:224 - 34; http://dx.doi.org/10.3201/eid0502.990206; PMID: 10221874
- Gentschev I, Dietrich G, Goebel W. The E. coli alpha-hemolysin secretion system and its use in vaccine development. Trends Microbiol 2002; 10:39 - 45; http://dx.doi.org/10.1016/S0966-842X(01)02259-4; PMID: 11755084
- Kuhnert P, Schlatter Y, Frey J. Characterization of the type I secretion system of the RTX toxin ApxII in “Actinobacillus porcitonsillarum”. Vet Microbiol 2005; 107:225 - 32; http://dx.doi.org/10.1016/j.vetmic.2005.01.020; PMID: 15863281
- Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, et al. Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A 2005; 102:3004 - 9; http://dx.doi.org/10.1073/pnas.0409900102; PMID: 15703294
- Cortajarena AL, Goni FM, Ostolaza H. A receptor-binding region in Escherichia coli alpha-haemolysin. J Biol Chem 2003; 278:19159 - 63; http://dx.doi.org/10.1074/jbc.M208552200; PMID: 12582172
- Sánchez-Magraner L, Cortajarena AL, Goñi FM, Ostolaza H. Membrane insertion of Escherichia coli alpha-hemolysin is independent from membrane lysis. J Biol Chem 2006; 281:5461 - 7; http://dx.doi.org/10.1074/jbc.M512897200; PMID: 16377616
- Rossignol G, Merieau A, Guerillon J, Veron W, Lesouhaitier O, Feuilloley MG, et al. Involvement of a phospholipase C in the hemolytic activity of a clinical strain of Pseudomonas fluorescens. BMC Microbiol 2008; 8:189; http://dx.doi.org/10.1186/1471-2180-8-189; PMID: 18973676
- Talarico S, Durmaz R, Yang Z. Insertion- and deletion-associated genetic diversity of Mycobacterium tuberculosis phospholipase C-encoding genes among 106 clinical isolates from Turkey. J Clin Microbiol 2005; 43:533 - 8; http://dx.doi.org/10.1128/JCM.43.2.533-538.2005; PMID: 15695641
- Alfano JR, Collmer A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu Rev Phytopathol 2004; 42:385 - 414; http://dx.doi.org/10.1146/annurev.phyto.42.040103.110731; PMID: 15283671
- Dale C, Plague GR, Wang B, Ochman H, Moran NA. Type III secretion systems and the evolution of mutualistic endosymbiosis. Proc Natl Acad Sci U S A 2002; 99:12397 - 402; http://dx.doi.org/10.1073/pnas.182213299; PMID: 12213957
- Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol 2006; 4:811 - 25; http://dx.doi.org/10.1038/nrmicro1526; PMID: 17041629
- Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol 2003; 1:137 - 49; http://dx.doi.org/10.1038/nrmicro753; PMID: 15035043
- Juhas M, Crook DW, Hood DW. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol 2008; 10:2377 - 86; http://dx.doi.org/10.1111/j.1462-5822.2008.01187.x; PMID: 18549454
- Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet 2005; 39:309 - 38; http://dx.doi.org/10.1146/annurev.genet.39.073003.114725; PMID: 16285863
- Heger A, Holm L. Exhaustive enumeration of protein domain families. J Mol Biol 2003; 328:749 - 67; http://dx.doi.org/10.1016/S0022-2836(03)00269-9; PMID: 12706730
- Gómez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med 2004; 10:842 - 8; http://dx.doi.org/10.1038/nm1079; PMID: 15247912
- Herwald H, Cramer H, Mörgelin M, Russell W, Sollenberg U, Norrby-Teglund A, et al. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell 2004; 116:367 - 79; http://dx.doi.org/10.1016/S0092-8674(04)00057-1; PMID: 15016372
- Rendón MA, Saldaña Z, Erdem AL, Monteiro-Neto V, Vázquez A, Kaper JB, et al. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc Natl Acad Sci U S A 2007; 104:10637 - 42; http://dx.doi.org/10.1073/pnas.0704104104; PMID: 17563352
- Howard JC, Heinemann C, Thatcher BJ, Martin B, Gan BS, Reid G. Identification of collagen-binding proteins in Lactobacillus spp. with surface-enhanced laser desorption/ionization-time of flight ProteinChip technology. Appl Environ Microbiol 2000; 66:4396 - 400; http://dx.doi.org/10.1128/AEM.66.10.4396-4400.2000; PMID: 11010889
- Attwood TK, Bradley P, Flower DR, Gaulton A, Maudling N, Mitchell AL, et al. PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res 2003; 31:400 - 2; http://dx.doi.org/10.1093/nar/gkg030; PMID: 12520033
- Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T. MvirDB--a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res 2007; 35:Database issue D391 - 4; http://dx.doi.org/10.1093/nar/gkl791; PMID: 17090593
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541 - 7; http://dx.doi.org/10.1038/nbt1360; PMID: 18464787
- Hirschman L, Clark C, Cohen KB, Mardis S, Luciano J, Kottmann R, et al, Novo Project. Habitat-Lite: a GSC case study based on free text terms for environmental metadata. OMICS 2008; 12:129 - 36; http://dx.doi.org/10.1089/omi.2008.0016; PMID: 18416669
- Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, et al. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:Database issue D346 - 54; http://dx.doi.org/10.1093/nar/gkp848; PMID: 19914934
- Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol 2009; 5:e1000262; http://dx.doi.org/10.1371/journal.pcbi.1000262; PMID: 19148271
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 2001; 29:22 - 8; http://dx.doi.org/10.1093/nar/29.1.22; PMID: 11125040
- Lee Y, Sultana R, Pertea G, Cho J, Karamycheva S, Tsai J, et al. Cross-referencing eukaryotic genomes: TIGR Orthologous Gene Alignments (TOGA). Genome Res 2002; 12:493 - 502; http://dx.doi.org/10.1101/gr.212002; PMID: 11875039
- Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 2001; 314:1041 - 52; http://dx.doi.org/10.1006/jmbi.2000.5197; PMID: 11743721
- O’Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 2005; 33:Database issue D476 - 80; http://dx.doi.org/10.1093/nar/gki107; PMID: 15608241
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, et al. The Pfam protein families database. Nucleic Acids Res 2008; 36:Database issue D281 - 8; http://dx.doi.org/10.1093/nar/gkm960; PMID: 18039703
- Zhong F, Yang D, Hao Y, Lin C, Jiang Y, Ying W, et al. Regular patterns for proteome-wide distribution of protein abundance across species. PLoS One 2012; 7:e32423; http://dx.doi.org/10.1371/journal.pone.0032423; PMID: 22427835
- Schulein R, Dehio C. The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol Microbiol 2002; 46:1053 - 67; http://dx.doi.org/10.1046/j.1365-2958.2002.03208.x; PMID: 12421311
- Cheung AM, Farizo KM, Burns DL. Analysis of relative levels of production of pertussis toxin subunits and Ptl proteins in Bordetella pertussis. Infect Immun 2004; 72:2057 - 66; http://dx.doi.org/10.1128/IAI.72.4.2057-2066.2004; PMID: 15039327
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5:1166 - 74; http://dx.doi.org/10.1038/ni1131; PMID: 15489856
- Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A 2004; 101:841 - 6; http://dx.doi.org/10.1073/pnas.0304916101; PMID: 14715899
- Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, et al. Legionella effectors that promote nonlytic release from protozoa. Science 2004; 303:1358 - 61; http://dx.doi.org/10.1126/science.1094226; PMID: 14988561
- Macnab RM. How bacteria assemble flagella. Annu Rev Microbiol 2003; 57:77 - 100; http://dx.doi.org/10.1146/annurev.micro.57.030502.090832; PMID: 12730325
- Cieslewicz MJ, Chaffin D, Glusman G, Kasper D, Madan A, Rodrigues S, et al. Structural and genetic diversity of group B streptococcus capsular polysaccharides. Infect Immun 2005; 73:3096 - 103; http://dx.doi.org/10.1128/IAI.73.5.3096-3103.2005; PMID: 15845517
- Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem 2002; 71:635 - 700; http://dx.doi.org/10.1146/annurev.biochem.71.110601.135414; PMID: 12045108
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117:285 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00343-5; PMID: 15109490
- Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner’s guide. Curr Opin Microbiol 2008; 11:3 - 8; http://dx.doi.org/10.1016/j.mib.2008.01.006; PMID: 18289922
- Johnson TL, Abendroth J, Hol WG, Sandkvist M. Type II secretion: from structure to function. FEMS Microbiol Lett 2006; 255:175 - 86; http://dx.doi.org/10.1111/j.1574-6968.2006.00102.x; PMID: 16448494