Abstract
The development of severe influenza has been attributed, in part, to a heightened innate immune response. Recent evidence suggests that endothelial activation, loss of barrier function, and consequent microvascular leak may also serve important mechanistic roles in the pathogenesis of severe influenza. The aim of this review is to summarize the current evidence in support of endothelial activation and dysfunction as a central feature preceding the development of severe influenza. We also discuss the effect of influenza on platelet–endothelial interactions.
Introduction
Influenza A viruses commonly infect the upper respiratory tract resulting in a self-limited infection with mild respiratory symptoms. In severe cases, which typically occur in elderly or immunocompromised patients, influenza may spread to the lower respiratory tract, causing viral pneumonia. This may progress to acute lung injury, a syndrome of increased pulmonary microvascular leakage leading to pulmonary edema, hypoxemia and respiratory failure.Citation1,Citation2 In this setting, the use of an antiviral drug for the specific treatment of influenza is only partially effective at reducing mortality,Citation3 underlining the urgent need for additional therapies.
The inadequacy of current therapeutic approaches is reinforced by the rapid development of viral resistance to anti-viral drugs and their limited efficacy when administered late in the course of disease. Instead, the lung microvascular leak that occurs in severe influenza virus infections may represent an attractive adjunctive therapeutic target. Major advances have emerged in our understanding of the role of the microvascular endothelium during infections, particularly in the field of sepsis.Citation4 Despite this, the contributions of the lung microvascular endothelium to the pathogenesis of severe influenza have been largely uninvestigated. The aim of this review is to summarize the literature on endothelial activation and dysfunction in severe influenza. Specifically, we will highlight the role of inflammation, permeability, and thrombosis as it pertains to the pulmonary microvascular endothelium during influenza virus infection. We conclude by discussing areas for future research in the field.
Endothelial Permeability and Activation during Influenza
Influenza A virus primarily binds the respiratory epithelium. Subsequent infection of epithelial cells can lead to pulmonary edema by interfering with epithelial sodium channel (ENaC) function, an important mediator of alveolar fluid clearance.Citation5-Citation7 However, endothelial dysfunction is thought to be the main factor involved in the development of pulmonary edema.Citation8 Thus, understanding how the influenza virus causes disruption of the endothelial barrier is critical.
Endothelial barrier dysfunction, a phenomenon that is particularly problematic in the lung due to the resultant alveolar flooding, may be a deleterious consequence of excessive cytokine production,. Indeed, many investigators have implicated a heterogenous subset of elevated cytokines and chemokines known as a “cytokine storm” in the pathogenesis of severe influenza.Citation9-Citation15 Tumor necrosis factor (TNF) is one of multiple cytokines implicated in the pathogenesis of influenzaCitation16,Citation17 that has been shown to mediate endothelial barrier dysfunction. Elevated TNF levels have been shown to increase Rho kinase-induced stress fiber formation leading to induction of endothelial apoptosis.Citation18-Citation20 Increased TNF, IL-6, and IL-1β following influenza virus infection has also been shown to upregulate trypsin resulting in the loss of the endothelial tight junction protein, zonula occludens-1 (ZO-1), and subsequent vascular hyperpermeability.Citation21 Elevated chemokine expression during influenza virus infection may also play a role in endothelial barrier dysfunction. For instance, upon avian influenza H5N1 virus infection, Chan and colleaguesCitation22 observed chemokine expression from epithelial cells at the basolateral surface, which would abut the microvascular endothelium in the alveolo-capillary membrane. Intriguingly, this effect was not observed with a low pathogenic H1N1 influenza virus, suggesting that chemokine-induced endothelial activation may contribute to the development of severe disease during H5N1 influenza infection.
Recent evidence suggests that the lung endothelium itself plays a previously unsuspected role in mediating the cytokine response. In particular, the endothelial sphingosine-1-phosphate receptor 1 (S1P1) was found to regulate the cytokine storm caused by severe influenza.Citation23 Influenza-infected mice that received an S1P1 specific agonist (CYM-5442) had significantly decreased early cytokine and chemokine levels as well as reduced inflammatory cell infiltrates in the lung. This resulted in a 60% improvement in survival. Teijaro and colleaguesCitation23 demonstrated that S1P1 located on the pulmonary endothelium was responsible for mediating this effect. While CYM-5442 attenuated leukocyte recruitment, this did not explain the reduction in cytokines. Instead, SIP1 agonism was shown to dampen global proinflammatory cytokine responses by blunting IFN-α production, an upstream regulator of early cytokine production. Unfortunately, the effects of SIP1 agonism on microvascular leak per se were not reported in this study. However, S1P is known to promote endothelial barrier integrity by inducing endothelial Rac activity, thereby increasing cortical actin formation and stabilizing the adherens junctions.Citation24,Citation25 Thus, the protective effect of the S1P1 agonist may have been due to both global cytokine repression and improved lung endothelial stability.
Cytokine production also leads to upregulation of adhesion molecules that facilitate leukocyte recruitment. Indeed, an influx of leukocytes into the lungs is critical to mounting a healthy innate immune response to infection. However, recent evidence in humans, murine models, and cell lines suggests an important contribution of neutrophil infiltration to the development of acute lung injury and the acute respiratory distress syndrome (ARDS) in influenza pneumonia.Citation26,Citation27 Activated neutrophils release neutrophil extracellular traps (NETs), macromolecular structures formed of extruded nuclear chromatin and bactericidal proteins.Citation28 While postulated to play a role in host defense against infection, NETs have been shown to exert cytotoxic effects upon endothelial cellsCitation29 and to contribute to lung damage in influenza-infected miceCitation26; A/PR/8 (H1N1)-infected mice developed lung damage with extensive focal NET formation within pulmonary lesions. In this study, NETs were found entangled with alveolar epithelium and small blood vessels in areas with hemorrhagic lesions, suggestive of NET-induced alveolo-capillary damage.Citation26 In addition, small airway occlusions were attributed in part to NET-endothelial or -epithelial cell interaction/attachment. Redox enzyme-dependent NET-enhanced endothelial cell damage was confirmed in vitro.
In addition to cytokine-mediated endothelial activation and endothelial damage from infiltrating leukocytes, direct invasion of the endothelium by influenza virus may activate the endothelium resulting in functional changes in endothelial protein expression, enhanced endothelial permeability, and significant vascular destruction. Viral replication within the alveolar epithelium leads to apoptosis, exposing the underlying basolateral surface of the pulmonary endothelium which expresses α2,6- linked sialic acid residues, the receptors for the human influenza virus.Citation30 In vitro, HPAIV H5N1 and H3N2 influenza subtypes have been shown to replicate within human lung microvascular endothelial cells.Citation22,Citation30-Citation32 However, in vivo, direct infection of the lung microvascular endothelium has only been demonstrated for avian influenza H5N1 viruses.Citation33 Influenza virus infection of the endothelium can lead to activation of the transcription factor NFκB resulting in upregulated cytokine and chemokine production giving rise to vascular leak.Citation21,Citation31 In the presence of the NFκB dominant negative mutant IKK2, Schmolke et al.Citation31 reported a 46% reduction in H5N1-induced mRNA expression in endothelial cells, including IFN-β. NFκB-dependent activation of influenza-inducible genes may be more pronounced during H5N1 infection of the endothelium compared with infection by other influenza subtypes.Citation34 While NFκB signaling in the endothelium may be important for influenza-induced cytokine production, NFκB signaling within the endothelium may play a more direct role in regulating vascular permeability in vivo.Citation35 For instance, in a murine model of E. coli sepsis, mice that overexpressed an endothelial-specific NFκB inhibitor had decreased endothelial permeability, decreased expression of markers of organ injury, and improved survival. Importantly, systemic and tissue inflammation were similar in the transgenic mice compared with wild-type mice.Citation35
In contrast, a recent report demonstrated a negligible role for NFκB in mediating endothelial stability.Citation36 Instead, Zhu and colleaguesCitation36 reported an IL-1β-induced MYD88-ARNO-ARF6 signaling pathway that regulates vascular stability independent of NFκB function. This cytokine-mediated pathway may also be of importance for influenza-induced vascular leak.Citation36,Citation37
Ultimately, an increase in endothelial permeability almost always reflects endothelial apoptosis or remodeling of endothelial cell–cell junctions (adherens junctions and tight junctions) (). Adherens junction proteins, notably VE-cadherin, possess an extracellular domain that connects endothelial cells, and an intracellular domain that connects to the actin cytoskeleton via catenin proteins.Citation38 Recently, London et al.Citation39 identified Slit and its cognate receptor, Robo-4, as important mediators of VE-cadherin retention at the plasmalemma. Increased cell surface VE-cadherin was mediated by enhanced association of p120-catenin to VE-cadherin, which prevented VE-cadherin internalization. Slit2N-mediated effects were abrogated in the presence of VE-cadherin antibody. Of key interest, in vivo administration of Slit2N, the biologically active component of Slit, significantly improved lung injury, lung endothelial integrity, and survival in H5N1-infected mice. Remarkably, these effects were observed in the absence of an effect on pulmonary inflammation, cytokine levels, and viral load, suggesting that strengthening the microvascular endothelial barrier may be sufficient to improve clinical outcome.
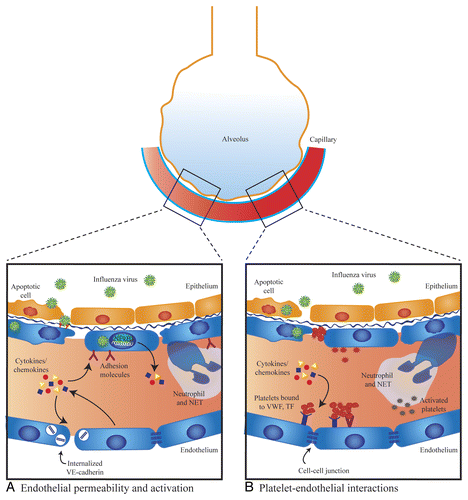
Figure 1. Mechanisms of endothelial dysfunction in influenza virus infection. (A) Endothelial permeability and activation. Elevated levels of pro-inflammatory cytokines/chemokines can directly induce endothelial leak through disruption of cell–cell junctions and may also cause endothelial cells to express elevated levels of adhesion molecules that promote leukocyte recruitment. Neutrophils release neutrophil extracellular traps (NETs), which can damage endothelial cells. There is in vitro evidence that influenza can directly infect lung endothelial cells and cause activation of NFκB, endothelial apoptosis, and loss of junctional proteins. In vivo, only avian H5N1 influenza has been shown to directly infect endothelial cells. (B) Platelet–endothelial interactions. Circulating cytokines/chemokines cause increased expression of platelet-binding receptors. Influenza virus can directly infect lung endothelium and induce endothelial apoptosis exposing the extracellular matrix, which has a high affinity for platelets. Influenza may directly induce platelet activation and activated platelets bind to endothelium. Activated platelets may interact with neutrophils triggering the production of NETs.
Alterations in tight junction proteinsCitation38 may also mediate endothelial leak during influenza infection.Citation21 Tight junction-mediated vascular leak may play a role in influenza pathogenesis, independent of adherens junction protein modification. Replication-deficient influenza A/X-31 virus induced degradation of the tight junction protein claudin-5 in vitro, thereby augmenting endothelial permeability in the absence of changes in adherens junctions.Citation32 This finding was demonstrated to be independent of influenza-induced endothelial apoptosis, indicating a specific involvement of claudin-5 tight junction proteins in influenza pathogenesis. Furthermore, formoterol, a β2-agonist that induces claudin-5 expression, attenuated influenza-induced pulmonary edema in a murine model of influenza.Citation32 As mentioned earlier, other investigators have proposed that influenza-induced cytokine production upregulates the human trypsin/hPRSS gene, which in turn mediates the loss of endothelial tight junction protein ZO-1, thereby increasing endothelial permeability.Citation21
Influenza and Platelet–Endothelial Interactions
In addition to leak, there is evidence to suggest that endothelial dysfunction following influenza virus infections may manifest as altered thrombogenicity. Healthy endothelial monolayers are anti-thrombogenic; during the H1N1 pandemic in 2009 (H1N1pdm09), there were multiple reports of flu-associated thrombosis. A retrospective review of 119 hospitalized patients found that 7 patients (5.9%) had thrombotic vascular events that were diagnosed or occurred during hospitalization.Citation40 Three of these patients had arterial thrombosis and four had venous thrombosis. A study in Michigan looked at 10 patients with H1N1pdm09 influenza who were admitted to the intensive care unit with ARDS.Citation41 Five had pulmonary emboli, while two others showed evidence of hypercoagulation. Whether these complications are directly attributable to the virus or simply reflect the overall severity of illness remains unclear.
Similarly, epidemiological evidence supporting a link between influenza and cardiovascular disease has been reported for decades.Citation42-Citation44 A temporal relationship between influenza infections and the incidence of cardiovascular disease has been reported by a number of groupsCitation42-Citation47 and several studies have found the influenza vaccine to be associated with a reduction in stroke, transient ischemic attack, and hospitalization due to cardiac disease.Citation48-Citation50
The link between influenza infection and cardiovascular disease has also been reported in animals. In one study, apoE−/− mice (an accepted murine model of atherosclerosis) were infected with influenza A/Hong Kong/68 (H3N2) and vascular histology of the aorta was compared with apoE−/− uninfected mice as well as infected wild-type mice. Infected apoE−/− mice showed increased subendothelial cellular infiltration in atherosclerotic plaques compared with uninfected apoE−/− mice. Infected wild-type mice showed no evidence of cellular infiltration of the vascular intima.Citation51 This group found clustered platelets on the plaques of the majority of infected animals, but none in uninfected animals. Thus, influenza may worsen existing vascular disease resulting in increased endothelial damage and platelet adhesion.
While a causal link between influenza and thrombotic disease has not yet been definitely established,Citation52 a variety of plausible mechanisms have been proposed that highlight the relationship between the influenza virus, platelet activation, and endothelial dysfunction. Elevated levels of circulating cytokines associated with influenza infectionCitation53 can induce endothelial activation leading to upregulation of cell surface adhesion molecules that favor platelet adhesion. In HUVECs, TNF and IL-1β have been shown to induce the expression of type 1 plasminogen activator inhibitor (PAI-1), which promotes platelet binding, while inhibiting the expression of tissue-type plasminogen activator (tPa) and thrombomodulin, which are anticoagulant.Citation54 Similarly, infusion of IL-1 into rabbits caused a time-dependent increase in tissue factor (TF) expression on aortic endothelial cellsCitation55; TF is known to be a key player in the coagulation process.Citation56 Endothelial cells also increase production of platelet activating factor (PAF) upon stimulation with TNF and IL-1α.Citation57 Cytokines can also induce endothelial cell retraction, exposing the pro-atherogenic extracellular matrix.
Influenza-induced lung injury itself may be an important factor promoting thrombosis. Patients with severe influenza require supplemental oxygen because of profound hypoxemia.Citation1,Citation2 Hypoxia has been shown to induce a pro-inflammatory state in the endothelium causing the increased release of IL-1, IL-6, PAF, ICAM-1, p-selectin, and VWF,Citation58 all of which are associated with platelet activation and adhesion.
There is also evidence that the influenza virus per se may directly affect the endothelium resulting in platelet adhesion. Influenza H3N2 virus has been shown to infect endothelial cells in vitro and to trigger endothelial cell apoptosis,Citation32 which is known to enhance platelet adhesion.Citation59 Endothelial cell death would cause exposure of the extracellular matrix to circulating blood, favoring platelet binding.Citation60 Cultured HUVEC monolayers infected with influenza have been shown to reduce clotting times by 55% after 3 h of infection and by 66% after 24 h of infection, compared with uninfected monolayers.Citation61 This was attributed to an increase in TF expression, but at least some of the effect may have been mediated through cytokines and/or the induction of apoptosis as neither was measured in this study.
In addition to affecting the endothelium, the influenza virus may have a direct effect on platelets. An H3N2 virus added directly to platelets was found to induce clumping of both human and rabbit platelets.Citation62 In this study, both live and dead virus were adsorbed by platelets and the adsorption period was linked to clumping. Infusion of influenza into rabbits induced rapid thrombocytopenia. Platelet activation by influenza has also been documented in humans. In one prospective study comparing patients with severe influenza (H1N1) and patients with severe bacterial pneumonia (all being treated for ARDS) to healthy controls, patients with influenza showed the greatest degree of baseline platelet activation as evidenced by increased formation of platelet-monocyte aggregates and increased binding of the PAC-1 antibody, which binds to the active conformation of αIIbβ3 integrin on platelets.Citation63 Platelets could also promote endothelial damage during influenza infection through their interaction with neutrophils. As mentioned earlier, excessive neutrophils have been associated with worse lung pathology in mice infected with H1N1 influenza, attributed to the formation of neutrophil extracellular traps.Citation26 Intriguingly, it has been reported that addition of activated platelets to neutrophils in vitro is sufficient to induce NET formation.Citation64 In a mouse model of transfusion-related lung injury, inhibition of platelets either with aspirin (acetylsalicylic acid) or a glycoprotein IIb/IIIa inhibitor reduced both NET formation and lung injury.Citation64 In a study of acid-induced lung injury in mice, platelet depletion reduced lung neutrophil infiltration, improved histological changes, and increased survival.Citation65 Depletion of platelets was also found to reduce neutrophil influx and pathological changes associated with LPS-induced lung injury.Citation66 Remarkably, however, little is known about platelet–endothelial interactions and their contribution to acute lung injury during severe influenza. This is the subject of ongoing work in our laboratory.
In summary, influenza virus, platelets, and endothelial cells may interact in a variety of ways to induce lung endothelial dysfunction. These include induction of pro-coagulant pathways, activation of platelets and the endothelium, and enhanced interaction between platelets and neutrophils leading to the breakdown of the lung endothelial barrier ().
Future Directions
Microvascular endothelial barrier activation/dysfunction due to influenza virus infection may contribute to the development of severe lung injury that occurs in a subset of patients with influenza. As mortality in this group remains high, novel therapeutic strategies that target the lung endothelium represent a promising therapeutic approach and may elicit a synergistic effect when combined with antiviral and supportive treatments.
A number of agents which enhance the endothelial barrier have been described (e.g., S1P and Slit2N) and may prove effective in combating influenza-induced lung injury.Citation39,Citation67-Citation69 As platelets may play a detrimental role in the pathogenesis of influenza, treatments that blunt platelet activation or prevent their interaction with endothelial cells also warrant investigation.
In conclusion, influenza virus may interact with the lung microvascular endothelium both directly and indirectly causing damage to the alveolo-capillary membrane resulting in severe disease. Targeting of the microvascular endothelium in conjunction with antiviral administration is thus an attractive therapeutic strategy to improve patient outcomes.
Acknowledgments
Research in WLL’s laboratory is supported by the Canadian Institutes of Health Research and an Early Researcher Award from the Government of Ontario.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Domínguez-Cherit G, Lapinsky SE, Macias AE, Pinto R, Espinosa-Perez L, de la Torre A, et al. Critically Ill patients with 2009 influenza A(H1N1) in Mexico. JAMA 2009; 302:1880 - 7; http://dx.doi.org/10.1001/jama.2009.1536; PMID: 19822626
- Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al, Canadian Critical Care Trials Group H1N1 Collaborative. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009; 302:1872 - 9; http://dx.doi.org/10.1001/jama.2009.1496; PMID: 19822627
- McGeer A, Green KA, Plevneshi A, Shigayeva A, Siddiqi N, Raboud J, et al, Toronto Invasive Bacterial Diseases Network. Antiviral therapy and outcomes of influenza requiring hospitalization in Ontario, Canada. Clin Infect Dis 2007; 45:1568 - 75; http://dx.doi.org/10.1086/523584; PMID: 18190317
- Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med 2011; 3:88ps25; http://dx.doi.org/10.1126/scitranslmed.3002011; PMID: 21697528
- Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, et al. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 2004; 287:L366 - 73; http://dx.doi.org/10.1152/ajplung.00011.2004; PMID: 15121635
- Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, et al. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med 2008; 178:969 - 76; http://dx.doi.org/10.1164/rccm.200803-455OC; PMID: 18689466
- Kunzelmann K, Beesley AH, King NJ, Karupiah G, Young JA, Cook DI. Influenza virus inhibits amiloride-sensitive Na+ channels in respiratory epithelia. Proc Natl Acad Sci U S A 2000; 97:10282 - 7; http://dx.doi.org/10.1073/pnas.160041997; PMID: 10920189
- Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 2008; 14:22 - 30; http://dx.doi.org/10.1097/MCC.0b013e3282f269b9; PMID: 18195622
- Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 2000; 156:1951 - 9; http://dx.doi.org/10.1016/S0002-9440(10)65068-7; PMID: 10854218
- Carey MA, Bradbury JA, Seubert JM, Langenbach R, Zeldin DC, Germolec DR. Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J Immunol 2005; 175:6878 - 84; PMID: 16272346
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007; 445:319 - 23; http://dx.doi.org/10.1038/nature05495; PMID: 17230189
- Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 2008; 4:e1000115; http://dx.doi.org/10.1371/journal.ppat.1000115; PMID: 18670648
- Perrone LA, Szretter KJ, Katz JM, Mizgerd JP, Tumpey TM. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J Infect Dis 2010; 202:1161 - 70; http://dx.doi.org/10.1086/656365; PMID: 20815704
- Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 2004; 363:617 - 9; http://dx.doi.org/10.1016/S0140-6736(04)15595-5; PMID: 14987888
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 2006; 12:1203 - 7; http://dx.doi.org/10.1038/nm1477; PMID: 16964257
- Van Reeth K, Nauwynck H, Pensaert M. Bronchoalveolar interferon-alpha, tumor necrosis factor-alpha, interleukin-1, and inflammation during acute influenza in pigs: a possible model for humans?. J Infect Dis 1998; 177:1076 - 9; http://dx.doi.org/10.1086/517398; PMID: 9534986
- Belisle SE, Tisoncik JR, Korth MJ, Carter VS, Proll SC, Swayne DE, et al. Genomic profiling of tumor necrosis factor alpha (TNF-alpha) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-alpha signaling and increased severity of 1918 pandemic influenza virus infection. J Virol 2010; 84:12576 - 88; http://dx.doi.org/10.1128/JVI.01310-10; PMID: 20926563
- Petrache I, Crow MT, Neuss M, Garcia JG. Central involvement of Rho family GTPases in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res Commun 2003; 306:244 - 9; http://dx.doi.org/10.1016/S0006-291X(03)00945-8; PMID: 12788095
- Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol 2003; 28:574 - 81; http://dx.doi.org/10.1165/rcmb.2002-0075OC; PMID: 12707013
- Petrache I, Birukov K, Zaiman AL, Crow MT, Deng H, Wadgaonkar R, et al. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. FASEB J 2003; 17:407 - 16; http://dx.doi.org/10.1096/fj.02-0672com; PMID: 12631580
- Wang S, Le TQ, Kurihara N, Chida J, Cisse Y, Yano M, et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis 2010; 202:991 - 1001; http://dx.doi.org/10.1086/656044; PMID: 20731583
- Chan MC, Chan RW, Yu WC, Ho CC, Chui WH, Lo CK, et al. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res 2009; 10:102; http://dx.doi.org/10.1186/1465-9921-10-102; PMID: 19874627
- Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011; 146:980 - 91; http://dx.doi.org/10.1016/j.cell.2011.08.015; PMID: 21925319
- Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 2005; 280:17320 - 8; http://dx.doi.org/10.1074/jbc.M411674200; PMID: 15728185
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 2001; 108:689 - 701; PMID: 11544274
- Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 2011; 179:199 - 210; http://dx.doi.org/10.1016/j.ajpath.2011.03.013; PMID: 21703402
- Kanchana S, Kanchana S, Vijitsopa T, Thammakumpee K, Yamwong S, Sawanyawisuth K. Clinical factors predictive of pneumonia caused by pandemic 2009 H1N1 influenza virus. Am J Trop Med Hyg 2013; 88:461 - 3; http://dx.doi.org/10.4269/ajtmh.12-0132; PMID: 23382162
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303:1532 - 5; http://dx.doi.org/10.1126/science.1092385; PMID: 15001782
- Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 2012; 7:e32366; http://dx.doi.org/10.1371/journal.pone.0032366; PMID: 22389696
- Zeng H, Pappas C, Belser JA, Houser KV, Zhong W, Wadford DA, et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol 2012; 86:667 - 78; http://dx.doi.org/10.1128/JVI.06348-11; PMID: 22072765
- Schmolke M, Viemann D, Roth J, Ludwig S. Essential impact of NF-kappaB signaling on the H5N1 influenza A virus-induced transcriptome. J Immunol 2009; 183:5180 - 9; http://dx.doi.org/10.4049/jimmunol.0804198; PMID: 19786538
- Armstrong SM, Wang C, Tigdi J, Si X, Dumpit C, Charles S, et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS One 2012; 7:e47323; http://dx.doi.org/10.1371/journal.pone.0047323; PMID: 23115643
- Feldmann A, Schäfer MK, Garten W, Klenk HD. Targeted infection of endothelial cells by avian influenza virus A/FPV/Rostock/34 (H7N1) in chicken embryos. J Virol 2000; 74:8018 - 27; http://dx.doi.org/10.1128/JVI.74.17.8018-8027.2000; PMID: 10933711
- Viemann D, Schmolke M, Lueken A, Boergeling Y, Friesenhagen J, Wittkowski H, et al. H5N1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J Immunol 2011; 186:164 - 73; http://dx.doi.org/10.4049/jimmunol.0904170; PMID: 21106851
- Xu H, Ye X, Steinberg H, Liu SF. Selective blockade of endothelial NF-kappaB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J Pathol 2010; 220:490 - 8; PMID: 20020511
- Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, et al. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature 2012; 492:252 - 5; http://dx.doi.org/10.1038/nature11603; PMID: 23143332
- Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am J Physiol 1989; 257:L399 - 410; PMID: 2610269
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006; 86:279 - 367; http://dx.doi.org/10.1152/physrev.00012.2005; PMID: 16371600
- London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med 2010; 2:23ra19; http://dx.doi.org/10.1126/scitranslmed.3000678; PMID: 20375003
- Bunce PE, High SM, Nadjafi M, Stanley K, Liles WC, Christian MD. Pandemic H1N1 influenza infection and vascular thrombosis. Clin Infect Dis 2011; 52:e14 - 7; http://dx.doi.org/10.1093/cid/ciq125; PMID: 21288835
- Centers for Disease Control and Prevention (CDC). Intensive-care patients with severe novel influenza A (H1N1) virus infection - Michigan, June 2009. MMWR Morb Mortal Wkly Rep 2009; 58:749 - 52; PMID: 19609249
- Eickhoff TC, Sherman IL, Serfling RE. Observations on excess mortality associated with epidemic influenza. JAMA 1961; 176:776 - 82; http://dx.doi.org/10.1001/jama.1961.03040220024005; PMID: 13726091
- Gordon T, Thom T. The recent decrease in CHD mortality. Prev Med 1975; 4:115 - 25; http://dx.doi.org/10.1016/0091-7435(75)90077-8; PMID: 1153392
- Soltero I, Liu K, Cooper R, Stamler J, Garside D. Trends in mortality from cerebrovascular diseases in the United States, 1960 to 1975. Stroke 1978; 9:549 - 58; http://dx.doi.org/10.1161/01.STR.9.6.549; PMID: 741484
- Field TS, Zhu H, Tarrant M, Mitchell JR, Hill MD. Relationship between supra-annual trends in influenza rates and stroke occurrence. Neuroepidemiology 2004; 23:228 - 35; http://dx.doi.org/10.1159/000079948; PMID: 15316249
- Spodick DH, Flessas AP, Johnson MM. Association of acute respiratory symptoms with onset of acute myocardial infarction: prospective investigation of 150 consecutive patients and matched control patients. Am J Cardiol 1984; 53:481 - 2; http://dx.doi.org/10.1016/0002-9149(84)90016-X; PMID: 6695777
- Mattila KJ. Viral and bacterial infections in patients with acute myocardial infarction. J Intern Med 1989; 225:293 - 6; http://dx.doi.org/10.1111/j.1365-2796.1989.tb00084.x; PMID: 2732669
- Lavallée P, Perchaud V, Gautier-Bertrand M, Grabli D, Amarenco P. Association between influenza vaccination and reduced risk of brain infarction. Stroke 2002; 33:513 - 8; http://dx.doi.org/10.1161/hs0202.102328; PMID: 11823662
- Grau AJ, Fischer B, Barth C, Ling P, Lichy C, Buggle F. Influenza vaccination is associated with a reduced risk of stroke. Stroke 2005; 36:1501 - 6; http://dx.doi.org/10.1161/01.STR.0000170674.45136.80; PMID: 15947266
- Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med 2003; 348:1322 - 32; http://dx.doi.org/10.1056/NEJMoa025028; PMID: 12672859
- Naghavi M, Wyde P, Litovsky S, Madjid M, Akhtar A, Naguib S, et al. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation 2003; 107:762 - 8; http://dx.doi.org/10.1161/01.CIR.0000048190.68071.2B; PMID: 12578882
- Smeeth L, Casas JP, Hingorani AD. The role of infection in cardiovascular disease: more support but many questions remain. Eur Heart J 2007; 28:1178 - 9; http://dx.doi.org/10.1093/eurheartj/ehm073; PMID: 17470675
- Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramírez P, et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care 2009; 13:R201; http://dx.doi.org/10.1186/cc8208; PMID: 20003352
- Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA Jr., Loskutoff DJ. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem 1988; 263:5797 - 803; PMID: 3128548
- Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A 1986; 83:3460 - 4; http://dx.doi.org/10.1073/pnas.83.10.3460; PMID: 3486418
- Camerer E, Kolstø AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res 1996; 81:1 - 41; http://dx.doi.org/10.1016/0049-3848(95)00209-X; PMID: 8747518
- Bussolino F, Camussi G, Baglioni C. Synthesis and release of platelet-activating factor by human vascular endothelial cells treated with tumor necrosis factor or interleukin 1 alpha. J Biol Chem 1988; 263:11856 - 61; PMID: 3261295
- Pinsky DJ, Naka Y, Liao H, Oz MC, Wagner DD, Mayadas TN, et al. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. A mechanism for rapid neutrophil recruitment after cardiac preservation. J Clin Invest 1996; 97:493 - 500; http://dx.doi.org/10.1172/JCI118440; PMID: 8567972
- Bombeli T, Schwartz BR, Harlan JM. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood 1999; 93:3831 - 8; PMID: 10339490
- Tabuchi A, Kuebler WM. Endothelium-platelet interactions in inflammatory lung disease. Vascul Pharmacol 2008; 49:141 - 50; http://dx.doi.org/10.1016/j.vph.2008.06.004; PMID: 18625343
- Visseren FL, Bouwman JJ, Bouter KP, Diepersloot RJ, de Groot PH, Erkelens DW. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb Haemost 2000; 84:319 - 24; PMID: 10959707
- Terada H, Baldini M, Ebbe S, Madoff MA. Interaction of influenza virus with blood platelets. Blood 1966; 28:213 - 28; PMID: 5913052
- Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest 2012; 141:1490 - 5; http://dx.doi.org/10.1378/chest.11-2860; PMID: 22383669
- Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 2012; 122:2661 - 71; http://dx.doi.org/10.1172/JCI61303; PMID: 22684106
- Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest 2006; 116:3211 - 9; http://dx.doi.org/10.1172/JCI29499; PMID: 17143330
- Grommes J, Alard JE, Drechsler M, Wantha S, Mörgelin M, Kuebler WM, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med 2012; 185:628 - 36; http://dx.doi.org/10.1164/rccm.201108-1533OC; PMID: 22246174
- Ng HH, Narasaraju T, Phoon MC, Sim MK, Seet JE, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol 2012; 92:287 - 95; http://dx.doi.org/10.1016/j.yexmp.2012.03.003; PMID: 22421441
- Birukova AA, Xing J, Fu P, Yakubov B, Dubrovskyi O, Fortune JA, et al. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol 2010; 299:L652 - 63; http://dx.doi.org/10.1152/ajplung.00202.2009; PMID: 20729389
- Walsh KB, Teijaro JR, Rosen H, Oldstone MB. Quelling the storm: utilization of sphingosine-1-phosphate receptor signaling to ameliorate influenza virus-induced cytokine storm. Immunol Res 2011; 51:15 - 25; http://dx.doi.org/10.1007/s12026-011-8240-z; PMID: 21901448