
Abstract
The term microbiome refers to the genetic material of the catalog of microbial taxa associated with humans. As in all ecosystems, the microbiota reaches a dynamic equilibrium in the human body, which can be altered by environmental factors and external stimuli. Metagenomics is a relatively new field of study of microbial genomes within diverse environmental samples, which is of increasing importance in microbiology. The introduction of this ecological perception of microbiology is the key to achieving real knowledge about the influence of the microbiota in human health and disease. The aim of this review is to summarize the link between the human microbiota (focusing on the intestinal, vaginal, skin, and airway body sites) and health from this ecological point of view, highlighting the contribution of metagenomics in the advance of this field.
Introduction
The term microbiota refers to the microbial population present within the human body,Citation1 including bacteria, viruses, archea, protozoans, and fungi. Every individual human harbors 10–100 trillion symbiotic microbial cells, with gut bacteria being the most abundant.Citation2 In this context, the microbiota is implicitly assumed to be similar to a multicelled organ. However, due to the abundance of microhabitats in the human body and the large number of interactions between the different species with the host and the external environment, microbiota can also be conceptualized as a dynamic ecological community ().Citation3 Indeed, each microbial community within the human body has its own structure, depending on the exact environment where it is localized.Citation4 Many essential body processes require the presence of diverse microorganisms, as they provide the host with essential nutrients, metabolize indigestible compounds, and defend against colonization by opportunistic pathogens, as well as possessing immunomodulatory properties (for more details see also ref. Citation5).
Figure 1. The human microbiome conceptualized as a dynamic ecological community. Inter-relations between all the components of the ecosystem lead to an equilibrium state required to maintain the health status of the host.
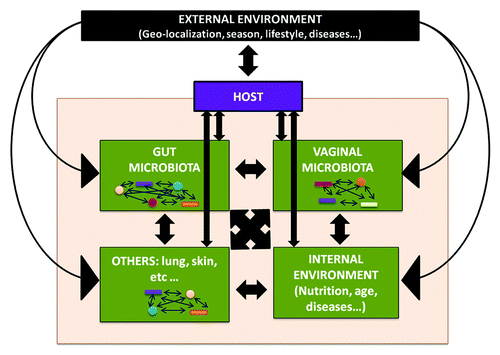
As with all ecosystems, a balance exists between the human body and the microbiota. However, this dynamic equilibrium can be altered at any time by environmental factors and external interferences, such as the use of antibiotics.Citation6 These alterations frequently result in microbial imbalances on or inside the body, a phenomenon also called dysbiosis. Thus, in some ecosystems, such as the gut, a high biodiversity is associated with a healthy status, while low biodiversity is more linked to pathological conditions.Citation7 On the contrary, in other ecosystems such as the vagina, high diversity is directly associated with illness such as vaginosis.Citation8 Nevertheless, a disruption of normal microbiota profile or biodiversity is frequently related with a physiopathological condition for the host. From this perspective, there is an increasing interest in the use of microorganisms (i.e., probiotics) to resolve dysbioses.
To achieve the study of this biodiversity, traditional methods were based for many years on culture dependent techniques. However, although the use of these techniques provided a large and interesting set of data, they also resulted in an erroneous view of the human microbiota composition in certain cases. Indeed, many microorganisms need special growth conditions, such as the extremely oxygen sensitive (EOS) bacteria, that makes their culturing and even their detection difficult;Citation9 whereas others have never been grown in culture and may require special, as yet unknown, growth conditions preventing their identification by culture-dependent methods.Citation4 Recently, several culture-independent techniques have been developed allowing for a qualitative and quantitative means of identification and are mostly based on PCR and DNA hybridization techniques. These simple methods have completely changed the notion of the human microbiota, opening the door to new and more complete fields such as metagenomics, which is the study of microbial genomes within diverse environmental samples.Citation10 Metagenomics was first introduced in 1998Citation11 and is now a widely used technique that has revolutionized the study of the microbiota as a result of its ability to generate a comprehensive catalog of microbial sequences present in various different ecological niches within a large host organism such as humans.
In the past, only single organisms were considered important regarding pathogenic interactions with humans. For this reason, only a few studies of either microbial communities or non-pathogenic bacteria were performed, since these bacterial types were believed not to impact on the well-being of humans. Fortunately, this incorrect concept and underestimation of the importance of the human microbiota has changed in the last few years.
Genomic Approaches to Study the Human Microbiota: Defining the Human Microbiome
The term microbiome refers to the genetic material of the catalog of microbial taxa associated with humans.Citation12 Being frequently confused with the term microbiota, the microbiome was first defined by Joshua Lederberg in 2001Citation13 and is sometimes referred to as our second genome.Citation14 Although great effort has been applied to the study of the human microbiota (e.g., the Human Microbiome Project, which was launched in 2008 with a funding of $157 million to determine whether there is a shared core microbiome among individuals),Citation15 the differences among individuals is huge compared with genomic variations.Citation12 This fascinating reality has potential implications in personal medicine, whereby microbiota composition analysis could be used routinely in clinical practice to diagnose dysbiosis-related disorders.Citation16
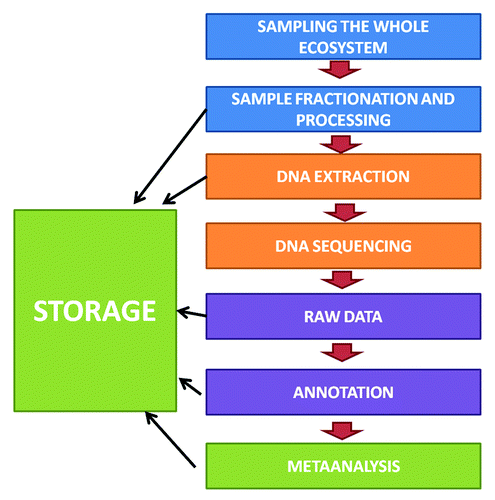
Although the term metagenomics was originally coined for the shotgun characterization of total DNA, it is also presently being applied for studies of marker genes such as the 16S rRNA.Citation12 Both shotgun characterization of total DNA and marker genes are used mainly to analyze community structure of the human microbiome. Sequencing the full-length 16S rRNA gene was performed classically by the Sanger dideoxy chain termination technique; today, next generation sequencing methodologies such as Ion Torrent PGM sequencing of 16S rRNA gene-based ampliconsCitation17 reduces the cost and increases the depth. Although a bacterial species is hard to define, the current definition requires a minimum of 97% identity in the 16S rRNA.Citation4 However, although 16S rRNA sequence is the best measure of low-abundance organismsCitation18 and it has been widely used allowing for cross-study comparisons,Citation15 more comprehensive results are found without focusing on target regions such as the 16S rRNA. Shotgun characterization allows either the cataloging of genes of organisms present in a communityCitation4 or the analysis of individual genomes in the ecosystem under study.Citation19 All these powerful tools provide information on community diversity and structureCitation20 even if caution is necessary in the sampling method. Huge variations can be introduced by methodological bias.Citation17 The flowchart diagram () briefly describes the main steps for metagenomic analysis. Several microecological processes have been defined using metagenomics such as microbiota establishment,Citation21 effect of diet on gut microbiome,Citation22 and microbiota changes in inflammatory bowel disease (IBD)Citation23 and obesity.Citation23,Citation24
Figure 2. Metagenomic approach general flowchart. After sampling the whole ecosystem and processing the samples, DNA is extracted and sequenced generating raw data. The sequences generated are annotated and submitted to metaanalysis. There are several points to store samples or data.
The Role of Microbiome in Human Health
As we can deduce from , where the human microbiota (microbiome by extension) has been represented as a collection of dynamic ecological communities, the perturbation of one of these communities has a direct impact on health and well-being of the host.Citation25,Citation26 As a result of recent technological advances, the vagina and the gastrointestinal tract (GIT) have been well characterized (and are to date the most studied human micro-ecosystems) representing low and high complexity communities, respectively.Citation16 Although dysbiosis has been mainly linked to the GIT, it can take place on any exposed surface or mucus membrane such as the vagina, the skin, or the respiratory system. Indeed, this kind of variation in the microbial population can strongly impact human health. Nowadays, the challenge of linking microbiome to human health and disease is being confronted by different research teams around the world. They are currently studying different disease states to identify potential correlations and ecological models of community structure and function in order to understand the dynamics of all ecosystems that comprise the human microbiome.
GIT ecosystem
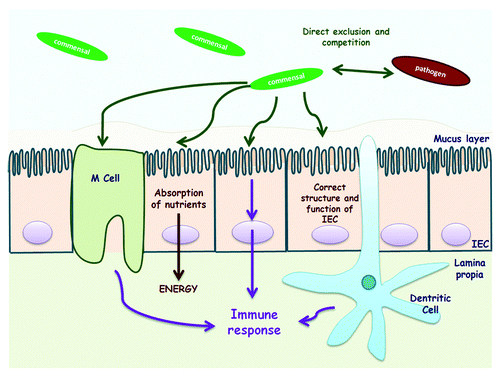
The composition of the human intestinal microbiome is less diverse than other bacterial ecosystems, such as those found in soil or water, presumably reflecting the harsh physico-chemical conditions of this niche. Most of the microorganisms colonizing humans are bacteria present in the GIT at a density of approximately 1013–1014 cells/g of fecal matter (particularly in the colon, 70% of total microbiota), and many of them are EOS.Citation27 The intestinal microbiome also contains a minority population of eukaryotic microorganisms (fungi, yeasts), viruses, and archae.Citation4 The human gut microbiome is considered to be beneficial for the host due to its key role in the stimulation and maturation of the immune system, promotion of mucosal structure and function, and providing colonization resistance against pathogen attack ().Citation5 Recently, microbiota functions have been reviewed thoroughly by Sommer et al.Citation28 and Serikov et al.Citation29
Figure 3. Commensal intestinal bacteria crosstalk with the host. Commensal bacteria supply the host with essential nutrients and defend the host against opportunistic pathogens. Commensals are involved in the development of the intestinal architecture as well as in immunomodulatory processes (Modified from Martín et al.).Citation5 IEC, intestinal epithelial cell.
Metagenomics approach to study the intestinal microbiome
The development of molecular ecology, with emphasis on 16S rDNA-based approaches, has dramatically changed our vision of the gut microbiome. Recent reviews have described the metagenomic exploration of the human intestinal microbiome through the availability of the reference gene catalog and mainly from the European project MetaHITCitation23 and the American Human Microbiome Project.Citation30,Citation31 The MetaHIT consortium has reported 3.3 million non-redundant genes in the human gut microbiome aloneCitation23 and much effort has focused on defining a core human gut microbiome (i.e., a set of features shared across all or the vast majority of gut microbiomes).Citation32 The average human’s intestinal microbiome is now better defined and comprises a huge diversity of bacterial species present in each individual (approximately 160).Citation23 Firmicutes and bacteroidetes are both dominant phyla representing 90% of the human microbiome. However, the microbiome composition differs along the GIT (from mouth to the rectum), as well as between individuals.Citation33 In spite of this relative heterogeneity, through fecal metagenomic analysis, it is possible to distinguish three main robust clusters named “enterotypes” in the gut microbiome, which are determined by species composition. Each of these three enterotypes is identifiable by the variation in the levels of one of three genera Bacteroides, Prevotella, and Ruminococcus.Citation34 Their abundance and proportions vary between individuals and is associated with long-term dietary habits.Citation35 However, the enterotype is quite a complicated concept because of the necessity to consider the intestinal microbiome according to different factors that could eventually impact its composition (e.g., aging, geographical origin, nutritional needs and habits, physiological variations and the impact of westernization, etc.).Citation36 Thus, the enterotype concept is one possible way to simplify the microbiome complexity.
Intestinal microbiome: a component of health
Intestinal bacteria were first qualified as commensals. This adjective comes from the Latin: cum (“with”) and mensa (“table”), which means sharing of the meal. Indeed, the host supplies the energy substrate for microorganisms via food intake. In exchange, the intestinal microbiome (through its large repertoire of enzymatic activities), constitutes a complementary metabolic directory of the human digestive system. The co-evolution of the GIT and the microbiome led to the selection of adapted bacteria developing a beneficial cohabitation with the human host.Citation27,Citation37,Citation38 The metagenomic approach allows a better knowledge of this symbiotic relationship. The intestinal microbiome (the collective genomes of the microorganisms that reside in our GIT) consists of 150-fold more genes than the human genome itself, suggesting the importance and the impact of our “second genome” on human physiology.Citation23 For instance, the understanding of the ability of the microbiome to metabolize fiber has made some progress, due to the exploration of functional libraries using Escherichia coli as host.Citation39 Besides, the composition of the intestinal microbiota is widely influenced by diet. For example, a study using high-throughput 16s rDNA and comparing the microbiota of children with a western diet to children with a fiber-rich diet (in a rural African village of Burkina Faso), demonstrated that bacterial composition was clearly different. Indeed, the bacterial microbiome of the inhabitants of Burkina Faso was more adapted to the degradation of cellulose.Citation22
The gut microbiota represents a real functional barrier allowing the inhibition of pathogen growth and colonization. Metagenomic sequencing has enabled systematic and unbiased characterization of microbial populations including the viral spectrum and it will enable the development of therapeutic strategies and/or vaccines in the near future.Citation40 Indeed, the use of this approach also allows the identification of new potential pathogens. For example, a novel virus (bat papillomavirus) was discovered and characterized using 454 sequencing from rectal swabs randomly collected from asymptomatic wild, food, and pet animals.Citation41 A modification of the GIT´s microbial barrier could be an influencing factor in diseases such as IBD, one of the most studied diseases in this field. Recent advances in “omics” approaches (i.e., genomics, transcriptomics, proteomics, and metabolomics) have opened the door for further investigation of the structure and function of the gut microbiome without the need to cultivate, identifying some promising approaches for future therapeutic and diagnostic applications.Citation42 For instance, the beneficial effects in the physiopathology of Crohn disease (CD, a type of IBD) patients of the commensal bacterium Faecalibacterium prausnitziiCitation43 on one side and the undesired effects of the opportunistic pathogen, adherent and invasive Escherichia coli (AIEC)Citation44 on the other, are two well-illustrated examples of the potential of these therapeutic and diagnostic applications. Indeed, F. prausnitzii could be considered as a sensor of human intestinal health, with different studies based on metagenomics methods reporting a reduction of this anti-inflammatory bacterium in CD patients and in other patients with intestinal disorder.Citation9 In contrast, it was recently shown by DNA pyrosequencing that in TLR5-deficient mice, AIEC colonization might induce lasting changes in the microbiota.Citation45 Metagenomic approaches to analyze microbiome composition in a genetically susceptible host, through the detection of a pathobiont in a developing microbiome can predict the development of chronic inflammation.Citation45 In the same way, the reduction in diversity of fecal microbiome in CD patients was revealed by a metagenomic approach.Citation7 Some authors have already proposed the identification of novel specific diagnostic targets for CD patients through integrated metagenomics/metaproteomics approaches.Citation46 IBD is not the only example of the link between disease and microbiome; metabolic disorders, celiac disease, irritable bowel syndrome (IBS), and colorectal cancer can also be cited.Citation9 It would be interesting to determine the link between enterotypes and pathologic phenotypes. In fact, if an enterotype is shown to be related to disease, long-term dietary interventions may allow modulation of an individual’s enterotype for improving health.Citation35
Vaginal ecosystem
The microbiome associated with the vagina has an important influence on human development, physiology, and immunity. This community of mutualistic bacteria constitutes the first line of defense for the host by excluding non-indigenous microbes that may cause sickness.Citation47 A mature microbiota is already established in early adolescence after the hormonal changes typical of this periodCitation48 and it includes some microorganisms also present in the GIT, even if the relative frequencies are different. The first microbiological study of the human vagina reported lactobacilli as the dominant microorganisms of this cavity,Citation49 being more than 70% of all microorganisms isolated from vaginal exudates of healthy and fertile women (and 100% in some cases).Citation47,Citation50,Citation51 Nevertheless, the species found may vary depending on the methodology used for the identification. In this context, following culture and phenotypic characterization, the most dominant species found are Lactobacillus acidophilus and/or L. fermentum,Citation52-Citation54 but when genetic methods are applied, the predominant species reported are L. crispatus, L. gasseri, and L. jensenii.Citation55-Citation58
Metagenomics to study the vaginal microbiome
Understanding bacterial composition and the interrelationships of constituent species is necessary to understand the role of the vaginal ecosystem, as well as the effect of different habits and practices. Although the vaginal ecosystem is dynamic as a result of its physiological function (menstrual cycle) and personal habits (contraception and hygiene practices), it remains stable over the long-term as a result of physiological and microbiological factors. In this sense, the Vaginal Human Microbiome Project (at the Virginia Commonwealth University) aims to investigate the complex vaginal microbiome and its link to human health and disease as well as its variability with different physiological conditions.Citation59 This fundamental knowledge is needed to diagnose and properly assess the risk of disease.
Just as it has been described by culture-dependent methods, metagenomic approaches also describe the vagina as an ecosystem rich in lactobacilli. While 20 species of lactobacilli have been isolated from the vagina, normally only one or two species predominate at the same time in each ecosystem, the most frequently reported being L. crispatus, L. inners, L. jensenii, and L. gasseri.Citation60-Citation66 Although the paradigm of the association between lactobacilli abundance and vaginal health seems to be true for the majority of women, it does not necessarily apply to all. In fact, it has been shown that a typical vaginal environment (i.e., pH > 4.5) is usually rich in lactobacilli. In this sense, vaginal ecosystems dominated by other lactic acid-producing bacteria, such as Bifidobacterium sp., Atopobium vaginae, Megasphaera sp., and Leptotrichia sp. have also been described.Citation57,Citation62,Citation67 A higher vaginal pH has been reported in some racial and ethnic groupsCitation68-Citation70 as well as the absence of lactobacilli and the presence of Gardnerella vaginalis, Prevotella sp., Pseudomonas sp., and/or Streptococcus sp. being predominant.Citation57 In fact, several studies have demonstrated the presence of different microbiome profiles named vagitypes, many of which are dominated by a single bacterial taxon.Citation59 However, caution has to be taken with this interpretation, since it is not possible to completely rule out a transition state between disease and health in these atypical microbiomes.Citation71,Citation72
Metagenomics has also identified differences in vaginal microbiome profiles depending on geographical origin,Citation67,Citation69,Citation70 as well as during the menstrual cycle and the period of a woman’s life, mainly due to hormonal changes.Citation48 Thus, estrogen deficiency (typical of post-menopause states) leads to a reduction in lactobacilli population.Citation73-Citation75 The vaginal microbiome also suffers changes due to other environmental factors and sexual practices.Citation76,Citation77 Genetic polymorphisms related to normal signaling of the innate immune system have also been associated with vaginal microbiota changes, promoting the presence of less healthy microbiota.Citation78-Citation80 During pregnancy, changes in the vaginal ecosystem (estrogen and progesterone levels, epithelium thickness, and extra glycogen production)Citation81,Citation82 lead to a significant reduction in diversity and richness of the vaginal microbiota.Citation83
Vaginal microbiome: a component of health
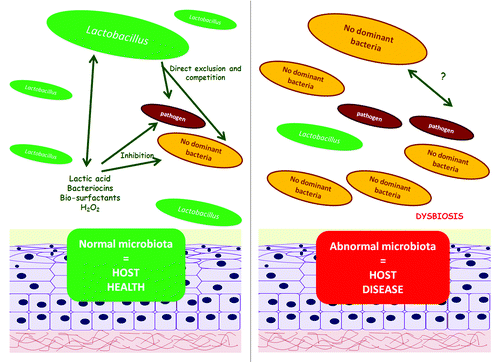
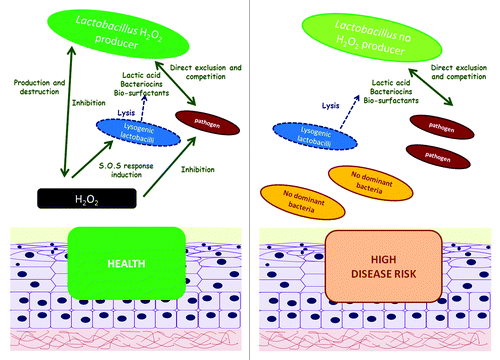
The vaginal microbiota, primarily lactobacilli, has been found to assert its beneficial effect against pathogens by two main mechanisms: (1) exclusion, driven by the competition for epithelial cell receptors and (2) inhibition of growth, due to generation of antimicrobial compoundsCitation84 (). The first mechanism results from the ability of lactobacilli to compete for receptors against urogenital pathogens such as group B Streptococcus (GBS), Staphylococcus aureus, Gardnerella vaginalis, Neisseria gonorrhoeae, Pseudomonas aeruginosa, Klebsiella pneumoniae, Candida albicans, and Actinomyces neuii.Citation85-Citation90 Impairment of adherence by treatment of lactobacilli or epithelial cells with proteases, lipases, or periodic acid suggests that the bacterial adhesins and cellular receptors are proteins, lipids, or polysaccharides respectively.Citation86,Citation90-Citation92 Furthermore, identification of the proteins anchored to the bacterial cell wall has provided a list of polypeptides putatively involved in mucous adherence.Citation93-Citation95 In addition, some lactobacilli can co-aggregate with potential pathogens, such as E. coli, C. albicans, and G. vaginalis, which may help in their clearance.Citation86,Citation88
Figure 4. Beneficial effect of lactobacilli on the vaginal ecosystem. Lactobacilli protect the host epithelium as a result of two main mechanisms: (1) exclusion, driven by the competition to epithelial cell receptors and (2) inhibition of growth, due to generation of antimicrobial compounds. When the vaginal microbiota is dominated by lactobacilli a health status is found in this ecosystem; alternatively, when no dominant species predominates in the vaginal ecosystem this dysbiosis normally leads to a disease state.
Regarding the second mechanism, lactobacilli are able to produce several antimicrobial compounds which are mainly organic acids produced from the fermentation of sugars, that lead to the typically low pH of the vagina which inhibits the growth of most pathogens.Citation96 Furthermore, vaginal lactobacilli are also able to produce bacteriocins, bio-surfactants,Citation97,Citation98 and hydrogen peroxide (H2O2).Citation99 At least three bacteriocins have been identified in vaginal Lactobacillus strains: Lactocine 160, Salivaricine CRL 1328, and L23.Citation99,Citation100-Citation103 Indeed, the prevalence of H2O2 producing strains has been correlated with reduced incidence of bacterial vaginosis (BV) and vaginal infections.Citation50,Citation77 However, the exclusion capacities of this compound are controversial, as the antimicrobial activity of H2O2 can be neutralized by semen and cervix–vaginal fluids.Citation104 Hydrogen peroxide has also been postulated to be simultaneously regulated by and regulate the lactobacilli population, due to the ability of some lactobacilli to destroy this moleculeCitation99 and the existence of a H2O2-mediated prophage induction mechanism that leads to lysis of the host lactobacillus.Citation105,Citation106 The role of H2O2 in this ecosystem is an example of how a single modification in one element of the ecosystem (including an environmental one) can alter the health status of the host in this dynamic ecological community ().
Figure 5. Role of hydrogen peroxide in the vaginal ecosystem. Hydrogen peroxide is an antimicrobial substance also known to counterbalance lactobacilli population due to a SOS response-mediated prophage induction in lysogenic lactobacilli.Citation105,Citation106 Some lactobacilli are able to destroy this substance.Citation99 An overall reduced number of H2O2 producing lactobacilli enhances the risk of disease.
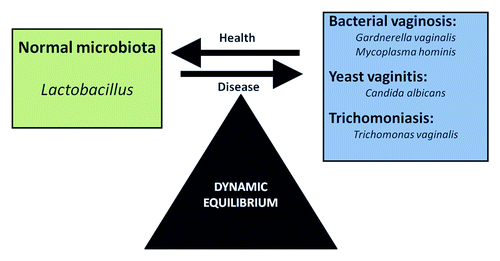
Abnormal vaginal microbiota can occur because of sexually transmitted pathogens or overgrowth of resident organisms.Citation107 The most common pathologies are BV, the proliferation of Candida sp. (mainly C. albicans) (candidiasis) and Trichomonas vaginalis (trichomoniasis)Citation47 (). BV is the most frequent vaginal imbalance and was shown by molecular methods to be associated with a high microbiota diversity.Citation60 and the presence of unfamiliar bacteria such as Mobiluncus sp., Atopobium sp., Megasphaera sp., and Ureaplasma urealyticum.Citation8,Citation57,Citation108-Citation110 Metagenomics has also been used recently to identify some uncultivable organisms associated with BV, the presence of which has been proposed as a diagnostic alternative to traditional culture-dependent methodologies.Citation8,Citation60,Citation107,Citation111 Epidemiologically, vaginal dysbiosis such as BV has been associated with preterm birth, development of pelvic inflammatory disease, and acquisition of sexually transmitted infections.Citation112
Figure 6. Vaginal equilibrium. A reduction in lactobacilli or overgrowth of some pathogens or no-dominant commensal bacteria can lead to a dysbiosis-related illness.
Skin microbiome
Skin represents a physical barrier to infection as a result of epidermis cohesion and more particularly to its cornified layer. Skin also harbors several physiological populations of microorganisms including commensal or symbiotic bacteria, fungi, parasites, and viruses known as the skin microbiota.Citation113 The presence of a complex ecosystem plays a well-documented role in preventing adherence and invasion by virulent pathogens through biological competition.Citation114 A better understanding of the skin microbiota’s roles requires investigation beyond the taxonomic catalog of bacteria for the characterization of specific activities associated with functional gene products.Citation115 Advances in molecular technologies allowed the identification of a much greater diversity of cutaneous microbiota than what was revealed previously using culture-based methods.Citation113,Citation116 Recently, the construction of shotgun metagenomic libraries provided access to the functions performed by dominant skin colonizing taxa, including Corynebacterium, Staphylococcus, and Propionibacterium.Citation115 This approach revealed the specific capabilities of skin microbiota to interact with and exploit compounds from the human skin.
Moreover, metagenomics techniques led to the identification of a new virus, the human polyomavirus, although there was no correlation between its presence on the skin and pathology. In fact, this virus is present on healthy skin and in the majority of merkel cell carcinomas.Citation117,Citation118 The characterization of the skin viral microbiota, using high throughput metagenomic sequencing (HTS) (a highly comprehensive method based on random sequencing of the entire DNA) would allow identification of microbiome patterns associated with particular skin conditions.Citation119
Airway microbiome
The human respiratory system, from the nose to the lung, is the ecological niche for many commensal microorganisms and for potential respiratory or invasive pathogens. In contrast to the GIT, the respiratory tract of healthy individuals harbors a homogenous microbiota that decreases in biomass from upper to lower tract.Citation120 Although the colonization by potential pathogens of the upper respiratory tract microbiome, especially the nasopharyngeal microbiome, induces identifiable disease in only a small percentage of people, colonization represents a major source of secretions containing bacteria that spreads between individuals.Citation121 For instance, in the case of children the impact of age, season, type of child day care, number of siblings, acute respiratory illness, diet, and sleeping position have been described; whereas in adults, other factors have been also implicated such as contact with children, chronic obstructive pulmonary disease, obesity, immunosuppression, allergic conditions, acute sinusitis, etc.Citation121 For instance, a metagenomic study on the detailed composition and variability in nasopharyngeal microbiota in samples from young children revealed that it differs between seasons.Citation122 During fall/winter which tends to be associated with increased incidence of respiratory and invasive infections, a predominance of Proteobacteria and Fusobacteria was observed. However, in spring, Bacteroidetes and Firmicutes were more abundant and, among them, (Brevi)bacillus and Lactobacillus species that can protect against respiratory or invasive infections. Another component of the nasopharyngeal microbiome are viruses and approximately 30% of all presumed viral cases fail diagnostic tests for etiologic agents.Citation123 Thus, metagenomics could allow the detection of known viruses in this specific environment, as well as the detection of new ones.Citation124 In the same way, some authors propose to define “the human virome project”, as a systematic exploration of the viruses that infect humans for an investigation of a novel pathogen, and provide a blueprint for comprehensive diagnosis of unexplained acute illnesses or outbreaks in clinical and public health settings.Citation125,Citation126 For example, in the case of the 2009 H1N1 influenza virus, this kind of strategy was shown to have the potential to replace conventional diagnostic tests.Citation126
Little is known concerning the recently described lower respiratory tract microbiome, even if it is likely to provide important pathogenic insights (cystic fibrosis, respiratory disease of the newborn, chronic obstructive pulmonary disease, and asthma).Citation127 Moreover, infectious agents are known to be or are suspected of having key roles in a number of chronic lung conditions. The bulk of published evidence demonstrates that phylogenetically diverse microbial communities in the lungs of healthy humans can be detected using high-throughput sequencing.Citation128-Citation130 Better characterization of the lung microbiome could help in understanding its role in preserving health or causing disease particularly in specific groups of patients, for example in smokers.Citation131
Concluding Remarks
Nowadays, we recognize the need to study the human microbiome as a whole ecosystem to better understand the relation between microbiota and host health or disease. For this reason, powerful methodologies are required to globally analyze these ecosystems, with metagenomic approaches being key for further analysis of the human microbiome. However, external influences, as well as methodological and sampling bias and inter-individual differences have to be taken into account in the data interpretation. For this reason, to define the average human microbiome, standard operating procedures are critically needed as well as metaanalysis studies, since it is reasonable to anticipate that communities would differ on the basis of the existence of inter-individual differences.
Due to the close relationship between the microbiome and health and the existence of biomarkers typical of different pathologies, the suggestion to modulate our microbiota sounds logical from a therapeutic point of view. Furthermore, the inter-individual differences and physiological parameters suggest personal medicine as a future treatment.
In the future, this kind of approach (i.e., metagenomics) would identify biomarkers of well-being that correspond to a general and more balanced microbiota. In addition, metagenomics can also provide us with a better understanding of the relationship between us and our microbiome and the role of this interaction with health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This review was a part of FPARIS collaborative project selected and supported by the Vitagora Competitive Cluster and funded by the French FUI (Fond Unique Interministériel; FUI: F1010012D), the FEDER (Fonds Européen de Développement Régional; Bourgogne: 34606), the Burgundy Region, the Conseil Général 21, and the Grand Dijon. This work was also supported by Merck Médication Familiale (Dijon, France) and Biovitis (Saint Etienne de Chomeil, France). R.M. and S.M. receive a salary from the same grants. Authors would like to thank Martin Beaumont for his bibliographic work during his stage in our laboratory.
References
- Bakhtiar SM, LeBlanc JG, Salvucci E, Ali A, Martin R, Langella P, Chatel JM, Miyoshi A, Bermúdez-Humarán LG, Azevedo V. Implications of the human microbiome in inflammatory bowel diseases. FEMS Microbiol Lett 2013; 342:10 - 7; http://dx.doi.org/10.1111/1574-6968.12111; PMID: 23431991
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804 - 10; http://dx.doi.org/10.1038/nature06244; PMID: 17943116
- Foxman B, Goldberg D, Murdock C, Xi C, Gilsdorf JR. Conceptualizing human microbiota: from multicelled organ to ecological community. Interdiscip Perspect Infect Dis 2008; 2008:613979; http://dx.doi.org/10.1155/2008/613979; PMID: 19259327
- Weinstock GM. Genomic approaches to studying the human microbiota. Nature 2012; 489:250 - 6; http://dx.doi.org/10.1038/nature11553; PMID: 22972298
- Martín R, Miquel S, Ulmer J, Kechaou N, Langella P, Bermúdez-Humarán LG. Role of commensal and probiotic bacteria in human health: a focus on inflammatory bowel disease. Microb Cell Fact 2013; 12:71; http://dx.doi.org/10.1186/1475-2859-12-71; PMID: 23876056
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 2008; 6:e280; http://dx.doi.org/10.1371/journal.pbio.0060280; PMID: 19018661
- Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006; 55:205 - 11; http://dx.doi.org/10.1136/gut.2005.073817; PMID: 16188921
- Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med 2005; 353:1899 - 911; http://dx.doi.org/10.1056/NEJMoa043802; PMID: 16267321
- Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, Thomas M, Wells JM, Langella P. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol 2013; 16:255 - 61; http://dx.doi.org/10.1016/j.mib.2013.06.003; PMID: 23831042
- Dimon MT, Wood HM, Rabbitts PH, Arron ST. IMSA: integrated metagenomic sequence analysis for identification of exogenous reads in a host genomic background. PLoS One 2013; 8:e64546; http://dx.doi.org/10.1371/journal.pone.0064546; PMID: 23717627
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 1998; 5:R245 - 9; http://dx.doi.org/10.1016/S1074-5521(98)90108-9; PMID: 9818143
- Ursell LK, Metcalf JL, Parfrey LW, Knight R. Defining the human microbiome. Nutr Rev 2012; 70:Suppl 1 S38 - 44; http://dx.doi.org/10.1111/j.1753-4887.2012.00493.x; PMID: 22861806
- Lederberg J, McCray AT. Ome sweet omics: a genealogical treasury of words. Scientist 2001; 15:8
- Brüls T, Weissenbach J. The human metagenome: our other genome?. Hum Mol Genet 2011; 20:R2 R142 - 8; http://dx.doi.org/10.1093/hmg/ddr353; PMID: 21840927
- Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al, NIH HMP Working Group. The NIH Human Microbiome Project. Genome Res 2009; 19:2317 - 23; http://dx.doi.org/10.1101/gr.096651.109; PMID: 19819907
- Eloe-Fadrosh EA, Rasko DA. The human microbiome: from symbiosis to pathogenesis. Annu Rev Med 2013; 64:145 - 63; http://dx.doi.org/10.1146/annurev-med-010312-133513; PMID: 23327521
- Milani C, Hevia A, Foroni E, Duranti S, Turroni F, Lugli GA, Sanchez B, Martín R, Gueimonde M, van Sinderen D, et al. Assessing the fecal microbiota: an optimized ion torrent 16S rRNA gene-based analysis protocol. PLoS One 2013; 8:e68739; http://dx.doi.org/10.1371/journal.pone.0068739; PMID: 23869230
- Böttger EC. Rapid determination of bacterial ribosomal RNA sequences by direct sequencing of enzymatically amplified DNA. FEMS Microbiol Lett 1989; 53:171 - 6; http://dx.doi.org/10.1111/j.1574-6968.1989.tb03617.x; PMID: 2482222
- Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, et al, Human Microbiome Jumpstart Reference Strains Consortium. A catalog of reference genomes from the human microbiome. Science 2010; 328:994 - 9; http://dx.doi.org/10.1126/science.1183605; PMID: 20489017
- Gans J, Wolinsky M, Dunbar J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005; 309:1387 - 90; http://dx.doi.org/10.1126/science.1112665; PMID: 16123304
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 2011; 108:Suppl 1 4578 - 85; http://dx.doi.org/10.1073/pnas.1000081107; PMID: 20668239
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010; 107:14691 - 6; http://dx.doi.org/10.1073/pnas.1005963107; PMID: 20679230
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al, MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59 - 65; http://dx.doi.org/10.1038/nature08821; PMID: 20203603
- Delzenne NM, Neyrinck AM, Bäckhed F, Cani PD. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol 2011; 7:639 - 46; http://dx.doi.org/10.1038/nrendo.2011.126; PMID: 21826100
- Antonio MA, Rabe LK, Hillier SL. Colonization of the rectum by Lactobacillus species and decreased risk of bacterial vaginosis. J Infect Dis 2005; 192:394 - 8; http://dx.doi.org/10.1086/430926; PMID: 15995952
- Reid G, Beuerman D, Heinemann C, Bruce AW. Probiotic Lactobacillus dose required to restore and maintain a normal vaginal flora. FEMS Immunol Med Microbiol 2001; 32:37 - 41; http://dx.doi.org/10.1111/j.1574-695X.2001.tb00531.x; PMID: 11750220
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444:1022 - 3; http://dx.doi.org/10.1038/4441022a; PMID: 17183309
- Sommer F, Bäckhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol 2013; 11:227 - 38; http://dx.doi.org/10.1038/nrmicro2974; PMID: 23435359
- Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev 2010; 90:859 - 904; http://dx.doi.org/10.1152/physrev.00045.2009; PMID: 20664075
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207 - 14; http://dx.doi.org/10.1038/nature11234; PMID: 22699609
- Blottière HM, de Vos WM, Ehrlich SD, Doré J. Human intestinal metagenomics: state of the art and future. Curr Opin Microbiol 2013; 16:232 - 9; http://dx.doi.org/10.1016/j.mib.2013.06.006; PMID: 23870802
- Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol 2009; 587:4153 - 8; http://dx.doi.org/10.1113/jphysiol.2009.174136; PMID: 19491241
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 308:1635 - 8; http://dx.doi.org/10.1126/science.1110591; PMID: 15831718
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al, MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011; 473:174 - 80; http://dx.doi.org/10.1038/nature09944; PMID: 21508958
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011; 334:105 - 8; http://dx.doi.org/10.1126/science.1208344; PMID: 21885731
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486:222 - 7; PMID: 22699611
- Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology 2009; 136:65 - 80; http://dx.doi.org/10.1053/j.gastro.2008.10.080; PMID: 19026645
- Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol 2013; 11:61; http://dx.doi.org/10.1186/1741-7007-11-61; PMID: 23692866
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012; 486:215 - 21; http://dx.doi.org/10.1038/nature11209; PMID: 22699610
- Finkbeiner SR, Allred AF, Tarr PI, Klein EJ, Kirkwood CD, Wang D. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog 2008; 4:e1000011; http://dx.doi.org/10.1371/journal.ppat.1000011; PMID: 18398449
- Tse H, Tsang AK, Tsoi HW, Leung AS, Ho CC, Lau SK, Woo PC, Yuen KY. Identification of a novel bat papillomavirus by metagenomics. PLoS One 2012; 7:e43986; http://dx.doi.org/10.1371/journal.pone.0043986; PMID: 22937142
- Lamendella R, VerBerkmoes N, Jansson JK. ‘Omics’ of the mammalian gut--new insights into function. Curr Opin Biotechnol 2012; 23:491 - 500; http://dx.doi.org/10.1016/j.copbio.2012.01.016; PMID: 22626866
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105:16731 - 6; http://dx.doi.org/10.1073/pnas.0804812105; PMID: 18936492
- Miquel S, Peyretaillade E, Claret L, de Vallée A, Dossat C, Vacherie B, Zineb H, Segurens B, Barbe V, Sauvanet P, et al. Complete genome sequence of Crohn’s disease-associated adherent-invasive E. coli strain LF82. PLoS One 2010; 5:e12714; http://dx.doi.org/10.1371/journal.pone.0012714; PMID: 20862302
- Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2013; Forthcoming http://dx.doi.org/10.1136/gutjnl-2013-304909; PMID: 23896971
- Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One 2012; 7:e49138; http://dx.doi.org/10.1371/journal.pone.0049138; PMID: 23209564
- Martín R, Soberón N, Vázquez F, Suárez JE. [Vaginal microbiota: composition, protective role, associated pathologies, and therapeutic perspectives]. Enferm Infecc Microbiol Clin 2008; 26:160 - 7; PMID: 18358215
- Yamamoto T, Zhou X, Williams CJ, Hochwalt A, Forney LJ. Bacterial populations in the vaginas of healthy adolescent women. J Pediatr Adolesc Gynecol 2009; 22:11 - 8; http://dx.doi.org/10.1016/j.jpag.2008.01.073; PMID: 19232297
- Doderlein ASG. Das Scheindensekret und seine bedeutung fur das puerperalfieber. Leipzig: O Durr 1982.
- Eschenbach DA, Davick PR, Williams BL, Klebanoff SJ, Young-Smith K, Critchlow CM, Holmes KK. Prevalence of hydrogen peroxide-producing Lactobacillus species in normal women and women with bacterial vaginosis. J Clin Microbiol 1989; 27:251 - 6; PMID: 2915019
- Redondo-Lopez V, Cook RL, Sobel JD. Emerging role of lactobacilli in the control and maintenance of the vaginal bacterial microflora. Rev Infect Dis 1990; 12:856 - 72; http://dx.doi.org/10.1093/clinids/12.5.856; PMID: 2237129
- Alvarez-Olmos MI, Barousse MM, Rajan L, Van Der Pol BJ, Fortenberry D, Orr D, Fidel PL Jr.. Vaginal lactobacilli in adolescents: presence and relationship to local and systemic immunity, and to bacterial vaginosis. Sex Transm Dis 2004; 31:393 - 400; http://dx.doi.org/10.1097/01.OLQ.0000130454.83883.E9; PMID: 15215693
- Boyd MA, Antonio MA, Hillier SL. Comparison of API 50 CH strips to whole-chromosomal DNA probes for identification of Lactobacillus species. J Clin Microbiol 2005; 43:5309 - 11; http://dx.doi.org/10.1128/JCM.43.10.5309-5311.2005; PMID: 16208005
- Song YL, Kato N, Matsumiya Y, Liu CX, Kato H, Watanabe K. Identification of Lactobacillus species of human origin by a commercial kit, API50CHL. Rinsho Biseibutshu Jinsoku Shindan Kenkyukai Shi 1999; 10:77 - 82; PMID: 10681709
- Vallor AC, Antonio MA, Hawes SE, Hillier SL. Factors associated with acquisition of, or persistent colonization by, vaginal lactobacilli: role of hydrogen peroxide production. J Infect Dis 2001; 184:1431 - 6; http://dx.doi.org/10.1086/324445; PMID: 11709785
- Matsumiya Y, Kato N, Watanabe K, Kato H. Molecular epidemiological study of vertical transmission of vaginal Lactobacillus species from mothers to newborn infants in Japanese, by arbitrarily primed polymerase chain reaction. J Infect Chemother 2002; 8:43 - 9; http://dx.doi.org/10.1007/s101560200005; PMID: 11957119
- Hyman RW, Fukushima M, Diamond L, Kumm J, Giudice LC, Davis RW. Microbes on the human vaginal epithelium. Proc Natl Acad Sci U S A 2005; 102:7952 - 7; http://dx.doi.org/10.1073/pnas.0503236102; PMID: 15911771
- Martín R, Soberón N, Vaneechoutte M, Flórez AB, Vázquez F, Suárez JE. Characterization of indigenous vaginal lactobacilli from healthy women as probiotic candidates. Int Microbiol 2008; 11:261 - 6; PMID: 19204898
- Fettweis JM, Serrano MG, Girerd PH, Jefferson KK, Buck GA. A new era of the vaginal microbiome: advances using next-generation sequencing. Chem Biodivers 2012; 9:965 - 76; http://dx.doi.org/10.1002/cbdv.201100359; PMID: 22589096
- Pavlova SI, Kilic AO, Kilic SS, So JS, Nader-Macias ME, Simoes JA, Tao L. Genetic diversity of vaginal lactobacilli from women in different countries based on 16S rRNA gene sequences. J Appl Microbiol 2002; 92:451 - 9; http://dx.doi.org/10.1046/j.1365-2672.2002.01547.x; PMID: 11872120
- Verhelst R, Verstraelen H, Claeys G, Verschraegen G, Delanghe J, Van Simaey L, De Ganck C, Temmerman M, Vaneechoutte M. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol 2004; 4:16; http://dx.doi.org/10.1186/1471-2180-4-16; PMID: 15102329
- Zhou X, Bent SJ, Schneider MG, Davis CC, Islam MR, Forney LJ. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 2004; 150:2565 - 73; http://dx.doi.org/10.1099/mic.0.26905-0; PMID: 15289553
- Song YL, Kato N, Matsumiya Y, Liu CX, Kato H, Watanabe K. Identification of and hydrogen peroxide production by fecal and vaginal lactobacilli isolated from Japanese women and newborn infants. J Clin Microbiol 1999; 37:3062 - 4; PMID: 10449509
- Wilks M, Wiggins R, Whiley A, Hennessy E, Warwick S, Porter H, Corfield A, Millar M. Identification and H(2)O(2) production of vaginal lactobacilli from pregnant women at high risk of preterm birth and relation with outcome. J Clin Microbiol 2004; 42:713 - 7; http://dx.doi.org/10.1128/JCM.42.2.713-717.2004; PMID: 14766841
- Biagi E, Vitali B, Pugliese C, Candela M, Donders GG, Brigidi P. Quantitative variations in the vaginal bacterial population associated with asymptomatic infections: a real-time polymerase chain reaction study. Eur J Clin Microbiol Infect Dis 2009; 28:281 - 5; http://dx.doi.org/10.1007/s10096-008-0617-0; PMID: 18762999
- Vitali B, Pugliese C, Biagi E, Candela M, Turroni S, Bellen G, Donders GG, Brigidi P. Dynamics of vaginal bacterial communities in women developing bacterial vaginosis, candidiasis, or no infection, analyzed by PCR-denaturing gradient gel electrophoresis and real-time PCR. Appl Environ Microbiol 2007; 73:5731 - 41; http://dx.doi.org/10.1128/AEM.01251-07; PMID: 17644631
- Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, Joyce P, Foster JA, Forney LJ. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J 2007; 1:121 - 33; http://dx.doi.org/10.1038/ismej.2007.12; PMID: 18043622
- Stevens-Simon C, Jamison J, McGregor JA, Douglas JM. Racial variation in vaginal pH among healthy sexually active adolescents. Sex Transm Dis 1994; 21:168 - 72; http://dx.doi.org/10.1097/00007435-199405000-00007; PMID: 8073345
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A 2011; 108:Suppl 1 4680 - 7; http://dx.doi.org/10.1073/pnas.1002611107; PMID: 20534435
- Fiscella K, Klebanoff MA. Are racial differences in vaginal pH explained by vaginal flora?. Am J Obstet Gynecol 2004; 191:747 - 50; http://dx.doi.org/10.1016/j.ajog.2004.03.032; PMID: 15467534
- Fredricks DN. Molecular methods to describe the spectrum and dynamics of the vaginal microbiota. Anaerobe 2011; 17:191 - 5; http://dx.doi.org/10.1016/j.anaerobe.2011.01.001; PMID: 21376827
- McCook A. The vagina catalogues. Nat Med 2011; 17:765 - 7; http://dx.doi.org/10.1038/nm0711-765; PMID: 21738144
- Heinemann C, Reid G. Vaginal microbial diversity among postmenopausal women with and without hormone replacement therapy. Can J Microbiol 2005; 51:777 - 81; http://dx.doi.org/10.1139/w05-070; PMID: 16391657
- Pabich WL, Fihn SD, Stamm WE, Scholes D, Boyko EJ, Gupta K. Prevalence and determinants of vaginal flora alterations in postmenopausal women. J Infect Dis 2003; 188:1054 - 8; http://dx.doi.org/10.1086/378203; PMID: 14513427
- Wilson JD, Lee RA, Balen AH, Rutherford AJ. Bacterial vaginal flora in relation to changing oestrogen levels. Int J STD AIDS 2007; 18:308 - 11; http://dx.doi.org/10.1258/095646207780749583; PMID: 17524189
- Cherpes TL, Hillier SL, Meyn LA, Busch JL, Krohn MA. A delicate balance: risk factors for acquisition of bacterial vaginosis include sexual activity, absence of hydrogen peroxide-producing lactobacilli, black race, and positive herpes simplex virus type 2 serology. Sex Transm Dis 2008; 35:78 - 83; http://dx.doi.org/10.1097/OLQ.0b013e318156a5d0; PMID: 17989585
- Hawes SE, Hillier SL, Benedetti J, Stevens CE, Koutsky LA, Wolner-Hanssen P, Holmes KK. Hydrogen peroxide-producing lactobacilli and acquisition of vaginal infections. J Infect Dis 1996; 174:1058 - 63; http://dx.doi.org/10.1093/infdis/174.5.1058; PMID: 8896509
- Genc MR, Onderdonk A. Endogenous bacterial flora in pregnant women and the influence of maternal genetic variation. BJOG 2011; 118:154 - 63; http://dx.doi.org/10.1111/j.1471-0528.2010.02772.x; PMID: 21054765
- Genc MR, Onderdonk AB, Vardhana S, Delaney ML, Norwitz ER, Tuomala RE, Paraskevas LR, Witkin SS, MAP Study Group. Polymorphism in intron 2 of the interleukin-1 receptor antagonist gene, local midtrimester cytokine response to vaginal flora, and subsequent preterm birth. Am J Obstet Gynecol 2004; 191:1324 - 30; http://dx.doi.org/10.1016/j.ajog.2004.05.074; PMID: 15507961
- Verstraelen H, Verhelst R, Nuytinck L, Roelens K, De Meester E, De Vos D, Van Thielen M, Rossau R, Delva W, De Backer E, et al. Gene polymorphisms of Toll-like and related recognition receptors in relation to the vaginal carriage of Gardnerella vaginalis and Atopobium vaginae. J Reprod Immunol 2009; 79:163 - 73; http://dx.doi.org/10.1016/j.jri.2008.10.006; PMID: 19200604
- Bourne A. Endocrines in gynaecology. Br Med J 1947; 1:652; http://dx.doi.org/10.1136/bmj.1.4505.652; PMID: 20248081
- Bourne A. Non-Gonococcal Vaginal Discharges. Br J Vener Dis 1943; 19:3 - 5; PMID: 21773334
- Aagaard K, Riehle K, Ma J, Segata N, Mistretta TA, Coarfa C, Raza S, Rosenbaum S, Van den Veyver I, Milosavljevic A, et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One 2012; 7:e36466; http://dx.doi.org/10.1371/journal.pone.0036466; PMID: 22719832
- Boris S, Barbés C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect 2000; 2:543 - 6; http://dx.doi.org/10.1016/S1286-4579(00)00313-0; PMID: 10865199
- Zárate G, Nader-Macias ME. Influence of probiotic vaginal lactobacilli on in vitro adhesion of urogenital pathogens to vaginal epithelial cells. Lett Appl Microbiol 2006; 43:174 - 80; http://dx.doi.org/10.1111/j.1472-765X.2006.01934.x; PMID: 16869901
- Boris S, Suárez JE, Vázquez F, Barbés C. Adherence of human vaginal lactobacilli to vaginal epithelial cells and interaction with uropathogens. Infect Immun 1998; 66:1985 - 9; PMID: 9573080
- Vielfort K, Sjölinder H, Roos S, Jonsson H, Aro H. Adherence of clinically isolated lactobacilli to human cervical cells in competition with Neisseria gonorrhoeae. Microbes Infect 2008; 10:1325 - 34; http://dx.doi.org/10.1016/j.micinf.2008.07.032; PMID: 18761100
- Osset J, Bartolomé RM, García E, Andreu A. Assessment of the capacity of Lactobacillus to inhibit the growth of uropathogens and block their adhesion to vaginal epithelial cells. J Infect Dis 2001; 183:485 - 91; http://dx.doi.org/10.1086/318070; PMID: 11133381
- Osset J, García E, Bartolomé RM, Andreu A. [Role of Lactobacillus as protector against vaginal candidiasis]. Med Clin (Barc) 2001; 117:285 - 8; http://dx.doi.org/10.1016/S0025-7753(01)72089-1; PMID: 11571120
- Martín R, Sánchez B, Suárez JE, Urdaci MC. Characterization of the adherence properties of human Lactobacilli strains to be used as vaginal probiotics. FEMS Microbiol Lett 2012; 328:166 - 73; http://dx.doi.org/10.1111/j.1574-6968.2011.02495.x; PMID: 22224921
- Boris S, Suárez JE, Barbés C. Characterization of the aggregation promoting factor from Lactobacillus gasseri, a vaginal isolate. J Appl Microbiol 1997; 83:413 - 20; http://dx.doi.org/10.1046/j.1365-2672.1997.00250.x; PMID: 9351223
- Vélez MP, De Keersmaecker SC, Vanderleyden J. Adherence factors of Lactobacillus in the human gastrointestinal tract. FEMS Microbiol Lett 2007; 276:140 - 8; http://dx.doi.org/10.1111/j.1574-6968.2007.00908.x; PMID: 17888009
- Sánchez B, Bressollier P, Urdaci MC. Exported proteins in probiotic bacteria: adhesion to intestinal surfaces, host immunomodulation and molecular cross-talking with the host. FEMS Immunol Med Microbiol 2008; 54:1 - 17; http://dx.doi.org/10.1111/j.1574-695X.2008.00454.x; PMID: 18631181
- Tjalsma H, Lambooy L, Hermans PW, Swinkels DW. Shedding & shaving: disclosure of proteomic expressions on a bacterial face. Proteomics 2008; 8:1415 - 28; http://dx.doi.org/10.1002/pmic.200700550; PMID: 18306176
- Muñoz-Provencio D, Pérez-Martínez G, Monedero V. Characterization of a fibronectin-binding protein from Lactobacillus casei BL23. J Appl Microbiol 2010; 108:1050 - 9; http://dx.doi.org/10.1111/j.1365-2672.2009.04508.x; PMID: 19735320
- Boskey ER, Cone RA, Whaley KJ, Moench TR. Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod 2001; 16:1809 - 13; http://dx.doi.org/10.1093/humrep/16.9.1809; PMID: 11527880
- Velraeds MM, van de Belt-Gritter B, van der Mei HC, Reid G, Busscher HJ. Interference in initial adhesion of uropathogenic bacteria and yeasts to silicone rubber by a Lactobacillus acidophilus biosurfactant. J Med Microbiol 1998; 47:1081 - 5; http://dx.doi.org/10.1099/00222615-47-12-1081; PMID: 9856644
- Velraeds MM, van de Belt-Gritter B, Busscher HJ, Reid G, van der Mei HC. Inhibition of uropathogenic biofilm growth on silicone rubber in human urine by lactobacilli--a teleologic approach. World J Urol 2000; 18:422 - 6; http://dx.doi.org/10.1007/PL00007084; PMID: 11204262
- Martín R, Suárez JE. Biosynthesis and degradation of H2O2 by vaginal lactobacilli. Appl Environ Microbiol 2010; 76:400 - 5; http://dx.doi.org/10.1128/AEM.01631-09; PMID: 19948869
- Aroutcheva AA, Simoes JA, Faro S. Antimicrobial protein produced by vaginal Lactobacillus acidophilus that inhibits Gardnerella vaginalis. Infect Dis Obstet Gynecol 2001; 9:33 - 9; http://dx.doi.org/10.1155/S1064744901000060; PMID: 11368257
- Simoes JA, Aroutcheva A, Heimler I, Shott S, Faro S. Bacteriocin susceptibility of Gardnerella vaginalis and its relationship to biotype, genotype, and metronidazole susceptibility. Am J Obstet Gynecol 2001; 185:1186 - 90; http://dx.doi.org/10.1067/mob.2001.118144; PMID: 11717655
- Ocaña VS, Pesce De Ruiz Holgado AA, Nader-Macías ME. Characterization of a bacteriocin-like substance produced by a vaginal Lactobacillus salivarius strain. Appl Environ Microbiol 1999; 65:5631 - 5; PMID: 10584033
- Pascual L, Ruiz F, Giordano W, Barberis IL. Vaginal colonization and activity of the probiotic bacterium Lactobacillus fermentum L23 in a murine model of vaginal tract infection. J Med Microbiol 2010; 59:360 - 4; http://dx.doi.org/10.1099/jmm.0.012583-0; PMID: 19926731
- O’Hanlon DE, Lanier BR, Moench TR, Cone RA. Cervicovaginal fluid and semen block the microbicidal activity of hydrogen peroxide produced by vaginal lactobacilli. BMC Infect Dis 2010; 10:120; http://dx.doi.org/10.1186/1471-2334-10-120; PMID: 20482854
- Martín R, Soberón N, Escobedo S, Suárez JE. Bacteriophage induction versus vaginal homeostasis: role of H(2)O(2) in the selection of Lactobacillus defective prophages. Int Microbiol 2009; 12:131 - 6; PMID: 19784933
- Pavlova SI, Kiliç AO, Mou SM, Tao L. Phage infection in vaginal lactobacilli: an in vitro study. Infect Dis Obstet Gynecol 1997; 5:36 - 44; PMID: 18476132
- Lamont RF, Sobel JD, Akins RA, Hassan SS, Chaiworapongsa T, Kusanovic JP, Romero R. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG 2011; 118:533 - 49; http://dx.doi.org/10.1111/j.1471-0528.2010.02840.x; PMID: 21251190
- Kazor CE, Mitchell PM, Lee AM, Stokes LN, Loesche WJ, Dewhirst FE, Paster BJ. Diversity of bacterial populations on the tongue dorsa of patients with halitosis and healthy patients. J Clin Microbiol 2003; 41:558 - 63; http://dx.doi.org/10.1128/JCM.41.2.558-563.2003; PMID: 12574246
- Doyle LM, McInerney JO, Mooney J, Powell R, Haikara A, Moran AP. Sequence of the gene encoding the 16S rRNA of the beer spoilage organism Megasphaera cerevisiae. J Ind Microbiol 1995; 15:67 - 70; http://dx.doi.org/10.1007/BF01569801; PMID: 7576462
- Haggerty CL, Totten PA, Ferris M, Martin DH, Hoferka S, Astete SG, Ondondo R, Norori J, Ness RB. Clinical characteristics of bacterial vaginosis among women testing positive for fastidious bacteria. Sex Transm Infect 2009; 85:242 - 8; http://dx.doi.org/10.1136/sti.2008.032821; PMID: 19004865
- Oakley BB, Fiedler TL, Marrazzo JM, Fredricks DN. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl Environ Microbiol 2008; 74:4898 - 909; http://dx.doi.org/10.1128/AEM.02884-07; PMID: 18487399
- Brotman RM. Vaginal microbiome and sexually transmitted infections: an epidemiologic perspective. J Clin Invest 2011; 121:4610 - 7; http://dx.doi.org/10.1172/JCI57172; PMID: 22133886
- Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol 2011; 9:244 - 53; http://dx.doi.org/10.1038/nrmicro2537; PMID: 21407241
- Cogen AL, Nizet V, Gallo RL. Skin microbiota: a source of disease or defence?. Br J Dermatol 2008; 158:442 - 55; http://dx.doi.org/10.1111/j.1365-2133.2008.08437.x; PMID: 18275522
- Mathieu A, Delmont TO, Vogel TM, Robe P, Nalin R, Simonet P. Life on human surfaces: skin metagenomics. PLoS One 2013; 8:e65288; http://dx.doi.org/10.1371/journal.pone.0065288; PMID: 23776466
- Grice EA, Kong HH, Renaud G, Young AC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA, NISC Comparative Sequencing Program. A diversity profile of the human skin microbiota. Genome Res 2008; 18:1043 - 50; http://dx.doi.org/10.1101/gr.075549.107; PMID: 18502944
- Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010; 7:509 - 15; http://dx.doi.org/10.1016/j.chom.2010.05.006; PMID: 20542254
- Wieland U, Mauch C, Kreuter A, Krieg T, Pfister H. Merkel cell polyomavirus DNA in persons without merkel cell carcinoma. Emerg Infect Dis 2009; 15:1496 - 8; http://dx.doi.org/10.3201/eid1509.081575; PMID: 19788824
- Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, Pariente K, Segondy M, Burguière A, Manuguerra JC, et al. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One 2012; 7:e38499; http://dx.doi.org/10.1371/journal.pone.0038499; PMID: 22723863
- Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med 2011; 184:957 - 63; http://dx.doi.org/10.1164/rccm.201104-0655OC; PMID: 21680950
- García-Rodríguez JA, Fresnadillo Martínez MJ. Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J Antimicrob Chemother 2002; 50:Suppl S2 59 - 73; http://dx.doi.org/10.1093/jac/dkf506; PMID: 12556435
- Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, Bruin J, Montijn R, Bonten M, Sanders E. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 2011; 6:e17035; http://dx.doi.org/10.1371/journal.pone.0017035; PMID: 21386965
- Heikkinen T, Järvinen A. The common cold. Lancet 2003; 361:51 - 9; http://dx.doi.org/10.1016/S0140-6736(03)12162-9; PMID: 12517470
- Lysholm F, Wetterbom A, Lindau C, Darban H, Bjerkner A, Fahlander K, Lindberg AM, Persson B, Allander T, Andersson B. Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS One 2012; 7:e30875; http://dx.doi.org/10.1371/journal.pone.0030875; PMID: 22355331
- Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A 2005; 102:12891 - 6; http://dx.doi.org/10.1073/pnas.0504666102; PMID: 16118271
- Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Yu G, Kim E, Pillai DR, Guyard C, Mazzulli T, et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One 2010; 5:e13381; http://dx.doi.org/10.1371/journal.pone.0013381; PMID: 20976137
- Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res 2012; 160:258 - 66; http://dx.doi.org/10.1016/j.trsl.2012.02.005; PMID: 22683412
- Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 2011; 6:e16384; http://dx.doi.org/10.1371/journal.pone.0016384; PMID: 21364979
- Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al. Disordered microbial communities in asthmatic airways. PLoS One 2010; 5:e8578; http://dx.doi.org/10.1371/journal.pone.0008578; PMID: 20052417
- Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, et al, National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 2011; 127:372 - 81, e1-3; http://dx.doi.org/10.1016/j.jaci.2010.10.048; PMID: 21194740
- Kiley JP. Advancing respiratory research. Chest 2011; 140:497 - 501; http://dx.doi.org/10.1378/chest.11-0774; PMID: 21813528