
Abstract
Metagenomics provides a means of assessing the total genetic pool of all the microbes in a particular environment, in a culture-independent manner. It has revealed unprecedented diversity in microbial community composition, which is further reflected in the encoded functional diversity of the genomes, a large proportion of which consists of novel genes. Herein, we review both sequence-based and functional metagenomic methods to uncover novel genes and outline some of the associated problems of each type of approach, as well as potential solutions. Furthermore, we discuss the potential for metagenomic biotherapeutic discovery, with a particular focus on the human gut microbiome and finally, we outline how the discovery of novel genes may be used to create bioengineered probiotics.
Introduction
Metagenomics is a term that describes both a field of scientific research and a set of techniques that enables the culture-independent analysis of a microbial community in any environmental sample.Citation1 The field has grown exponentially in the last 10 to 15 years; allowing researchers unparalleled access to the “uncultured majority”Citation2 of our microbial counterparts in our environments and closer to home, on and in our own bodies. This review aims to provide a concise overview of the principal methods of metagenomic analysis and how metagenomics can be used for the discovery of novel genetic loci. We will also discuss the potential uses of such genes for the creation of bioengineered probiotics, as well as the potential for the discovery of novel biotherapeutics for human medicine. A wealth of information has been gained in the relatively short history of metagenomics and thus, it is timely to discuss the future directions of this ever expanding field of research.
It has been estimated that there are more than 1030 microbial cells on Earth; 9 orders of magnitude greater than the number of known stars in the universe.Citation3,Citation4 It is estimated that over 99% of microbes in many environments are, as yet, uncultured,Citation5 while cultured prokaryotic representatives are overwhelmingly from just four phyla: Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria.Citation6,Citation7 Metagenomics provides a means to access, characterize, and quantify this untapped diversity of microbes and discover novel genes, metabolic pathways, and important products with biotechnological, pharmaceutical, and medical relevance.
Metagenomic analysis can take a sequence-based or a functional approach (or a combination of both) to study a complex microbial community, but always begins with the isolation of DNA from the environment of interest.
Sample DNA Isolation
Careful and considered isolation of total metagenomic DNA is the key step to ensure the sample obtained is truly representative of the community as a whole and is of high-quality, high-purity, and as unbiased as possible. Due to the obvious heterogeneity of environmental samples it is difficult to lyse cells of all species with equal efficacy. Harsh lysis methods may result in physical damage to DNA, while a more gentle approach is likely to result in an under-representation of DNA from more difficult to lyse gram-positive and archaeal members of the community.Citation8 This is a particular issue with regard to recovery of DNA from low abundance species.
Different methods can be used to access the DNA, such as one or a combination of mechanical, enzymatic, chemical, or temperature-based cell lysis. Extraction can either be direct or indirect. Direct extraction is performed from the environmental sample of interest by cell lysis and subsequent separation of the DNA from sample particles and cell debris,Citation9 while indirect extraction first involves the separation of cells from the environmental sample followed by subsequent lysis.Citation10,Citation11 There are numerous methods to extract metagenomic DNA that have been optimized to suit the nature of the particular sample in question and which take into consideration the physico-chemical conditions, microbial make-up, and origin of the environmental sample.Citation11-Citation14 For example, Salonen and coworkersCitation14 compared 4 different methods of metagenomic DNA extraction from a human fecal sample and found that the average DNA yield varied from approximately 40 µg to 1000 µg, depending on the method used, while variations of 10- to 20-fold between methods were observed for yields of archaeal DNA. Two methods were studied in further detail and a method using repeated bead-beating and sequential precipitation steps was deemed most suitable. Despite a lower initial DNA yield, it was noted that the effectiveness of cell breakage was the main factor affecting the final microbial composition and level of diversity.Citation14
Strategies to Identify Novel Genes
Sequence-based metagenomics
Traditional metagenomic sequencing relied upon Sanger sequencing, which produces relatively long reads of greater than 700 base pairs (bp) with a low error rate.Citation15 However, the development of next-generation sequencing (NGS) technologies coupled with significant reductions in sequencing costs has resulted in a movement away from Sanger sequencing as the gold standard, primarily due to the relative high cost and intensive cloning required by this approach compared with NGS methods such as 454 and Illumina. One of the first large scale metagenomic projects employed Sanger sequencing to characterize the microbial population of the Sargasso Sea.Citation13 Over 1 billion bp of DNA was sequenced and more than 1.2 million novel genes identified, including over 700 that encoded bacterial rhodopsin proteins (proteorhodopsins). Proteorhodopsins are retinal-binding membrane proteins that function as light-activated proton pumps. At the time, proteorhodopsins had been recently discovered in bacteria using metagenomics and were previously thought to exist only in halophilic archaea (bacteriorhodopsins).Citation16 This discovery prompted a major rethink about carbon and energy flux in the world’s oceans, as it became apparent that bacteria were able to harness light energy to generate ATP non-photosynthetically.Citation17
Next-generation sequencing (NGS) technologies
Ever improving sequencing technologies coupled with dramatically decreasing costs means that a “sequence everything” approach can be applied to diverse environmental samples, followed by assembly and annotation to gain a comprehensive picture of the abundance and encoded functional potential of the microbial community members. Some of the most widely used NGS platforms include the GS-FLX 454 pyrosequencer (Roche), MiSeq, HiSeq, and Genome Analyzer II platforms (Illumina), SOLiD system (Life Technologies/Applied Biosystems), Ion Torrent and Ion Proton (Life Technologies), and the PacBio RS II (Pacific Biosciences). For detailed information and technical aspects of these technologies the reader is directed to comprehensive reviews elsewhere.Citation18-Citation21
Large-scale sequencing projects employing such technologies generate huge datasets that can consist of millions of gene sequences, and owing to the nature of metagenomes, a large proportion of these will be novel, but with no known function. This initial, untargeted sequencing approach can be subsequently used in combination with a more targeted approach which aims to identify a specific type of gene or gene family. It should be noted that we mean untargeted in relation to identifying specific genes initially, but focused rather on the overall community composition, phylogeny, and/or functional capacity. In addition, metagenome sequencing using NGS technologies followed by annotation, homology searches, and phylogenetic clustering will uncover genes that are more divergent and more interesting than the consensus genes with known sequences used to design probes or primers for initial gene-focused studies.Citation22 For example, Hess and coworkers sequenced 286 gigabytes (Gb) of cow rumen metagenomic DNA using Illumina GAIIx and HiSeq 2000 platforms. They then searched for carbohydrate active enzyme (CAZymes) sequences bioinformatically and minimized the dependence on known CAZyme sequences by searching for individual functional domains of such enzymes rather than overall sequence similarity. This approach revealed 27 755 potential candidate genes from over 2.5 million predicted open reading frames (ORFs). Interestingly 24% of the genes were most similar to “hypothetical proteins” or “predicted proteins” in the non-redundant NCBI database. The sheer volume of candidate genes identified and the novelty of many sequences validates the initial high-throughput bioinformatic search. A sub-group of genes were subsequently amplified by PCR and the activity of their encoded proteins was assessed through functional heterologous expression.Citation23
Gene targeting or metagene analysis
Identification of novel genes for particular gene families or enzyme classes can be achieved by using DNA probes or PCR. First a multiple sequence alignment of many sequences of the type of gene of interest is generated to identify the most conserved regions. Consensus primers are then designed and used to amplify target sequences from isolated metagenomic DNA. Subsequently, gene-specific primers can be designed and genome-walking can be performed to retrieve the full gene sequence and those of neighboring genes if desired.Citation24 This method is effective as large libraries do not need to be constructed and screened and novel genes can be identified irrespective of gene expression, which is a major hurdle to function-based screening (discussed later). Furthermore, full-length sequences are not required to identify genes initially, although they will have to be determined at a later stage.Citation25 This approach suffers from bias due to primer and probe design being limited to known sequences of similar gene family members as well as prejudices due to over-representation of dominant species in the metagenomic library. This will lead to PCR bias and less chance of amplification of genes from less abundant community members. Furthermore, it is unlikely that functionally homologous genes that have arisen due to convergent evolution will be identified.Citation26 While a significant limitation of this approach is its inability to uncover completely novel, non-homologous genes, this approach has nonetheless been used successfully to identify genes that encode biotechnologically and industrially relevant enzymes such as esterases, lipases, and oxidoreductases.Citation9,Citation27,Citation28
Of therapeutic relevance, a number of sequence-based screens have focused on identifying novel antimicrobial compounds. Type I and type II polyketide synthases (PKS) which are encoded on operons for biosynthesis of antibiotics and anti-cancer compounds are often the target genes. In one study, a soil metagenomic library was screened by PCR for type I PKS genes using primers that target regions flanking the highly conserved active site of the ketoacyl synthetase domain. Eleven novel type I PKS sequences were identified, with amino acid identity to known sequences ranging from 46% to 61%.Citation29 Feng and coworkers used degenerate primers to amplify ketosynthase β chain length factor (KSβ) genes from a metagenomic library and used the amplicons as probes to identify PKS containing clones. Functional analysis of the clones revealed both known and unknown antimicrobial compounds, some of which had potent activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE).Citation30 In a recent extensive study, conserved domains within non-ribosomal peptide synthetase (NRPS) and PKS genes were used as targets to screen a >15 million member cosmid mega-library. Multiplex sequencing of PCR products, followed by clustering and BLAST analyses to identify sequences of most interest, revealed 18 gene clusters predicted to encode novel glycopeptide- and lipopeptide-like antibiotics, as well as anti-cancer and immunosuppressive compounds.Citation31 A PCR-based approach was used to identify genes encoding OxyC enzymes, which are often found on biosynthetic gene clusters for glycopeptide antibiotics such as vancomycin and teicoplanin. In a twist on the previous examples, modifying enzymes found on these gene clusters were used to create 15 new sulfated glycopeptide antibiotic derivatives.Citation32,Citation33 Development of analogs, which could substitute for last-line antibiotics such as vancomycin is an important research finding. Finally, Iwai et al. used a number of sets of degenerate primers for PCR, coupled with 454 pyrosequencing to identify genes encoding aromatic dioxygenases. This approach provides a more comprehensive view of the diversity of sequences present in the environment and this sequence information can be used to design probes to recover full-length genes subsequently.Citation34
Data mining and “synthetic metagenomics”
An innovative approach to identify novel genes has been described in recent years which involves mining existing sequence databases and/or metagenomic data sets for sequences of interest, followed by chemical synthesis of selected genes.Citation35 The authors name this process “synthetic metagenomics” and successfully applied it to identify novel methyl halide transferase (MHT) enzymes, which are important in agriculture and industrial applications for more efficient production of biofuels. Eighty-nine putative MHTs were identified, with amino acid identities to a known MHT as low as 18% and an average of 28% amino acid identity between sequences. The genes included 61 bacterial, 13 fungal, 1 archaeal, and 14 from plants. The genes were codon optimized for heterologous expression in Escherichia coli and yeast cells and then chemically synthesized. Only 6% of the synthesized genes showed no MHT activity, which is remarkable considering only one was actually annotated as a MHT and only 55% were annotated as generic methyltransferases.Citation35 A similar study has also applied this approach to glycoside hydrolases.Citation36 Both demonstrate the utility of “synthetic metagenomics”, which of course could be applied to any gene of interest.
Plasmid and integron capture
Horizontal gene transfer (HGT), mediated by mobile genetic elements such as plasmids and integrons play a crucial role in bacterial evolution, adaptation, and survival.Citation37 Plasmids are likely to contain genes necessary for niche colonization and encode functions important in that environment. Furthermore, genes encoding antibiotic resistance, virulence and antimicrobial production can often be found on plasmids. Developed to capture plasmids from the human gut mobile metagenome, the TRACA (transposon-aided capture) method identified a number of novel plasmids of both gram-positive and gram-negative origin.Citation38,Citation39 TRACA could be applied to any environment once sufficient quantities of DNA can be isolated and would provide a valuable means to identify novel mobile genes within specific environments. Indeed, TRACA has been recently used to identify plasmids from bacteria in the human oral cavity and activated sludge.Citation40,Citation41
Integrons are gene recombination and expression systems. They encode an integrase protein and contain a integration site (attI) to capture genes from what are known as mobile gene cassettes which contain a conserved site-specific recombination sequence called a 59-be (base element).Citation42 By targeting conserved sequences associated with integron-gene cassettes using PCR one can retrieve full-length, novel genes from metagenomes in the absence of any other sequence information.Citation43 The overwhelming majority of integron-associated genes lack known homologs or cannot be assigned a function. A recent study found that 85% of Treponema-associated integron genes from the Human Microbiome Project (HMP) data set were of unknown function.Citation44 Furthermore, proteins with novel structure and folds have been identified among integron-gene cassettes which belong to as yet uncharacterized protein families.Citation45
Suppressive subtractive hybridization
Suppressive subtractive hybridization (SSH) is a PCR-based approach that can be used to compare and identify differences in the genomes of closely related bacterial species or differentially expressed mRNA transcripts within a cell.Citation46,Citation47 Differences between “driver” (control) and “tester” sequences are highlighted by the removal of sequences or transcripts of similar abundance. This approach has been used to identify novel sequences from complex metagenomic samples. Ninety-six bacterial and archaeal DNA fragments were identified from a rumen metagenome using this approach, of which 39 had no significant similarity to any database sequences.Citation48
More recently, SSH has been adapted to recover full-length, novel genes from metagenomes.Citation49 Degenerate primers are first used to generate multiple target-gene amplicons (“driver DNA”), which are then used as hybridization probes to capture full-length genes. “Driver DNA” is biotinylated and immobilised on streptavidin-coated magnetic beads. Metagenomic DNA (“tester DNA”) is then added to the coated beads to capture full-length genes of interest. This approach has greater specificity and less non-specific amplification compared with other PCR-based techniques, but does require the creation of a genomic DNA library. Nevertheless, this approach can retrieve numerous gene targets in a single reaction.Citation49
Functional metagenomics
The second main area of metagenomic analysis is functional metagenomics. The oft-stated, broad aims of metagenomic analyses are to discover “who is there?” and “what are they doing?” Large scale NGS projects can tell us about the former, but is limited with regard to the latter, due to the dependence on known sequences already in the databases. Functional metagenomics on the other hand suffers no such reliance on previous sequence information and is a powerful approach with the potential to discover new functions for known genes and also, completely novel genes, gene families, and their encoded proteins.
Functional metagenomics requires the creation of a metagenomic library containing fragments of community metagenomic DNA. The process follows the same general steps for the creation of a genomic DNA library; enzymatic digestion or random shearing of DNA, ligation to a suitable vector and transformation to a heterologous host.Citation50 One of the biggest technical challenges is constructing a library with sufficient coverage of the community metagenome and requires an extremely large number of clones, especially for complex environments. This is compounded further when one considers a library would need to contain 100- to 1000-fold coverage in order to possess a significant proportion of clones carrying sequences form rare (<1%) community species.Citation51 Nonetheless, it is achievable; cosmid megalibraries have been created from soil which contain >1.5 × 107 unique clones. At clone densities such as this, it is believed the library approaches saturation.Citation31
Insert size and vector
Two types of library can be constructed depending on the type of screen to be performed and the desired library size. Libraries can be small insert libraries, created using plasmids and containing inserts of <10 kb, or large insert libraries, created using fosmid (25–45 kb inserts) or cosmid (15–40 kb inserts) vectors, or BAC (bacterial artificial chromosome; 100–200 kb inserts) vectors.Citation52 Small insert libraries are maintained on high copy number plasmids with strong promoters, which are usually used in activity screens where a single gene is responsible for the activity. Large insert libraries are suitable for the identification of multi-gene encoded products, operons, and entire biochemical pathways and usually utilize low-copy number or inducible vectors. Inducible vectors are advantageous in that the library may be stably maintained at low copy, but can be induced to high copy for downstream applications such as transposon mutagenesis, as well as DNA extraction, cloning, and sequencing. Large inserts also facilitate gene neighborhood analysis and increase the probability of phylogenetic and taxonomic assignment of sequences.Citation53,Citation54
Cloning host
To date, E. coli has been the cloning host of choice for the vast majority of metagenomic projects. E. coli possesses a number of desirable attributes that make it the host of choice; in depth knowledge of its physiology and biochemistry following decades of intensive research being primary among them. Furthermore, E. coli (1) has a high transformation efficiency, (2) is somewhat promiscuous with regard to the diversity of foreign expression signals it recognizes, (3) lacks genes for restriction modification and homologous recombination, and (4) is capable of translating mRNA with diverse translation signals because the normal translational dependency on the degree of complementarity of the 3′ terminus of the 16S RNA and the Shine–Dalgarno sequence does not apply in E. coli.Citation55,Citation56 Despite these advantages, E. coli, like any expression host, is unable to express all foreign DNA due to differences in the transcriptional, translational, and posttranslational machinery of the originating organism. For example, cis-acting factors such as a promoter and a ribosome binding site (RBS) compatible with the host machinery, as well as factors that may need to be supplied in trans by the host cell, such as chaperones, transcription factors, or a compatible secretion system, are all potentially negative effectors of efficient expression.
Mathematical formulae have been developed to predict the chance of a given gene of interest being expressed in E. coli. Thirty-two sequenced bacterial genomes from three different phyla (Actinobacteria, Firmicutes, and Proteobacteria), as well as 10 archaeal genomes were used in the analysis. Approximately 40% of genes in total were predicted to be functional in E. coli, but this ranged from 7% of actinobacterial genes, to 73% of genes from Firmicutes.Citation57
Hit rates
The hit rates (i.e., the chances of identifying a gene of interest through functional metagenomic expression) are traditionally quite low and can vary widely. Several factors influence the hit rate such as the source of the metagenomic DNA, the size of the gene of interest, its abundance in the metagenome and consequently the library, the vector system and host of choice, the screen itself, and the ability of the host to successfully express the gene.Citation58 Hit rates have been shown to range from one positive hit per 2.7 megabases (Mb) of DNA screened to 1 per 3979.5 Mb.Citation58-Citation60
Strategies to Improve Heterologous Expression
Alternative or dual hosts
The use of different cloning hosts or the use of one host to maintain the library followed by transfer of the library to a different host (usually from a different phylum or genera) for screening have been shown to be successful, while demonstrating the “different host, different hit” effect.Citation55 Considering the environment of study in choosing a suitable host can increase the success of a functional metagenomic screen. Streptomyces species are commonly found in soil, are genetically amenable and produce a diverse range of medically relevant secondary metabolites such as antibiotics. Thus, numerous studies have employed Streptomyces species as screening hosts. McMahon and coworkers modified an integrative E. coli–Streptomyces cosmid vector to increase its recoverability and used it to create a cosmid metagenomic library. Initially the library was created in E. coli and then transferred via conjugation to a Streptomyces lividans mutant strain, which was defective in pigmented antibiotic production (increasing the ability to identify active clones). Screens for hemolytic activity and the production of secondary metabolites and pigments identified 12 biologically active clones due to functional expression of metagenomic DNA. The key observation however, is that none of the phenotypes were detectable in E. coli, demonstrating the utility of using a different host system.Citation61 A functional metagenomic screen to identify antibiotic production, altered colony morphology, and pigmentation identified a number of clones expressing the desired traits using a cosmid library maintained in six different proteobacterial hosts (E. coli, Pseudomonas putida, Burkholderia graminis, Caulobacter vibrioides, Ralstonia metallidurans, and Agrobacterium tumefaciens). Interestingly, there was little overlap between the active clones, revealing substantial interphylum variations in gene expression, even among more closely related species.Citation62 Other alternative hosts include Bacillus subtilis, Rhizobium leguminosarum, and a dual E. coli–Thermus thermophilus system for screening extremophilic microorganisms.Citation63-Citation65 Development of different hosts and molecular tools to make them genetically malleable will expand the repertoire and diversity of novel genes that can be recognized and heterologously expressed.
Modified vectors
The use of alternative hosts is often coupled with the use or creation of novel expression vectors that can function in multiple hosts or maximize the chances of expression in said hosts. One example of a broad host-range fosmid and BAC vector is pRS44. Created for metagenomic screening, pRS44 contains two origins of replication, can be induced from low- to high-copy with l-arabinose and contains an origin of transfer to allow conjugation to additional hosts, as well as a stabilization element (parDE). A 20 000 member clone library with insert sizes of up to 200 kb was successfully created and subsequently transferred to two other putative host organisms, Pseudomonas fluorescens and Xanthomonas campestris.Citation66 Similarly, Kakirde et al. (2010) created a BAC vector capable of replication and expression in diverse genera of gram-negative bacteria such as Escherichia, Salmonella, Enterobacter, Pseudomonas, Serratia, and Vibrio.Citation67 Recently, viral gene expression elements have been used to increase the expression and hit rate of metagenomic clones. Using phage T7 RNA polymerase to drive transcription and an inducible phage anti-termination protein to bypass many transcriptional terminators present on the insert, a 6-fold increase in the number of carbenicillin-resistant clones identified in the screen was observed.Citation68 Furthermore, the use of a vector with dual-orientation promoters allows for bi-directional transcription and the possibility to significantly increase the hit rate. Such expression does not rely on the presence of a native, insert-borne promoter, or on the orientation of the cloned insert.Citation59
Codon optimization
The majority of organisms have a particular preference for certain translation initiation codons and for overall codon usage, which is known as codon usage bias.Citation69 E. coli for example uses AUG as a start codon to initiate approximately 90% of translation, therefore non-AUG start codons such as GUG and UUG may not be efficiently recognized.Citation58 Foreign genes highly transcribed in E. coli have been shown to possess similar promoter sequences that bind the sigma factor RpoD (σ70) of E. coli.Citation70 The effect of codon usage bias on expression has been investigated by introducing synonymous mutations to the third base position of the gene encoding green fluorescent protein (GFP). Expression levels between the 154 protein variants differed by up to 250-fold. Variable expression however, did not correlate with codon bias but rather, with the stability of mRNA folding near the ribosomal binding site. Codon bias on the other hand influenced the overall translation efficiency and cellular fitness.Citation71 Interestingly a recent study suggests (on the basis of analyzing 11 diverse metagenomes) that regardless of phylogeny, microbes in the same environmental niche share common preferences for synonymous codon usage at the community level. These codon usage signatures differ between different metagenomes and can be used to predict functionally relevant genes for the community as a whole and also identify and characterize unknown genes with similar codon usage patterns to those genes.Citation72
Knowledge of such issues and data mining large metagenomic data sets will enable the design of the most suitable expression systems with the greatest chance of success and also enable the creation of novel metagenome-derived synthetic genes or operons that are optimized for expression in E. coli or another relevant host.Citation35,Citation36 An overview of the main methods to identify novel genes through metagenomics is presented in .
Figure 1. An overview of the main methods of novel gene discovery using metagenomics. A sample from the environment of interest (e.g., soil, human gastrointestinal tract, or ocean) is collected (A), total metagenomic DNA is isolated directly or indirectly from the sample either by harsh or gentle lysis (B); metagenomic DNA is subsequently subjected to sequence-based analysis (C), which can include creating a clone library followed by random Sanger sequencing or direct sequencing of metagenomic DNA using next generation (NGS) methods (C-1), gene targeting using degenerate primers designed from conserved regions of homologous sequences followed by PCR amplification (C-2), data mining of existing metagenomic sequence data sets for genes or conserved domains of interest, followed by complete synthesis of host-optimized genes (C-3); capture of novel genes from plasmids using the TRACA (transposon-aided capture) method or integron-associated genes using PCR with primers that target conserved integration site sequences (C-4). Functional metagenomics (D) involves the creation of a small- or large-insert library using a specific type of vector (plasmid, fosmid, cosmid, or bacterial artificial chromosome [BAC]) depending on the needs of the user or aims of the project (D-1), choosing a suitable heterologous host for expression of metagenomic DNA. The most widely used host is E. coli, but species of Streptomyces and Bacillus, for example, have also been utilized. The library is maintained in 96- or 384-well micro-titer plates and stored at –80 °C (D-2). Clones containing metagenomic DNA can be screened to identify a phenotype of interest by replicating all or a portion of the library into new micro-titer plates or onto agar plates. Growth of library clones can be assessed for production of antimicrobials, pigmentation, altered morphology, or increased resistance to various stressors relative to control strains carrying empty vectors (D-3). Transposon mutagenesis may be performed on positive clones to identify the gene or genes of interest (D-4), which may be subsequently cloned as an isolated gene(s) to confirm the phenotype (D-5). Novel genes are discovered using sequence-based or functional metagenomic strategies, or a combination of both (E).
![Figure 1. An overview of the main methods of novel gene discovery using metagenomics. A sample from the environment of interest (e.g., soil, human gastrointestinal tract, or ocean) is collected (A), total metagenomic DNA is isolated directly or indirectly from the sample either by harsh or gentle lysis (B); metagenomic DNA is subsequently subjected to sequence-based analysis (C), which can include creating a clone library followed by random Sanger sequencing or direct sequencing of metagenomic DNA using next generation (NGS) methods (C-1), gene targeting using degenerate primers designed from conserved regions of homologous sequences followed by PCR amplification (C-2), data mining of existing metagenomic sequence data sets for genes or conserved domains of interest, followed by complete synthesis of host-optimized genes (C-3); capture of novel genes from plasmids using the TRACA (transposon-aided capture) method or integron-associated genes using PCR with primers that target conserved integration site sequences (C-4). Functional metagenomics (D) involves the creation of a small- or large-insert library using a specific type of vector (plasmid, fosmid, cosmid, or bacterial artificial chromosome [BAC]) depending on the needs of the user or aims of the project (D-1), choosing a suitable heterologous host for expression of metagenomic DNA. The most widely used host is E. coli, but species of Streptomyces and Bacillus, for example, have also been utilized. The library is maintained in 96- or 384-well micro-titer plates and stored at –80 °C (D-2). Clones containing metagenomic DNA can be screened to identify a phenotype of interest by replicating all or a portion of the library into new micro-titer plates or onto agar plates. Growth of library clones can be assessed for production of antimicrobials, pigmentation, altered morphology, or increased resistance to various stressors relative to control strains carrying empty vectors (D-3). Transposon mutagenesis may be performed on positive clones to identify the gene or genes of interest (D-4), which may be subsequently cloned as an isolated gene(s) to confirm the phenotype (D-5). Novel genes are discovered using sequence-based or functional metagenomic strategies, or a combination of both (E).](/cms/asset/a8f91555-4619-4b3a-b9ac-37f11fb870cf/kvir_a_10927208_f0001.gif)
Functional Metagenomic Discovery of Therapeutically Relevant Compounds
The vast majority of microbes are as yet uncultured and many will harbor the ability to produce therapeutically relevant novel compounds. By utilizing one or more of the strategies mentioned above to increase the chances of successful heterologous expression, there is potential to discover such novel compounds from uncultured members of well-known producers such as the actinomycetes and also from novel microbial species. Nevertheless, novel compounds have been successfully discovered using functional metagenomics. The antibiotics turbomycin A and B were discovered, somewhat serendipitously, following a functional screen for hemolytic clones in an E. coli metagenomic library. Turbomycin A had been reported previously as a fungal metabolite, but not bacterial, while turbomycin B was a novel compound. The production of both antibiotics is proposed to occur via a single gene on the insert coupled with endogenous indole production from the E. coli host. Both antibiotics displayed broad-spectrum activity against a number of gram-positive and gram-negative pathogens.Citation73
Gene clusters for the biosynthesis of novel compounds have often been found to be silent in both culturable organisms and also when cloned and expressed in a heterologous host. To overcome this, clones found to harbor PKS genes were subsequently screened for the presence of putative transcription factors in the neighboring DNA, which usually tightly regulate such gene clusters. By cloning the genes encoding the transcription factors (under the control of the strong, constitutive promoter of the erythromycin resistance gene ermE) and subsequently transforming the corresponding PKS clone with the expression plasmid, a dramatic increase in expression was observed and a clone producing an antibacterial metabolite was identified. The compound, tetarimycin A, is a gram-positive specific antibiotic with potent activity against MRSA. The authors suggest that there are likely to be more members of this class of novel antibiotic encoded by similar gene clusters present in the environment.Citation74 A combination of sequence-based metagenomics and heterologous expression using a transcription factor (similar to the example above) has identified a compound with inhibitory activity against disease-related protein kinases, while another study identified a compound with potent anti-tumor activity.Citation75,Citation76
Therapeutics from Within: “Bioprospecting” the Human Microbiome
A microbiome is the totality of all microbes, their genes and interactions in a particular environment. The sequencing of the human genome was a historic scientific achievement and heralded exciting opportunities to decipher many of the intricacies of human diseases and conditions. It soon became apparent, however, that studying the human genome and organism as a single entity gave an incomplete picture without considering the role of the human microbiome.Citation77,Citation78 While the previous examples have dealt with the identification of novel genes that encode secondary metabolites such as antibiotics, the majority of these have been identified in the external environment. Large-scale metagenomic research projects, such as Metagenomics of the Human Intestinal Tract (MetaHit), the Human Microbiome Project (HMP), and ELDERMET, among others,Citation79-Citation83 have focused on looking within the human body, not only for novel genes and compounds, but also for novel species and for changes in the abundance and diversity of extant species of the human microbiome and how these may be “mined” or manipulated in a positive way for human health.
Thuricin CD was discovered from a screen of more than 30 000 colonies from human faecal samples. Produced by Bacillus thuringiensis, thuricin CD is a novel bacteriocin with narrow-spectrum, potent antimicrobial activity against clinical isolates of Clostridium difficile. Importantly thuricin is as effective as the clinically-indicated antibiotics, vancomycin, and metronidazole, in killing C. difficile and causes negligible “collateral damage” to other members of the gut microbiota in an ex vivo colon model, making it an attractive therapeutic compound to pursue for clinical application. While thuricin was not identified using a metagenomics approach, its discovery and its desirable properties indicate that applying sequence-based or functional metagenomic approaches (or a combination of both) could identify similar and also other novel antimicrobials.Citation84,Citation85 Indeed, similar to the in silico data-mining approach mentioned previously, gene clusters similar to that of thuricin have been identified in genomic and metagenomic sequence data sets.Citation86 Using the amino acid sequences of the radical S’-adenosylmethionine (SAM) proteins (TrnC and TrnD) encoded on the thuricin gene cluster as driver sequences, 100 TrnC and 54 TrnD homologs were identified in the genomes of 112 unique microbial strains, none of which had been previously associated with bacteriocin production. Further analysis revealed 15 novel, putative thuricin gene clusters. In addition, 365 TrnC and 151 TrnD homologs were found in metagenomic data sets from diverse environments indicating the widespread presence of such genes in the environment. At the time of publication in 2011, the HMP data set would have been unavailable, but it would be interesting to investigate the abundance of such gene clusters in the different anatomical human microbiomes, particularly the gut microbiome.
Discovery of novel antimicrobials has become of paramount importance due to significant increases in global antibiotic resistance and the emergence of multiantibiotic-resistant “superbugs”.Citation87 In a recently published annual report on infection and antimicrobial resistance, England’s Chief Medical Officer, Professor Dame Sally Davies, highlights the seriousness of the problem, makes a number of recommendations and states that, “it [antimicrobial resistance] should be placed on the national risk register [specifically, the National Security Risk Assessment] and the Government should campaign for it to be given higher priority internationally, including collaborations to ensure the development of new antimicrobials and vaccines, such as Private Public Partnerships.”Citation88 The full report can be downloaded at https://www.gov.uk/government/publications/chief-medical-officer-annual-report-volume-2. Furthermore, overall societal costs due to antimicrobial-resistant infections in the European Union, Iceland, and Norway is estimated to be approximately €1.5 billion annually.Citation89 Research to identify novel antimicrobials or targets thereof has been scaled down or completely abandoned by some pharmaceutical companies.Citation90 Payne and coworkers give an insight from the perspective of a pharmaceutical company of the difficulties and costs associated with such research and the poor returns, both financial and of novel products.Citation91 This is compounded by the fact that only two new classes of antibiotics have been marketed since 1962.Citation92 Overall, these facts emphasize the need for novel and alternative strategies to combat antimicrobial resistance.
In a recent study, Eggerthella lenta, a common actinobacterial member of the gut microbiota, was shown to be solely responsible for the inactivation of the cardiac drug digoxin.Citation93 A highly upregulated, two-gene (cgr1 and 2) operon was likely responsible for reducing digoxin to an inactive form, while arginine inhibited expression and strains lacking the operon were unable to reduce digoxin. Germ-free mice colonized with E. lenta had lower serum and urine digoxin compared with mice colonized with E. lenta cgr-negative strains. Furthermore, mice fed a high protein diet had increased serum and urine digoxin indicating a reduction in digoxin inactivation, likely through increased dietary arginine.Citation93
Such studies open up new avenues for future therapeutic strategies. Profiling fecal samples for E. lenta in patients requiring digoxin could result in treatment interventions such as dietary supplementation with increased protein or arginine. The continual reduction in sequencing costs may see metagenomic sequencing become part of a personalized medicine treatment strategy or to ascertain the microbiome composition of subjects for clinical trials or before, during, and after a particular drug regimen. Bacterial β-glucuronidases of the human gut microbiota can reactivate CPT-11 (a chemotherapeutic agent used to treat colon cancer) in the human gut, causing severe diarrhea and limiting the dose intensification. Inhibitors of these β-glucuronidases have been identified that could increase therapeutic efficacy.Citation94 The opposite of the E. lenta example could also be exploited; identification of a novel probiotic species that positively correlates with drug metabolism (such as activation of a pro-drug) or detoxification that could be co-administered as a probiotic with the drug. Metagenomics can increase our understanding of the structure, function, diversity, and interactions of our microbiota which will aid the development of personalized medicine programs and the identification of novel drug targets and compounds. Furthermore, metagenomics in combination with in vitro assays and animal models may be used to identify specific microbes or combinations of microbes responsible for different drug interactions.Citation95 However, the inter-individual variability of the microbiome, even between healthy subjects remains a challenge for targeted therapies.Citation96,Citation97
Bioengineered Probiotics
“Let food be thy medicine and medicine be thy food” —Hippocrates
There has been an increasing interest in the development of bioengineered (also termed designer or recombinant) probiotics in recent years.Citation98-Citation106 Probiotics are defined as “live microorganisms which when administered in adequate amounts confer a health benefit on the host”.Citation107 Numerous research groups have engineered probiotic strains that successfully target specific pathogens and toxins, as well as developing probiotics as vaccine and drug delivery platforms.
Paton’s group has used host-cell receptor mimicry to bioengineer probiotics that bind and neutralize toxins produced by shigatoxigenic and enterotoxigenic E. coli (STEC and ETEC, respectively), as well as Vibrio cholerae. Recombinant bacteria expressing different oligosaccharide toxin receptor mimics in their lipopolysaccharide (LPS) outer core were engineered and subsequently shown to bind the toxins (shiga toxin and cholera toxin). This proved to be an extremely successful approach with up to 100% survival rates in murine models infected with virulent STEC or V. cholerae compared with controls.Citation108-Citation110 A key advantage of this approach is that development of resistance is very unlikely as a selective pressure is not imposed on the pathogen.Citation111 Identification of genes that encode specific glycosyl transferases and sugar precursors are required to create different structural receptor mimics. Metagenomics may be useful to identify novel variants of such genes for the construction of diverse mimics that can target a broader range of pathogens and toxins. Other recombinant probiotics include those that exert immunomodulatory effects and target cancer and HIV (for a review, see Paton, Morona, and PatonCitation112), that modulate the fatty acid composition of host adipose tissue by producing trans-10, cis-12 conjugated linoleic acid (CLA), and that promote in vitro intestinal wound healing mediated by probiotic secretion of human epidermal growth factor (EGF).Citation113,Citation114
The above examples demonstrate the diverse therapeutic potential of bioengineered probiotics, which could be made even more diverse through the use of food-grade lactococci and lactobacilli for receptor mimicryCitation111 and the identification and cloning of novel, metagenome-derived genes in probiotic microbes.
Metabiotechnology: application of metagenome-derived genes to bioengineered probiotics
We have previously coined the term “metabiotechnology” to describe the use of metagenomics as a robust method of identifying novel genes for use in biotechnology and the creation of bioengineered probiotics.Citation115,Citation116 This is an extension of the pathobiotechnology concept which aims to exploit stress response, host evasion, and virulence determinants of pathogenic microbes to enhance both physiological and technological robustness and stress resistance, as well as the clinical efficacy of probiotics.Citation101,Citation105,Citation106,Citation117 An increased ability to overcome the many host-imposed stresses of the gastrointestinal tract (low pH, bile, low iron concentrations, and increased osmolarity) is a desirable trait in what are sometimes promising, but physiologically or technologically fragile probiotic strains.Citation118
Listeria monocytogenes has been used as the model organism to successfully demonstrate proof-of-concept for pathobiotechnology. The betL gene (which encodes a betaine transporter involved in osmotic stress resistance and virulenceCitation119-Citation122) was cloned in Lactobacillus salivarius UCC118, resulting in a significant increase in tolerance to numerous in vitro and technological stress conditions.Citation123 Furthermore, when expressed in Bifidobacterium breve UCC2003, betL also increased low pH and osmotic stress resistance in vitro, while also increasing gastrointestinal persistence and protecting against L. monocytogenes infection in a murine model.Citation124 Using a similar approach, the bile exclusion system (BilE) of L. monocytogenes was used to increase the bile tolerance and gastrointestinal persistence of both L. lactis NZ9000 and B. breve UCC2003, while B. breve alone reduced the listerial load in murine livers.Citation100
The identification of similar genes to those mentioned above from the symbiotic gut microbiota using metagenomics could provide a large suite of genes for creating novel therapeutic probiotics and concerns regarding the use of genes from pathogens may be somewhat alleviated by these “self” genes. It is also likely that certain genes from the permanent inhabitants of the human gut microbiota will provide more robust protection against various GI stresses, as gut microbes will be better adapted for survival in this niche. Examples of genes discovered through metagenomics which could be used to create bioengineered probiotics are outlined below.
The ability to colonize the gastrointestinal tract is one of a number of important characteristics for a probiotic strain to possess.Citation125 A functional metagenomics approach has been used to identify novel genes which conferred enhanced colonization potential when expressed in E. coli.Citation126 An initial screen of a BAC library created from the murine gut metagenome identified a number of clones with enhanced ability to form biofilms. Further analysis of two clones in detail revealed 4- and 3-gene operons respectively responsible for the phenotype. The operons were cloned in isolation and expressed in E. coli. The clones exhibited between 4- and 17-fold enhanced adherence to HT-29 human cell lines in vitro, as well as enhanced intestinal colonization of antibiotic pre-treated mice, with approximately 6- to 28-fold more colony-forming units (CFUs) recovered from plated intestinal homogenates.Citation126 Such genes could be especially useful for lactococcal-based engineered probiotics; while these strains are able to reach the intestine, they cannot usually colonize.Citation127
The gastric juice in the stomach is the first major hurdle to overcome for potential probiotics. The pH of gastric juice ranges from pH 3 to pH 5, but can be as low as pH 1.5 during fasting.Citation128 The ability to survive this initial challenge significantly influences microbial numbers that reach the intestine. Metagenomic analysis of extreme environments has proved fruitful in identifying novel stress resistance genes. Fifteen novel acid resistance genes from 11 clones were identified in a small-insert E. coli metagenomic library from the extremely acidic Tinto River.Citation129 Some clones were conferred with extreme acid resistance to pH 1.8, with up to 53% survival after 60 min treatment. The various genes responsible for the phenotype were both functionally and phylogenetically diverse and some could confer broad-host range acid resistance to Pseudomonas putida and B. subtilis, in addition to E. coli. Increasing the acid resistance of probiotics thus has the potential to enable greater persistence and enhanced therapeutic effect.
Recent work in our laboratory has identified novel salt tolerance genes from the human gut microbiota using functional metagenomics.Citation130 From a screen of over 20 000 clones and subsequent transposon mutagenesis and cloning, 5 genes were identified that could confer increased salt tolerance to E. coli. The ability to respond and adapt to increases in external osmolarity caused by salt and other solutes is an important survival determinant.Citation131 Like the examples with the betL salt tolerance locus mentioned above,Citation123,Citation124 these metagenome-derived genes could be used in a similar manner to increase the stress resistance of probiotic strains.
Bile resistance is also a desirable trait in a probiotic.Citation125 In addition to bile exclusion systems, some microorganisms possess bile salt hydrolase (BSH) genes which convert conjugated bile acids into their unconjugated form.Citation132 Bile acids, and consequently BSH, can influence host physiology in the form of lipid absorption and cholesterol homeostasis.Citation132 A comprehensive metagenomic study of the human gut microbiota revealed that BSH activity is highly conserved in this environment, being found in all major bacteria and archaeal divisions in the human gut. Furthermore, BSH activity increased bile resistance in vitro and gastrointestinal survival in vivo.Citation133 Consequently probiotics and BSH have been investigated as potential cholesterol-lowering therapeutics.Citation134,Citation135 Discovery of novel BSH genes and engineering probiotics with increased BSH activity may therefore be a viable and attractive therapeutic goal.
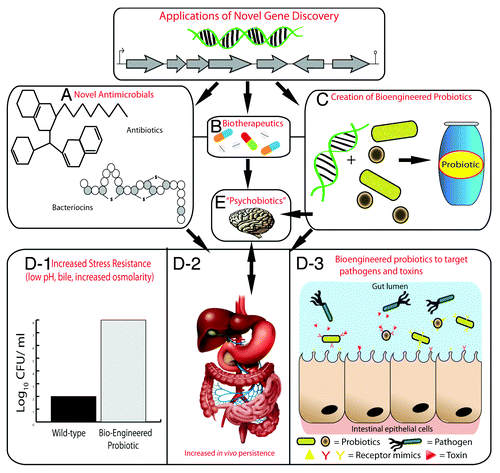
Finally, the gut–brain axis and the increasing evidence that the gut microbiota play a role in influencing processes in the brain has received much attention of late.Citation136-Citation139 In addition, some microbes are capable of producing many neuroactive compounds such as gamma-aminobutyric acid (GABA), norepinephrine, serotonin, and dopamine.Citation140 A recent study has shown that mice administered with a probiotic strain of Lactobacillus rhamnosus exhibited reduced anxiety and differential expression of GABA receptors in the brain. Animals that underwent vagotomy surgery (effectively cutting the main neural link between the gut and brain) did not display any of the effects seen in non-vagotomised animals when administered L. rhamnosus.Citation141 Such probiotics have recently been termed “psychobiotics”, a live organism that, when ingested in adequate amounts, produces a health benefit in patients suffering from psychiatric illness.Citation142 While much of the data are pre-clinical, it is certainly an exciting area to monitor in the future for further research and clinical trials. Perhaps metagenomics could be used to (1) identify genes from pathways that produce neuroactive compounds in the gut metagenome, (2) reveal the differences in the abundance of microbes capable of producing these compounds in the general population, (3) design functional assays to detect clones that produce known or novel neuroactive compounds, or (4) create bioengineered psychobiotics for specific psychiatric illnesses. Some of the potential applications of metagenomic novel gene discovery can be seen in .
Figure 2. Applications of novel genes discovered by metagenomics. Novel genes or gene clusters discovered through metagenomics will encode proteins for the production of novel antimicrobial compounds such as antibiotics and bacteriocins (A) and biotherapeutics (B). Novel genes may also be used to create bioengineered probiotic strains (C). Bioengineered probiotics can be created that are more resistant host-associated stresses of the gastrointestinal tract such as low pH, bile, and increased osmolarity (D-1), which will lead to increased survival and persistence in the gastrointestinal tract (D-2). This will ultimately increase their therapeutic efficacy; using them to target specific pathogens or toxins by expressing receptor-mimic structures on their surface, thus preventing infection (D-3). The use of probiotics that have a health benefit to persons with a psychiatric illness have been termed “psychobiotics”. The creation of bioengineered psychobiotics that can produce neuroactive compounds at an increased level or for a specific illness may be possible by identifying novel genes through metagenomics (E).
Biological Containment
The potential use of bioengineered probiotics essentially involves the use of a genetically modified organism (GMO) and raises genuine concerns regarding what would be the deliberate release of a GMO into the environment when used as a biotherapeutic. A number of containment strategies have been developed and can be categorized as either passive or active containment. With passive containment, an auxotrophic mutant is created which cannot grow without the exogenous provision of an essential metabolite. The most successful example of this is thymidine auxotrophy. Steidler et al. replaced the essential thyA gene (encoding thymidylate synthase) in L. lactis, with a gene encoding human interleukin 10 (IL-10) by chromosomal integration.Citation143 The mutant strain cannot grow in the absence of thymidine or thymine, which are absent in the environment, and cell death is induced by the phenomenon of “thymineless death”.Citation144 This strain has been used in a human clinical trial for the treatment of IBD.Citation145 Active containment strategies are usually more complex and specific for the microorganism in question, involving many gene deletions, tightly regulated gene expression, and an inherent “suicide” system.Citation146,Citation147
Conclusions and Outlook
As we have outlined, metagenomic analyses enable comprehensive investigations of microbial communities and provide unprecedented access to the genetic diversity therein. An incredible amount of information has been gained in a relatively short time with regard to the taxonomic, phylogenetic, and genetic novelty within diverse metagenomes. The development of new technologies and the continual reduction in sequencing costs will expand this at an ever increasing rate in the future, while the development of novel hosts and expression systems will increase hit rates and the variety of novel genes that can be discovered, thus helping to overcome some of the limitations of metagenomics. Development of novel and innovative screening assays will facilitate the discovery of previously unknown gene functions and novel therapeutic compounds. Greater emphasis will need to be placed on correct functional annotation of genes, as this already greatly lags behind the generation and deposition of metagenome sequences, which hampers subsequent analyses due to incorrect or absent database annotations. Synthetic biology is likely to come to the fore in the near future,Citation148,Citation149 which will be coupled with decreasing costs and new opportunities to combine metagenomics and synthetic biology, possibly to synthesize optimized collections of genes mined from metagenome data sets for functional experiments. Identification of novel species or information from metagenomes will give clues to the metabolic and physiological requirements to enable the development of culture conditions to grow novel species. Bioengineered probiotics face tough challenges if they are ever to be realized as clinical therapeutics; the creation and use of GMOs is a sensitive issue and acceptance by consumers would require undeniable demonstrations of biological containment, safety, and ultimately, efficacy in large, double-blind, placebo controlled clinical trials.Citation150 Nevertheless, the extreme seriousness of increasing rates of antibiotic resistance and dissemination means that now is the time to think outside the box and intensively search out novel antimicrobials and therapeutics. It would be unfortunate to reach a stage where consumer acceptance of such novel therapies is based solely on a lack of alternatives. Metagenomics is likely to play a key role in identifying and developing such novel therapeutics.
| Abbreviations: | ||
| DNA | = | deoxyribonucleic acid |
| bp | = | base pair |
| NGS | = | next generation sequencing |
| ATP | = | adenosine triphosphate |
| Gb | = | gigabase |
| CAZyme | = | carbohydrate-active enzyme |
| ORF | = | open reading frame |
| NCBI | = | National Centre for Biotechnology Information |
| PCR | = | polymerase chain reaction |
| PKS | = | polyketide synthase |
| KSβ | = | ketosynthase beta/chain length factor |
| MRSA | = | methicillin-resistant Staphylococcus aureus |
| VRE | = | vancomycin-resistant Enterococcus |
| NRPS | = | non-ribosomal peptide synthetase |
| MHT | = | methyl halide transferase |
| HGT | = | horizontal gene transfer |
| TRACA | = | transposon-aided capture |
| 59-be | = | 59-base element |
| HMP | = | Human Microbiome Project |
| SSH | = | suppressive subtractive hybridization |
| BAC | = | bacterial artificial chromosome |
| RBS | = | ribosome binding site |
| GFP | = | green fluorescent protein |
| Mb | = | megabases |
| SAM protein | = | S’-adenosylmethionine protein |
| MetaHit | = | Metagenomics of the Human Intestinal Tract |
| IBD | = | inflammatory bowel disease |
| STEC | = | shigatoxigenic E. coli |
| ETEC | = | enterotoxigenic E. coli |
| LPS | = | lipopolysaccharide |
| HIV | = | human immunodeficiency virus |
| CLA | = | conjugated linoleic acid |
| EGF | = | epidermal growth factor |
| CFU | = | colony forming unit |
| BSH | = | bile salt hydrolase |
| GABA | = | gamma-aminobutyric acid |
| GMO | = | genetically modified organism |
| IL-10 | = | interleukin 10 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Alimentary Pharmabiotic Centre is a research center funded by Science Foundation Ireland (SFI 07/CE/B1368). We acknowledge the continued financial assistance of the Alimentary Pharmabiotic Centre, funded by Science Foundation Ireland. J.R.M. acknowledges funding from The Royal Society which supports the bioinformatic cluster (Hive) at Cardiff University, School of Biosciences. R.D.S. is an ESCMID Research Fellow and Coordinator of ClouDx-i an FP7-PEOPLE-2012-IAPP project
References
- Sleator RD, Shortall C, Hill C. Metagenomics. Lett Appl Microbiol 2008; 47:361 - 6; http://dx.doi.org/10.1111/j.1472-765X.2008.02444.x; PMID: 19146522
- Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol 2003; 57:369 - 94; http://dx.doi.org/10.1146/annurev.micro.57.030502.090759; PMID: 14527284
- Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, van der Lelie D, Meyer F, Stevens R, et al. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 2012; 30:513 - 20; http://dx.doi.org/10.1038/nbt.2235; PMID: 22678395
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 1998; 95:6578 - 83; http://dx.doi.org/10.1073/pnas.95.12.6578; PMID: 9618454
- Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 1995; 59:143 - 69; PMID: 7535888
- Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome Biol 2002; 3:S0003; http://dx.doi.org/10.1186/gb-2002-3-2-reviews0003; PMID: 11864374
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013; 499:431 - 7; http://dx.doi.org/10.1038/nature12352; PMID: 23851394
- Furrie E. A molecular revolution in the study of intestinal microflora. Gut 2006; 55:141 - 3; http://dx.doi.org/10.1136/gut.2005.081695; PMID: 16407377
- Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR. Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol 2003; 69:6235 - 42; http://dx.doi.org/10.1128/AEM.69.10.6235-6242.2003; PMID: 14532085
- Santosa DA. Rapid extraction and purification of environmental DNA for molecular cloning applications and molecular diversity studies. Mol Biotechnol 2001; 17:59 - 64; http://dx.doi.org/10.1385/MB:17:1:59; PMID: 11280931
- Purohit MK, Singh SP. Assessment of various methods for extraction of metagenomic DNA from saline habitats of coastal Gujarat (India) to explore molecular diversity. Lett Appl Microbiol 2009; 49:338 - 44; http://dx.doi.org/10.1111/j.1472-765X.2009.02663.x; PMID: 19712192
- Delmont TO, Robe P, Clark I, Simonet P, Vogel TM. Metagenomic comparison of direct and indirect soil DNA extraction approaches. J Microbiol Methods 2011; 86:397 - 400; http://dx.doi.org/10.1016/j.mimet.2011.06.013; PMID: 21723887
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004; 304:66 - 74; http://dx.doi.org/10.1126/science.1093857; PMID: 15001713
- Salonen A, Nikkilä J, Jalanka-Tuovinen J, Immonen O, Rajilić-Stojanović M, Kekkonen RA, Palva A, de Vos WM. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods 2010; 81:127 - 34; http://dx.doi.org/10.1016/j.mimet.2010.02.007; PMID: 20171997
- Thomas T, Gilbert J, Meyer F. Metagenomics - a guide from sampling to data analysis. Microb Inform Exp 2012; 2:3; http://dx.doi.org/10.1186/2042-5783-2-3; PMID: 22587947
- Béjà O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 2000; 289:1902 - 6; http://dx.doi.org/10.1126/science.289.5486.1902; PMID: 10988064
- Béjà O, Spudich EN, Spudich JL, Leclerc M, DeLong EF. Proteorhodopsin phototrophy in the ocean. Nature 2001; 411:786 - 9; http://dx.doi.org/10.1038/35081051; PMID: 11459054
- Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012; 2012:251364; http://dx.doi.org/10.1155/2012/251364; PMID: 22829749
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 2012; 30:434 - 9; http://dx.doi.org/10.1038/nbt.2198; PMID: 22522955
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010; 11:31 - 46; http://dx.doi.org/10.1038/nrg2626; PMID: 19997069
- Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet 2011; 52:413 - 35; http://dx.doi.org/10.1007/s13353-011-0057-x; PMID: 21698376
- Schloss PD, Handelsman J. Biotechnological prospects from metagenomics. Curr Opin Biotechnol 2003; 14:303 - 10; http://dx.doi.org/10.1016/S0958-1669(03)00067-3; PMID: 12849784
- Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011; 331:463 - 7; http://dx.doi.org/10.1126/science.1200387; PMID: 21273488
- Kotik M. Novel genes retrieved from environmental DNA by polymerase chain reaction: current genome-walking techniques for future metagenome applications. J Biotechnol 2009; 144:75 - 82; http://dx.doi.org/10.1016/j.jbiotec.2009.08.013; PMID: 19712711
- Tuffin M, Anderson D, Heath C, Cowan DA. Metagenomic gene discovery: how far have we moved into novel sequence space?. Biotechnol J 2009; 4:1671 - 83; http://dx.doi.org/10.1002/biot.200900235; PMID: 19946882
- Singh B, Gautam SK, Verma V, Kumar M, Singh B. Metagenomics in animal gastrointestinal ecosystem: Potential biotechnological prospects. Anaerobe 2008; 14:138 - 44; http://dx.doi.org/10.1016/j.anaerobe.2008.03.002; PMID: 18457965
- Bell PJ, Sunna A, Gibbs MD, Curach NC, Nevalainen H, Bergquist PL. Prospecting for novel lipase genes using PCR. Microbiology 2002; 148:2283 - 91; PMID: 12177322
- Knietsch A, Waschkowitz T, Bowien S, Henne A, Daniel R. Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol 2003; 69:1408 - 16; http://dx.doi.org/10.1128/AEM.69.3.1408-1416.2003; PMID: 12620823
- Courtois S, Cappellano CM, Ball M, Francou FX, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, et al. Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol 2003; 69:49 - 55; http://dx.doi.org/10.1128/AEM.69.1.49-55.2003; PMID: 12513976
- Feng Z, Kallifidas D, Brady SF. Functional analysis of environmental DNA-derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proc Natl Acad Sci U S A 2011; 108:12629 - 34; http://dx.doi.org/10.1073/pnas.1103921108; PMID: 21768346
- Owen JG, Reddy BV, Ternei MA, Charlop-Powers Z, Calle PY, Kim JH, Brady SF. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc Natl Acad Sci U S A 2013; 110:11797 - 802; http://dx.doi.org/10.1073/pnas.1222159110; PMID: 23824289
- Banik JJ, Brady SF. Cloning and characterization of new glycopeptide gene clusters found in an environmental DNA megalibrary. Proc Natl Acad Sci U S A 2008; 105:17273 - 7; http://dx.doi.org/10.1073/pnas.0807564105; PMID: 18987322
- Banik JJ, Craig JW, Calle PY, Brady SF. Tailoring enzyme-rich environmental DNA clones: a source of enzymes for generating libraries of unnatural natural products. J Am Chem Soc 2010; 132:15661 - 70; http://dx.doi.org/10.1021/ja105825a; PMID: 20945895
- Iwai S, Chai B, Sul WJ, Cole JR, Hashsham SA, Tiedje JM. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J 2010; 4:279 - 85; http://dx.doi.org/10.1038/ismej.2009.104; PMID: 19776767
- Bayer TS, Widmaier DM, Temme K, Mirsky EA, Santi DV, Voigt CA. Synthesis of methyl halides from biomass using engineered microbes. J Am Chem Soc 2009; 131:6508 - 15; http://dx.doi.org/10.1021/ja809461u; PMID: 19378995
- Allgaier M, Reddy A, Park JI, Ivanova N, D’haeseleer P, Lowry S, Sapra R, Hazen TC, Simmons BA, VanderGheynst JS, et al. Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS One 2010; 5:e8812; http://dx.doi.org/10.1371/journal.pone.0008812; PMID: 20098679
- Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 2005; 3:722 - 32; http://dx.doi.org/10.1038/nrmicro1235; PMID: 16138100
- Jones BV, Marchesi JR. Transposon-aided capture (TRACA) of plasmids resident in the human gut mobile metagenome. Nat Methods 2007; 4:55 - 61; http://dx.doi.org/10.1038/nmeth964; PMID: 17128268
- Jones BV, Sun F, Marchesi JR. Comparative metagenomic analysis of plasmid encoded functions in the human gut microbiome. BMC Genomics 2010; 11:46; http://dx.doi.org/10.1186/1471-2164-11-46; PMID: 20085629
- Warburton PJ, Allan E, Hunter S, Ward J, Booth V, Wade WG, Mullany P. Isolation of bacterial extrachromosomal DNA from human dental plaque associated with periodontal disease, using transposon-aided capture (TRACA). FEMS Microbiol Ecol 2011; 78:349 - 54; http://dx.doi.org/10.1111/j.1574-6941.2011.01166.x; PMID: 21711368
- Zhang T, Zhang XX, Ye L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS One 2011; 6:e26041; http://dx.doi.org/10.1371/journal.pone.0026041; PMID: 22016806
- Recchia GD, Hall RM. Gene cassettes: a new class of mobile element. Microbiology 1995; 141:3015 - 27; http://dx.doi.org/10.1099/13500872-141-12-3015; PMID: 8574395
- Stokes HW, Holmes AJ, Nield BS, Holley MP, Nevalainen KM, Mabbutt BC, Gillings MR. Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA. Appl Environ Microbiol 2001; 67:5240 - 6; http://dx.doi.org/10.1128/AEM.67.11.5240-5246.2001; PMID: 11679351
- Wu YW, Rho M, Doak TG, Ye Y. Oral spirochetes implicated in dental diseases are widespread in normal human subjects and carry extremely diverse integron gene cassettes. Appl Environ Microbiol 2012; 78:5288 - 96; http://dx.doi.org/10.1128/AEM.00564-12; PMID: 22635997
- Sureshan V, Deshpande CN, Boucher Y, Koenig JE, Stokes HW, Harrop SJ, Curmi PM, Mabbutt BC, Midwest Center for Structural Genomics. Integron gene cassettes: a repository of novel protein folds with distinct interaction sites. PLoS One 2013; 8:e52934; http://dx.doi.org/10.1371/journal.pone.0052934; PMID: 23349695
- Akopyants NS, Fradkov A, Diatchenko L, Hill JE, Siebert PD, Lukyanov SA, Sverdlov ED, Berg DE. PCR-based subtractive hybridization and differences in gene content among strains of Helicobacter pylori. Proc Natl Acad Sci U S A 1998; 95:13108 - 13; http://dx.doi.org/10.1073/pnas.95.22.13108; PMID: 9789049
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, et al. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci U S A 1996; 93:6025 - 30; http://dx.doi.org/10.1073/pnas.93.12.6025; PMID: 8650213
- Galbraith EA, Antonopoulos DA, White BA. Suppressive subtractive hybridization as a tool for identifying genetic diversity in an environmental metagenome: the rumen as a model. Environ Microbiol 2004; 6:928 - 37; http://dx.doi.org/10.1111/j.1462-2920.2004.00575.x; PMID: 15305918
- Meyer QC, Burton SG, Cowan DA. Subtractive hybridization magnetic bead capture: a new technique for the recovery of full-length ORFs from the metagenome. Biotechnol J 2007; 2:36 - 40; http://dx.doi.org/10.1002/biot.200600156; PMID: 17124705
- Simon C, Daniel R. Construction of small-insert and large-insert metagenomic libraries. In: Streit, WR and Daniel, R, ed(s). Metagenomics - Methods and Protocols New York: Humana Press, 2010.
- Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 2004; 38:525 - 52; http://dx.doi.org/10.1146/annurev.genet.38.072902.091216; PMID: 15568985
- Ekkers DM, Cretoiu MS, Kielak AM, Elsas JD. The great screen anomaly--a new frontier in product discovery through functional metagenomics. Appl Microbiol Biotechnol 2012; 93:1005 - 20; http://dx.doi.org/10.1007/s00253-011-3804-3; PMID: 22189864
- Sleator RD. A beginner’s guide to phylogenetics. Microb Ecol 2013; 66:1 - 4; http://dx.doi.org/10.1007/s00248-013-0236-x; PMID: 23624570
- Sleator RD. Phylogenetics. Arch Microbiol 2011; 193:235 - 9; http://dx.doi.org/10.1007/s00203-011-0677-x; PMID: 21249334
- Gabor E, Liebeton K, Niehaus F, Eck J, Lorenz P. Updating the metagenomics toolbox. Biotechnol J 2007; 2:201 - 6; http://dx.doi.org/10.1002/biot.200600250; PMID: 17294408
- Boni IV. [Diverse molecular mechanisms for translation initiation in prokaryotes]. Mol Biol (Mosk) 2006; 40:658 - 68; http://dx.doi.org/10.1134/S002689330604011X; PMID: 16913225
- Gabor EM, Alkema WB, Janssen DB. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol 2004; 6:879 - 86; http://dx.doi.org/10.1111/j.1462-2920.2004.00640.x; PMID: 15305913
- Uchiyama T, Miyazaki K. Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 2009; 20:616 - 22; http://dx.doi.org/10.1016/j.copbio.2009.09.010; PMID: 19850467
- Lämmle K, Zipper H, Breuer M, Hauer B, Buta C, Brunner H, Rupp S. Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 2007; 127:575 - 92; http://dx.doi.org/10.1016/j.jbiotec.2006.07.036; PMID: 16963141
- Chung EJ, Lim HK, Kim JC, Choi GJ, Park EJ, Lee MH, Chung YR, Lee SW. Forest soil metagenome gene cluster involved in antifungal activity expression in Escherichia coli. Appl Environ Microbiol 2008; 74:723 - 30; http://dx.doi.org/10.1128/AEM.01911-07; PMID: 18065615
- McMahon MD, Guan C, Handelsman J, Thomas MG. Metagenomic analysis of Streptomyces lividans reveals host-dependent functional expression. Appl Environ Microbiol 2012; 78:3622 - 9; http://dx.doi.org/10.1128/AEM.00044-12; PMID: 22427497
- Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microbiol 2010; 76:1633 - 41; http://dx.doi.org/10.1128/AEM.02169-09; PMID: 20081001
- Li Y, Wexler M, Richardson DJ, Bond PL, Johnston AW. Screening a wide host-range, waste-water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol 2005; 7:1927 - 36; http://dx.doi.org/10.1111/j.1462-2920.2005.00853.x; PMID: 16309391
- Angelov A, Mientus M, Liebl S, Liebl W. A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst Appl Microbiol 2009; 32:177 - 85; http://dx.doi.org/10.1016/j.syapm.2008.01.003; PMID: 19285378
- Dobrijevic D, Di Liberto G, Tanaka K, de Wouters T, Dervyn R, Boudebbouze S, Binesse J, Blottière HM, Jamet A, Maguin E, et al. High-throughput system for the presentation of secreted and surface-exposed proteins from Gram-positive bacteria in functional metagenomics studies. PLoS One 2013; 8:e65956; http://dx.doi.org/10.1371/journal.pone.0065956; PMID: 23799065
- Aakvik T, Degnes KF, Dahlsrud R, Schmidt F, Dam R, Yu L, Völker U, Ellingsen TE, Valla S. A plasmid RK2-based broad-host-range cloning vector useful for transfer of metagenomic libraries to a variety of bacterial species. FEMS Microbiol Lett 2009; 296:149 - 58; http://dx.doi.org/10.1111/j.1574-6968.2009.01639.x; PMID: 19459950
- Kakirde KS, Wild J, Godiska R, Mead DA, Wiggins AG, Goodman RM, Szybalski W, Liles MR. Gram negative shuttle BAC vector for heterologous expression of metagenomic libraries. Gene 2011; 475:57 - 62; http://dx.doi.org/10.1016/j.gene.2010.11.004; PMID: 21112378
- Terrón-González L, Medina C, Limón-Mortés MC, Santero E. Heterologous viral expression systems in fosmid vectors increase the functional analysis potential of metagenomic libraries. Sci Rep 2013; 3:1107; http://dx.doi.org/10.1038/srep01107; PMID: 23346364
- Johnston C, Douarre PE, Soulimane T, Pletzer D, Weingart H, MacSharry J, Coffey A, Sleator RD, O’Mahony J. Codon optimisation to improve expression of a Mycobacterium avium ssp. paratuberculosis-specific membrane-associated antigen by Lactobacillus salivarius. Pathog Dis 2013; 68:27 - 38; http://dx.doi.org/10.1111/2049-632X.12040; PMID: 23620276
- Warren RL, Freeman JD, Levesque RC, Smailus DE, Flibotte S, Holt RA. Transcription of foreign DNA in Escherichia coli. Genome Res 2008; 18:1798 - 805; http://dx.doi.org/10.1101/gr.080358.108; PMID: 18701636
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009; 324:255 - 8; http://dx.doi.org/10.1126/science.1170160; PMID: 19359587
- Roller M, Lucic V, Nagy I, Perica T, Vlahovicek K. Environmental shaping of codon usage and functional adaptation across microbial communities. Nucleic Acids Res 2013; 41:8842 - 52; http://dx.doi.org/10.1093/nar/gkt673; PMID: 23921637
- Gillespie DE, Brady SF, Bettermann AD, Cianciotto NP, Liles MR, Rondon MR, Clardy J, Goodman RM, Handelsman J. Isolation of antibiotics turbomycin a and B from a metagenomic library of soil microbial DNA. Appl Environ Microbiol 2002; 68:4301 - 6; http://dx.doi.org/10.1128/AEM.68.9.4301-4306.2002; PMID: 12200279
- Kallifidas D, Kang HS, Brady SF. Tetarimycin A, an MRSA-active antibiotic identified through induced expression of environmental DNA gene clusters. J Am Chem Soc 2012; 134:19552 - 5; http://dx.doi.org/10.1021/ja3093828; PMID: 23157252
- Chang FY, Brady SF. Cloning and characterization of an environmental DNA-derived gene cluster that encodes the biosynthesis of the antitumor substance BE-54017. J Am Chem Soc 2011; 133:9996 - 9; http://dx.doi.org/10.1021/ja2022653; PMID: 21542592
- Chang FY, Brady SF. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc Natl Acad Sci U S A 2013; 110:2478 - 83; http://dx.doi.org/10.1073/pnas.1218073110; PMID: 23302687
- Feeney A, Sleator RD. The human gut microbiome: the ghost in the machine. Future Microbiol 2012; 7:1235 - 7; http://dx.doi.org/10.2217/fmb.12.105; PMID: 23075440
- Sleator RD. The human superorganism - of microbes and men. Med Hypotheses 2010; 74:214 - 5; http://dx.doi.org/10.1016/j.mehy.2009.08.047; PMID: 19836146
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al, MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011; 473:174 - 80; http://dx.doi.org/10.1038/nature09944; PMID: 21508958
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207 - 14; http://dx.doi.org/10.1038/nature11234; PMID: 22699609
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 2007; 14:169 - 81; http://dx.doi.org/10.1093/dnares/dsm018; PMID: 17916580
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al, MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59 - 65; http://dx.doi.org/10.1038/nature08821; PMID: 20203603
- Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O’Sullivan O, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012; 488:178 - 84; http://dx.doi.org/10.1038/nature11319; PMID: 22797518
- Rea MC, Dobson A, O’Sullivan O, Crispie F, Fouhy F, Cotter PD, Shanahan F, Kiely B, Hill C, Ross RP. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci U S A 2011; 108:Suppl 1 4639 - 44; http://dx.doi.org/10.1073/pnas.1001224107; PMID: 20616009
- Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A 2010; 107:9352 - 7; http://dx.doi.org/10.1073/pnas.0913554107; PMID: 20435915
- Murphy K, O’Sullivan O, Rea MC, Cotter PD, Ross RP, Hill C. Genome mining for radical SAM protein determinants reveals multiple sactibiotic-like gene clusters. PLoS One 2011; 6:e20852; http://dx.doi.org/10.1371/journal.pone.0020852; PMID: 21760885
- Nature E. The antibiotic alarm. Nature 2013; 495:141; http://dx.doi.org/10.1038/495141a; PMID: 23495392
- Davies SC. Annual Report of the Chief Medical Officer: Volume Two, 2011; Infections and the rise of antimicrobial resistance Department of Health, London 2013.
- European Medicines Agency. European Centre for Disease Prevention and Control. Joint technical report: the bacterial challenge—time to react. http://ecdc.europa.eu/en/publications/Publications/0909_TER_The_Bacterial_Challenge_Time_to_React.pdf 2009.
- Alanis AJ. Resistance to antibiotics: are we in the post-antibiotic era?. Arch Med Res 2005; 36:697 - 705; http://dx.doi.org/10.1016/j.arcmed.2005.06.009; PMID: 16216651
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 2007; 6:29 - 40; http://dx.doi.org/10.1038/nrd2201; PMID: 17159923
- Coates AR, Halls G, Hu Y. Novel classes of antibiotics or more of the same?. Br J Pharmacol 2011; 163:184 - 94; http://dx.doi.org/10.1111/j.1476-5381.2011.01250.x; PMID: 21323894
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013; 341:295 - 8; http://dx.doi.org/10.1126/science.1235872; PMID: 23869020
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010; 330:831 - 5; http://dx.doi.org/10.1126/science.1191175; PMID: 21051639
- Haiser HJ, Turnbaugh PJ. Is it time for a metagenomic basis of therapeutics?. Science 2012; 336:1253 - 5; http://dx.doi.org/10.1126/science.1224396; PMID: 22674325
- Roeselers G, Bouwman J, Venema K, Montijn R. The human gastrointestinal microbiota--an unexplored frontier for pharmaceutical discovery. Pharmacol Res 2012; 66:443 - 7; http://dx.doi.org/10.1016/j.phrs.2012.09.007; PMID: 23000437
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 308:1635 - 8; http://dx.doi.org/10.1126/science.1110591; PMID: 15831718
- Sleator RD. Probiotics -- a viable therapeutic alternative for enteric infections especially in the developing world. Discov Med 2010; 10:119 - 24; PMID: 20807472
- Sleator RD, Hill C. Rational design of improved pharmabiotics. J Biomed Biotechnol 2009; 2009:275287; http://dx.doi.org/10.1155/2009/275287; PMID: 19753318
- Watson D, Sleator RD, Hill C, Gahan CG. Enhancing bile tolerance improves survival and persistence of Bifidobacterium and Lactococcus in the murine gastrointestinal tract. BMC Microbiol 2008; 8:176; http://dx.doi.org/10.1186/1471-2180-8-176; PMID: 18844989
- Sleator RD, Hill C. ‘Bioengineered Bugs’ - a patho-biotechnology approach to probiotic research and applications. Med Hypotheses 2008; 70:167 - 9; http://dx.doi.org/10.1016/j.mehy.2007.03.008; PMID: 17452084
- Sleator RD, Hill C. New frontiers in probiotic research. Lett Appl Microbiol 2008; 46:143 - 7; http://dx.doi.org/10.1111/j.1472-765X.2007.02293.x; PMID: 18028323
- Sleator RD, Hill C. Designer probiotics: a potential therapeutic for Clostridium difficile?. J Med Microbiol 2008; 57:793 - 4; http://dx.doi.org/10.1099/jmm.0.47697-0; PMID: 18480340
- Sleator RD, Hill C. Battle of the bugs. Science 2008; 321:1294 - 5; http://dx.doi.org/10.1126/science.321.5894.1294b; PMID: 18772416
- Sleator RD, Hill C. Patho-biotechnology; using bad bugs to make good bugs better. Sci Prog 2007; 90:1 - 14; http://dx.doi.org/10.3184/003685007780440530; PMID: 17455762
- Sleator RD, Hill C. Patho-biotechnology: using bad bugs to do good things. Curr Opin Biotechnol 2006; 17:211 - 6; http://dx.doi.org/10.1016/j.copbio.2006.01.006; PMID: 16459072
- FAO/WHO. Expert consultation on evaluation of health and nutritional properties of probiotics in food including milk powder with live lactic acid bacteria. FAO/WHO (Food and Agriculture Organization/World Health Organization) Cordoba, Argentina: WHO 2001.
- Paton AW, Morona R, Paton JC. A new biological agent for treatment of Shiga toxigenic Escherichia coli infections and dysentery in humans. Nat Med 2000; 6:265 - 70; http://dx.doi.org/10.1038/73111; PMID: 10700227
- Focareta A, Paton JC, Morona R, Cook J, Paton AW. A recombinant probiotic for treatment and prevention of cholera. Gastroenterology 2006; 130:1688 - 95; http://dx.doi.org/10.1053/j.gastro.2006.02.005; PMID: 16697733
- Paton AW, Jennings MP, Morona R, Wang H, Focareta A, Roddam LF, Paton JC. Recombinant probiotics for treatment and prevention of enterotoxigenic Escherichia coli diarrhea. Gastroenterology 2005; 128:1219 - 28; http://dx.doi.org/10.1053/j.gastro.2005.01.050; PMID: 15887106
- Paton AW, Morona R, Paton JC. Designer probiotics for prevention of enteric infections. Nat Rev Microbiol 2006; 4:193 - 200; http://dx.doi.org/10.1038/nrmicro1349; PMID: 16462752
- Paton AW, Morona R, Paton JC. Bioengineered microbes in disease therapy. Trends Mol Med 2012; 18:417 - 25; http://dx.doi.org/10.1016/j.molmed.2012.05.006; PMID: 22721939
- Rosberg-Cody E, Stanton C, O’Mahony L, Wall R, Shanahan F, Quigley EM, Fitzgerald GF, Ross RP. Recombinant lactobacilli expressing linoleic acid isomerase can modulate the fatty acid composition of host adipose tissue in mice. Microbiology 2011; 157:609 - 15; http://dx.doi.org/10.1099/mic.0.043406-0; PMID: 21178166
- Choi HJ, Ahn JH, Park SH, Do KH, Kim J, Moon Y. Enhanced wound healing by recombinant Escherichia coli Nissle 1917 via human epidermal growth factor receptor in human intestinal epithelial cells: therapeutic implication using recombinant probiotics. Infect Immun 2012; 80:1079 - 87; http://dx.doi.org/10.1128/IAI.05820-11; PMID: 22184415
- Culligan EP, Hill C, Sleator RD. Probiotics and gastrointestinal disease: successes, problems and future prospects. Gut Pathog 2009; 1:19; http://dx.doi.org/10.1186/1757-4749-1-19; PMID: 19930635
- Culligan EP, Marchesi JR, Hill C, Sleator RD. Mining the human gut microbiome for novel stress resistance genes. Gut Microbes 2012; 3:394 - 7; http://dx.doi.org/10.4161/gmic.20984; PMID: 22688726
- Sleator RD, Hill C. Engineered pharmabiotics with improved therapeutic potential. Hum Vaccin 2008; 4:271 - 4; http://dx.doi.org/10.4161/hv.4.4.6315; PMID: 18682694
- Mattila-Sandholm T, Myllärinen P, Crittenden R, Mogensen G, Fondén R, Saarela M. Technological challenges for future probiotic foods. Int Dairy J 2002; 12:173 - 82; http://dx.doi.org/10.1016/S0958-6946(01)00099-1
- Sleator RD, Francis GA, O’Beirne D, Gahan CG, Hill C. Betaine and carnitine uptake systems in Listeria monocytogenes affect growth and survival in foods and during infection. J Appl Microbiol 2003; 95:839 - 46; http://dx.doi.org/10.1046/j.1365-2672.2003.02056.x; PMID: 12969299
- Sleator RD, Gahan CG, Abee T, Hill C. Identification and disruption of BetL, a secondary glycine betaine transport system linked to the salt tolerance of Listeria monocytogenes LO28. Appl Environ Microbiol 1999; 65:2078 - 83; PMID: 10224004
- Sleator RD, O’Driscoll B, Hill C, Gahan CGM. Analysis of the role of betL in contributing to the growth and survival of Listeria monocytogenes LO28. Int J Food Microbiol 2000; 60:261 - 8; http://dx.doi.org/10.1016/S0168-1605(00)00316-0; PMID: 11016615
- Hoffmann RF, McLernon S, Feeney A, Hill C, Sleator RD. A single point mutation in the listerial betL σ (A) -dependent promoter leads to improved osmo- and chill-tolerance and a morphological shift at elevated osmolarity. Bioengineered 2013; 4:4; http://dx.doi.org/10.4161/bioe.24094; PMID: 23478432
- Sheehan VM, Sleator RD, Fitzgerald GF, Hill C. Heterologous expression of BetL, a betaine uptake system, enhances the stress tolerance of Lactobacillus salivarius UCC118. Appl Environ Microbiol 2006; 72:2170 - 7; http://dx.doi.org/10.1128/AEM.72.3.2170-2177.2006; PMID: 16517668
- Sheehan VM, Sleator RD, Hill C, Fitzgerald GF. Improving gastric transit, gastrointestinal persistence and therapeutic efficacy of the probiotic strain Bifidobacterium breve UCC2003. Microbiology 2007; 153:3563 - 71; http://dx.doi.org/10.1099/mic.0.2007/006510-0; PMID: 17906153
- FAO/WHO. Working Group on Drafting Guidelines for the Evaluation of Probiotics in Food. 2002. Available from: http://www.who.int/foodsafety/fs_management/en/probiotic_guidelines.pdf
- Yoon MY, Lee KM, Yoon Y, Go J, Park Y, Cho YJ, Tannock GW, Yoon SS. Functional screening of a metagenomic library reveals operons responsible for enhanced intestinal colonization by gut commensal microbes. Appl Environ Microbiol 2013; 79:3829 - 38; http://dx.doi.org/10.1128/AEM.00581-13; PMID: 23584783
- Kimoto H, Nomura M, Kobayashi M, Mizumachi K, Okamoto T. Survival of lactococci during passage through mouse digestive tract. Can J Microbiol 2003; 49:707 - 11; http://dx.doi.org/10.1139/w03-092; PMID: 14735220
- Cotter PD, Hill C. Surviving the acid test: responses of gram-positive bacteria to low pH. [table of contents.] Microbiol Mol Biol Rev 2003; 67:429 - 53; http://dx.doi.org/10.1128/MMBR.67.3.429-453.2003; PMID: 12966143
- Guazzaroni ME, Morgante V, Mirete S, González-Pastor JE. Novel acid resistance genes from the metagenome of the Tinto River, an extremely acidic environment. Environ Microbiol 2013; 15:1088 - 102; http://dx.doi.org/10.1111/1462-2920.12021; PMID: 23145860
- Culligan EP, Sleator RD, Marchesi JR, Hill C. Functional metagenomics reveals novel salt tolerance loci from the human gut microbiome. ISME J 2012; 6:1916 - 25; http://dx.doi.org/10.1038/ismej.2012.38; PMID: 22534607
- Sleator RD, Hill C. Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol Rev 2002; 26:49 - 71; http://dx.doi.org/10.1111/j.1574-6976.2002.tb00598.x; PMID: 12007642
- Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006; 47:241 - 59; http://dx.doi.org/10.1194/jlr.R500013-JLR200; PMID: 16299351
- Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A 2008; 105:13580 - 5; http://dx.doi.org/10.1073/pnas.0804437105; PMID: 18757757
- Jones ML, Tomaro-Duchesneau C, Martoni CJ, Prakash S. Cholesterol lowering with bile salt hydrolase-active probiotic bacteria, mechanism of action, clinical evidence, and future direction for heart health applications. Expert Opin Biol Ther 2013; 13:631 - 42; http://dx.doi.org/10.1517/14712598.2013.758706; PMID: 23350815
- Kumar M, Nagpal R, Kumar R, Hemalatha R, Verma V, Kumar A, Chakraborty C, Singh B, Marotta F, Jain S, Yadav H.. Cholesterol-lowering probiotics as potential biotherapeutics for metabolic diseases. Exp Diabetes Res. 2012; 2012:902917
- Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 2012; 13:701 - 12; http://dx.doi.org/10.1038/nrn3346; PMID: 22968153
- Barrett E, Ross RP, O’Toole PW, Fitzgerald GF, Stanton C. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol 2012; 113:411 - 7; http://dx.doi.org/10.1111/j.1365-2672.2012.05344.x; PMID: 22612585
- Collins SM, Bercik P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 2009; 136:2003 - 14; http://dx.doi.org/10.1053/j.gastro.2009.01.075; PMID: 19457424
- Desbonnet L, Garrett L, Clarke G, Bienenstock J, Dinan TG. The probiotic Bifidobacteria infantis: An assessment of potential antidepressant properties in the rat. J Psychiatr Res 2008; 43:164 - 74; http://dx.doi.org/10.1016/j.jpsychires.2008.03.009; PMID: 18456279
- Lyte M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. Bioessays 2011; 33:574 - 81; http://dx.doi.org/10.1002/bies.201100024; PMID: 21732396
- Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, Bienenstock J, Cryan JF. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A 2011; 108:16050 - 5; http://dx.doi.org/10.1073/pnas.1102999108; PMID: 21876150
- Dinan TG, Stanton C, Cryan JF. Psychobiotics: a novel class of psychotropic. Biol Psychiatry 2013; 74:720 - 6; http://dx.doi.org/10.1016/j.biopsych.2013.05.001; PMID: 23759244
- Steidler L, Neirynck S, Huyghebaert N, Snoeck V, Vermeire A, Goddeeris B, Cox E, Remon JP, Remaut E. Biological containment of genetically modified Lactococcus lactis for intestinal delivery of human interleukin 10. Nat Biotechnol 2003; 21:785 - 9; http://dx.doi.org/10.1038/nbt840; PMID: 12808464
- Ahmad SI, Kirk SH, Eisenstark A. Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu Rev Microbiol 1998; 52:591 - 625; http://dx.doi.org/10.1146/annurev.micro.52.1.591; PMID: 9891809
- Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, van Deventer SJ, Neirynck S, Peppelenbosch MP, Steidler L. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin Gastroenterol Hepatol 2006; 4:754 - 9; http://dx.doi.org/10.1016/j.cgh.2006.03.028; PMID: 16716759
- Kong W, Brovold M, Koeneman BA, Clark-Curtiss J, Curtiss R 3rd. Turning self-destructing Salmonella into a universal DNA vaccine delivery platform. Proc Natl Acad Sci U S A 2012; 109:19414 - 9; http://dx.doi.org/10.1073/pnas.1217554109; PMID: 23129620
- Bahey-El-Din M. Lactococcus lactis-based vaccines from laboratory bench to human use: an overview. Vaccine 2012; 30:685 - 90; http://dx.doi.org/10.1016/j.vaccine.2011.11.098; PMID: 22154771
- O’ Driscoll A, Sleator RD. Synthetic DNA: the next generation of big data storage. Bioengineered 2013; 4:123 - 5; http://dx.doi.org/10.4161/bioe.24296; PMID: 23514938
- Sleator RD. Digital biology: a new era has begun. Bioengineered 2012; 3:311 - 2; http://dx.doi.org/10.4161/bioe.22367; PMID: 23099453
- Chakrabarty AM. Bioengineered bugs, drugs and contentious issues in patenting. Bioeng Bugs 2010; 1:2 - 8; http://dx.doi.org/10.4161/bbug.1.1.9850; PMID: 21327122