
Abstract
A hallmark of the antiviral response is the induction of interferons. First discovered in 1957 by Issac and Lindeman, interferons are noted for their ability to interfere with viral replication. Interferons act via autocrine and paracrine pathways to induce an antiviral state in infected cells and in neighboring cells containing interferon receptors. Interferons are the frontline defenders against viral infection and their primary function is to locally restrict viral propagation. Viruses have evolved mechanisms to escape the host interferon response, thus gaining a replicative advantage in host cells. This review will discuss recent findings on the mechanisms viruses use to evade the host interferon response. This knowledge is important because the treatment of viral infections is a challenge of global proportions and a better understanding of the mechanisms viruses use to persist in the host may uncover valuable insights applicable to the discovery of novel drug targets.
Introduction
A significant body of work conducted over the past three decades has helped gain valuable insight into the evasive maneuvers used by viruses to counteract the host antiviral response. It is now clearly established through observations made by various researchers that virally derived products target specific proteins and signaling mediators in the host which mediate antiviral activities.Citation1 This degree of specificity in host–pathogen interactions has generated much interest and intrigue among the virology and infectious disease communities. The current working model that attempts to reconcile these observations is the co-evolution interplay. In this model, only viruses that attain survival-associated mutations persist in the host.Citation2,Citation3 Survival-associated mutations result in the development of adaptation strategies, ultimately benefiting viral persistence. These adaptation strategies include attenuated molecular recognition of viruses by host receptors; altered expression of surface proteins, resulting in a curbed immune response;Citation4 and shifts in codon bias (or codon usage) that facilitate viral assembly and propagation.Citation5
During the initial stages of a viral infection, there exists a balance between virus promoting and virus inhibiting mechanisms.Citation6 Often, the virus inhibiting mechanisms are able to clear the infection. However, some viral subspecies can emerge, harboring mutations that can evade the host interferon response. The interferon response is a network of signaling pathways that help host cells fight off viruses and can be thought of as occurring in two phases: an intracellular phase where infected cells produce interferons and an intercellular phase where infection-induced interferons are secreted into the extracellular environment. Secreted interferons bind to interferon receptors on surrounding cells, leading to the synthesis of antiviral proteins and more interferons.Citation1,Citation6,Citation7
The mechanisms used by various RNA and DNA viruses to inhibit the interferon system involve: (1) mechanisms that interfere with the upstream mediators of viral recognition,Citation8 (2) mechanisms that circumvent the signaling pathways leading to interferon productionCitation9-Citation11 and (3) mechanisms that inhibit interferon-induced antiviral proteins.Citation12 This review will highlight what we currently know about the aforementioned mechanisms.
Viral Mechanisms That Inhibit the Upstream Mediators of Interferon Production
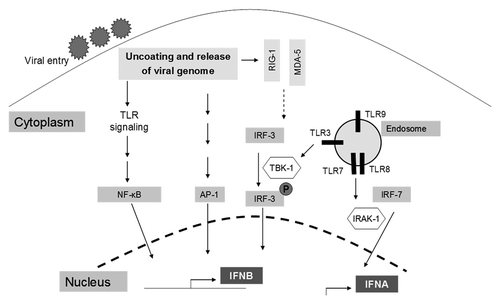
The mediators of viral recognition that led to the production of interferons consists of a group of receptors located either in the cytoplasm or on the surface of endosomes; areas that allow these receptors to efficiently detect viral invasion. These receptors include RIG-1, MDA-5, and TLRs 3, 7, 8, and 9 (summarized in ). Multiple viral proteins have evolved to specifically inhibit these cell intrinsic viral sensors.
Figure 1. A summary of signaling pathways leading to the induction of interferons. Viral entry is followed by the release of the viral genome into host cells, triggering the activation of TLRs, or cytoplasmic viral sensors (RIG1 and MDA5), or the activation of the NFκB, AP-1, IRF-3, and IRF-7 signaling pathways. Collectively, these pathways lead to the induction of interferons α and β, which are encoded by the IFNA and IFNB genes respectively.
RIG-1 is a cytoplasmic helicase that recognizes dsRNA and is activated by retinoic acid, interferons or viral infection.Citation13 Inactive RIG-1 contains a CARD domain at its N-terminal region. The enzyme TRIM25 ubiquitinates the CARD domain, thus activating RIG-1. Active RIG-1 signals downstream to NFκB and IRF-3—transcription factors necessary for the synthesis of IFN-β.Citation14 The nonstructural 1 (NS1) protein from the influenza A virus has been shown to inhibit the TRIM25-CARD domain interaction ().Citation14 A similar finding relating to RIG-1 targeting has been reported by Zhou et al.,Citation15 whose work demonstrates that the nucleoproteins of the Arenavirus binds RIG-1 and inhibit downstream signaling. They also demonstrate that the Z proteins of the New world Arenavirus inactivate RIG-1 through a direct interaction. In addition, Z proteins inhibit the activation and nuclear localization of NFκB. They also inhibit the dimerization and concomitant nuclear translocation of IRF-3; both events necessary for the transcriptional activation of IFN-β.Citation15,Citation16 Gori-Savellini et al. have shown that the Toscana virus-derived NSs protein interacts with RIG-1, leading to its proteasomal degradation.Citation17
Figure 2. A summary of viral mechanisms that inhibit the upstream mediators of interferon induction. NS1 and Z proteins target RIG-1, V proteins target MDA-5, and RSV targets TLRs 7 and 8.
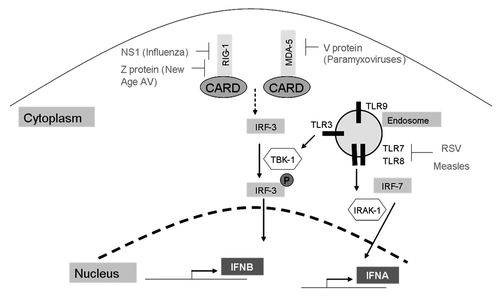
MDA-5 is a cytoplasmic virus sensor that recognizes ssRNA and like RIG-1, also contains a CARD domain that relays signals leading to IRF-3 activation and subsequently, IFN-β production. MDA-5 is inactivated by the V protein form Paramyxoviruses ().Citation18 Andrejeva et al.Citation18 have reported that the ectopic expression of the V protein to varying degrees can lead to suppression in both MDA-5 activity as well as IFN-β promoter activity, highlighting the influence of the V protein in inhibiting IFN-β signaling at multiple levels.
TLRs are an essential component of innate immunity and are the frontline detectors of bacterial and viral products.Citation19 First described in D. melanogaster, there are now 10 documented human TLRs and 13 murine TLRs.Citation20 Well known for their ability to recognize PAMPs, TLRs are also involved in inflammation (AP-1 and NFκB-mediated upregulation of pro-inflammatory cytokines) and T-cell activation largely by IRF-3-mediated transactivation of T-cell co-stimulatory genes.Citation19 In the context of a virus infection, TLRs 3, 7, 8, and 9 are important players. TLR7 binds ssRNA and TLR9 binds CpG DNA. Schlender et al. have demonstrated in a plasmacytoid DC model that respiratory syncytial virus (RSV) stain A2 or the measles virus induces a transient shut down of type-I IFN production.Citation8 Using TLR7 and TLR9 agonists, they demonstrated that RSV-driven shut down of type-I IFN production occurs through the inhibition of TLR7 and TLR9 signaling ().
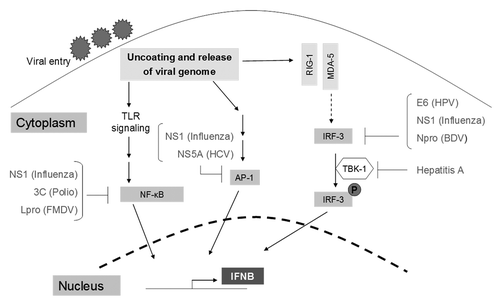
Figure 3. A summary of viral mechanisms that target the IRF-3, NFκB, and AP-1 signaling pathways. E6, NS1, and Npro target IRF-3; NS1, 3C, and L(pro) target NFκB; NS1 and NS5A target AP-1.
Mechanisms That Circumvent the Signaling Pathways Leading to Interferon Production
An abundance of data has been obtained concerning the involvement of virally derived proteins that evade host recognition and prevent interferon production. This is perhaps the most widely studied mechanism in the fields of virology and immunology. This mechanism will therefore be discussed under the following sub-categories: (1) virally derived proteins that inhibit the IRF-3, NFκB, and AP-1 signaling pathways and (2) virally derived proteins that inhibit the JAK-STAT signaling pathway.
Virally derived proteins that inhibit the IRF-3, NFκB, and AP-1 signaling pathways
Transcription factors involved in interferon production are activated when a virus is initially detected. These factors include IRF-3, NKκB, the stress activated transcription factor AP-1, and CBP/p300, the latter being associated with chromatin remodeling and concomitant transcriptional activation. Activated transcription factors subsequently translocate to the nucleus and interact with interferon promoter sequences, leading to the upregulation of interferon genes.
It has been shown that IRF-3 is a target for the E6 viral protein from human papilloma virus (HPV).Citation21 IRF-3 becomes activated during viral invasion when virally derived dsRNA is present in the cytoplasm. IRF-3, when activated, initiates INF-β production through its association with p300/CBP in the nucleus.Citation22 Ronco et al. have shown that the E6 viral protein interacts with IRF-3 and hampers its ability to transcriptionally activate IFN-β productionCitation21 (). IRF-3 inhibition however, is not limited to HPV. Talon et al. have previously reported that the influenza A virus attenuates the type-I IFN response.Citation23 Influenza A encodes NS1, a protein capable of interacting with dsRNA.Citation24 Talon et al. further demonstrated through NS1 knockout studies that the influenza A virus prevents IRF-3 activation through an NS1-dependent mechanism (), thus explaining the lack of IFN-β production. However, IFN-α production was also observed to be attenuated in a series of mammalian primary cells that were tested, leading the authors to hypothesize that NS1 may have broader target specificities than previously thought. Wang et al. have shown that NS1 has an inhibitory effect on the NFκB pathway.Citation25 There have also been reports of NS1 inhibiting JNK—a stress kinase acting upstream of the transcription factor AP-1.Citation26 A current model proposes that NS1 binds and sequesters dsRNA, thereby protecting it from cell intrinsic viral RNA detection mechanisms.Citation16
Animal viruses have also evolved to evade the cellular interferon response. Chen et al. report that the bovine diarrhea virus (BDV) derived protein Npro interacts with IRF-3, resulting in its polyubiquitination and degradation in HEK293 cells ().Citation27 I329L is a recently discovered protein encoded by the African swine fever virus and has been shown to act as a TLR3 inhibitor.Citation28 The blue tongue virus-derived NS3 protein inhibits the transcriptional activation of IFN-β.Citation29 Bao et al. have shown that the porcine reproductive and respiratory virus significantly curbs both interferon and TNF-α driven responses in porcine macrophages.Citation30 The investigators further demonstrate that this occurs through the transcriptional inhibition of genes coding for IRF-1, IRF-3, and NFκB. Pestivirus infections are either transient or persistent. Persistent infection is dependent on immune evasion strategies such as Npro-mediated IRF-3 degradation.Citation31 An additional strategy is the evasion of viral recognition by host PAMSs—a mechanism made possible by Ems, a virally derived RNase.Citation31
Certain viruses, such as the Kaposi sarcoma-associated herpes virus (KSHV), express their own IRF proteins. These virally derived IRF proteins have a high degree of homology to cellular IRF proteins.Citation22 KSHV expresses viral IRFs 1 and 2, which inhibit IRF-3 and subsequently, IFN-β production. An indirect mechanism of IRF-3 inactivation was reported by Paulmann et al., whose group demonstrated that hepatitis A virus infection inactivated TBK-1—a kinase critical for IRF-3 activation.Citation32 TBK-1 can also activate IRF-7, leading to the production of IFN-α. They demonstrate that the virally derived proteins 2B and 3ABC interact with a MAVS protein and block TBK1 activity. Their findings also suggest an interaction between 2B and IKKε, a kinase important for NFκB activation.
The NFκB transcription factor is essential for mounting innate immune responses against viruses. When activated, NFκB translocates to the nucleus and transcriptionally activates the synthesis of interferons and a host of pro-inflammatory cytokine genes.Citation33,Citation34 In the inactive state, the negative regulator IκB localizes NFκB to the cytoplasm. The NFκB transcription factor family in mammals is composed of the subunits RelA, RelB, c-Rel, and the precursor proteins p100 and p105.Citation35 Dimers of these subunits are involved in DNA binding and transcriptional activation through the recruitment of HAT proteins (such as CBP and p300) and HDACs 1 and 3.Citation35 It was recently reported that the 3C viral protein from the polio virus, a member of the piconovirus family, is capable of proteolytically cleaving the RelA subunit and functionally inactivating NFκB in HeLa cellsCitation36 (). Subsequently, RelA cleavage was demonstrated for two other viruses belonging to the piconovirus family, prompting the investigators to suggest that RelA cleavage could be a conserved feature shared by this family of viruses to evade host defense. Subsequently, RelA was shown to be a viral target and a report by de los Santos et al. showed reduced levels of the NFκB-responsive genes TNF-α and RANTES in cells infected with the foot-and-mouth disease virus (FMDV). This work demonstrated that the FMDV-derived protein L(pro) led to the degradation of RelA ().Citation37
The activation of the AP-1 transcription factor is an innate antiviral mechanism. Macdonald et al. explored the potential mechanisms through which the hepatitis C virus (HCV) inhibits AP-1 activation in Huh-7 cells. They found that the virally derived protein NS5A inhibits AP-1 mediated downstream events by interacting with Grb2—a signaling intermediate required for the nuclear localization of AP-1.Citation38 Further analysis revealed that a previously unidentified sequence in the C-terminal region of NS5A was required for the inhibition of AP-1 function. Intriguingly, the abrupt appearance of this sequence on NS5A has led to speculations that this could represent a proviral mechanism that has recently evolved.
Virally derived proteins that inhibit the JAK-STAT signaling pathway
The JAK-STAT signaling pathway is composed of the JAK and TYK2 kinases, IRF-9, and STAT proteins.Citation34 JAK1 and TYK2 are associated with the IFN α/β receptor and JAK2 is associated with the IFN γ receptor together with JAK1. Phosphorylation of these receptor associated kinases and STAT proteins is a crucial step in transmitting the interferon induced signal from the cell surface to the nucleus.Citation33,Citation34 The important function played by the JAK and TYK2 kinases and STAT proteins in the antiviral response makes them attractive targets for viral proteins.
A study conducted by Lin et al. found that the Japanese encephalitis virus (JEV) prevented the nuclear entry of STAT proteins in baby hamster kidney (BHK-21) cells, making them unresponsive to IFN-α.Citation39 This inhibitory function was shown to be regulated by NS5. Site directed mutagenesis studies revealed that the N-terminal region of NS5 was essential for this effect. Furthermore, the authors observed that NS5 marginally reduced the phosphorylation of STAT1 (on tyrosine 701) and greatly reduced the phosphorylation of TYK2 (on tyrosine 1054 and on tyrosine 1055). In addition, NS5 was demonstrated to have phosphatase activity ().Citation39 The specificity of NS5 in inhibiting IFN-α but not IFN-γ signaling is intriguing. A study by Mazzon et al. demonstrated that NS5 does not affect IFN-γ mediated gene expression.Citation40
Figure 4. A summary of viral mechanisms that target the JAK-STAT signaling pathway. NS5 and V proteins prevent the activation of TYK2 and JAK1 respectively; V proteins are also involved in STAT sequestration. NS5 and SV5 degrade STAT proteins; C proteins prevent the formation of activated STAT dimers and HCV core proteins induce SOCS expression. ISRE, interferon stimulated response element; GAS, gamma interferon activation site.
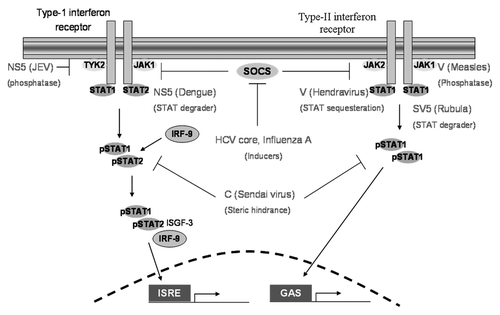
Targeting the JAK-STAT pathway appears to be a commonly used strategy, as evidenced by the diverse group of viruses expressing V proteins and C proteins. Although there are sequential differences among these viral proteins, the major motifs are conserved. The Paramyxovirus family for example (a group of enveloped ssRNA viruses) includes the measles, mumps and hendra viruses, all express V proteins capable of blocking the induction of IFN-β.Citation41 V proteins are not exclusive to Paramyxoviruses. The Rubella virus SV5 product (a V protein) has been shown to reduce the half-life of STAT proteins ().Citation42 Interestingly, the ability of SV5 to degrade STAT1 (and STAT3 in the case of mumps virus) is dependent on the presence of certain host proteins acting cooperatively with the viral protein.Citation43 The hendravirus V protein is able to sequester STAT proteins in large cytoplasmic complexes and limit their nuclear translocation.Citation44,Citation45 Cells infected with the measles virus have been shown to have very low levels of STAT proteins in the nucleus. Measles virus infected cells were found to be unresponsive to INF-α but still remained responsive to IFN-γ.Citation46 Yokota et al. further investigated this observation and demonstrated that the V protein prevents JAK1 phosphorylation and the C protein binds and incapacitates the IFN-α receptor 1 (IFNAR1), thus preventing IFN-α mediated downstream signaling events.Citation46
Like the V proteins, C proteins also target the JAK-STAT pathway. The Sendai virus C protein for example, has been show to inhibit STAT1 phosphorylation.Citation47 Studies have also shown that the Sendai virus C protein makes cells unresponsive to both type I and type II interferons.Citation48,Citation49 This was further investigated by Gotoh et al. who found that C proteins interact with the phosphorylated forms of STAT1 and STAT2, reducing their ability to form homodimers and heterodimers ().Citation49
Mechanisms That Inhibit Interferon-Induced Antiviral Proteins
Interferon-induced proteins help establish an antiviral state in host cells following a viral insult. This ensures immediate protection to cells containing interferon receptors that surround the site of infection. SOCS proteins are well known for their ability to negatively regulate the JAK-STAT signaling pathway. A study by Bode et al. showed that in HepG2 cells, the hepatitis C virus (HCV) core protein induced SOCS3 expression ().Citation50 A study conducted with the herpes simplex virus in human amnion cells showed SOCS3 induction as early as 2 h after cells were exposed to the virus.Citation51 Pothlichet et al. demonstrated that in respiratory epithelial cells infected with the influenza A virus, SOCS1 and SOCS3 expression were induced. Furthermore, SOCS protein expression was only observed when the RIG-1 and IFNAR1 signaling networks were intact, suggesting that this mechanism takes effect at a later stage of infection.Citation52
The PKR and ADAR1 enzymes are interferon inducible gene products.Citation53 Once activated, PKR phosphorylates eIF2α, resulting in translational inhibition in host cell. ADAR1 catalyzes the conversion of adenosine to inosine, an RNA editing mechanism. ADAR1 has recently emerged as a player that possesses proviral properties.Citation54 Studies by Toth et al. and Li et al. showed that ADAR1, acting synergistically with virally derived C and V proteins, decreased apoptosis by suppressing PKR, a proviral maneuver that extends the persistence of viruses in host cells. In addition, IFN-β-induced ADAR1 has been shown to affect the biogenesis of microRNA-142 and microRNA-376 through its ability to edit the precursor forms of these microRNAs.Citation55,Citation56 This editing, when present within the seed region of the microRNA, may impact mRNA target selectivity. Whether this phenomenon is a proviral or antiviral mechanism still remains to be elucidated.
Unlike type-I interferons that are produced as a result of viral entry, IFN-γ is mainly produced by activated T cells, NK cells, and NK-T cells. IFN-γ induces the upregulation of MHC class II molecules on infected macrophages. Interestingly, virally infected murine and human cells are found to reduce their expression of MHC class I molecules, making them better targets for NK cells. The murine cytomegalovirus (MCMV) however is capable of reducing MHC class II expression on murine bone marrow macrophages. This finding was further investigated by Heise et al. whose analysis showed that MCMV impaired IFN-γ production in a dose-dependent manner.Citation57 Poxviruses secrete proteins that interfere with the host interferon response. Upton et al. have shown that the myxoma virus secretes a protein that binds extracellular IFN-γ, thus sequestering it away from its receptor on host cells.Citation58
Additional Examples Illustrating Viral Inhibition of Interferon Pathways
The Vaccinia virus has a large DNA genome and exerts considerable regulatory control over the host innate immune response. Specifically, Vaccinia viruses evade the host interferon response at nearly every stage: blocking the induction of interferons, preventing the activation of NFκB and IRF-3 pathways, preventing the activation of the JAK-STAT pathway, and inhibiting antiviral mechanisms (such as PKR- and OAS-driven mechanisms) from taking effect.Citation59 These broad effects are orchestrated by Vaccinia virus-derived proteins. D9 and D10 proteins prevent the translation of interferon mRNAs; the E3 protein prevents RNA polymerase III from sensing viral DNA, preventing interferon production. E3 also inhibits the activation of PKR and OAS. C4, N1 as well as a myriad of other Vaccinia-derived proteins prevent the activation of NFκB at nearly every step of the NFκB pathway. The C6 protein prevents TBK1-driven activation of IRF-3 and IRF-7. The B18 and B8 proteins function as decoy receptors, acting as sinks for type-1 and type-2 interferons respectively. Vaccinia-derived VH1 is a phosphatase that dephosphorylates STAT1 and STAT2.Citation59
Like the Vaccinia virus, HCMV has a large DNA genome and codes for proteins that have evolved to specifically target host antiviral mechanisms. In addition to the commonly observed mechanisms such as the inhibition of interferon production, transcription factor inactivation and evasion of host PRRs, there are two additional mechanisms that require mention: (1) virally derived IL-10 and (2) the exploitation of antiviral mechanisms to sustain viral persistence.Citation60 HCMV borrows the coding region of the IL-10 gene from the host and incorporates it into its own arsenal. By doing so, the virus is able to use CmvIL-10 (i.e., virally derived IL-10) to limit the release of IFN-α. Given the anti-inflammatory effects of IL-10, it had been postulated that this mechanism has evolved to prevent the paracrine “warning” that neighboring cells typically receive during a viral infection. HCMV infection also stimulates the release of interferon gamma-inducible protein 16, bone marrow stromal cell antigen 2, viperin, and cyclooxygenase-2—all of these are interferon inducible factors.Citation60 Intriguingly, these factors have been observed to facilitate viral replication and/or propagation.
In the context of infections by RNA viruses, RIG-1 and MDA-5 are the gatekeepers of the cell. RNA viruses have developed methods to evade detection and prevent signaling events downstream of these receptors. Zinzula et al. have reviewed the three major mechanisms through which RNA viruses evade detection: by compartmentalizing their genomes to specific locations within the cytoplasm to prevent detection by cytoplasmic RNA sensors; by removing specific regions of their genomes that serve as recognition “hotspots”; and by physically interacting with cytoplasmic RNA sensors for the purpose of attenuating downstream signaling events.Citation61
Karim et al. observed that human keratinocytes persistently infected with human papillomavirus induced the production of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1). UCHL1 is a cell-intrinsic ubiquitin ligase that was observed to prevent the induction of type-1 interferons in response to HPV infection. The investigators went on to demonstrate that UCHL1 interfered with critical ubiquitination events upstream of IRF-3 and NFκB activation, resulting in reduced levels of IRF-3 phosphorylation and NFκB nuclear localization.Citation62
The recently discovered human metapneumovirus has developed mechanisms to inhibit TLR4-mediated NFκB activation, RIG-1-mediated IRF-3 activation, as well as type-1 interferon receptor-mediated JAK-STAT pathway activation. Specifically, virally derived G protein inhibits TLR4 activation and RIG-1 recognition of the viral genome; P proteins hide viral genetic material in cytoplasmic inclusion bodies; the SH protein prevents NFκB activation; and the hMPV protein prevents the activation of Tyk2 and STAT1.Citation63
The Marburg virus and the Ebola virus contain the evolutionarily conserved virulence protein VP35. VP35 binds the backbone region as well as the blunt ends of viral dsRNA genomes, preventing viral recognition by the PRRs RIG-1 and MDA-5. Ramanan et al. show that in the Marburg virus, VP35–backbone interactions prevent PRR recognition while VP35–blunt end interactions do not. They also show that both these interactions prevent PRR recognition in the case of Ebola virus, thus highlighting functional differences in the same virulence factor among different viruses.Citation64
NS proteins of the Flavivirus family have been shown to act at various levels of the type-1 interferons pathway, resulting in the delay and/or inhibition of cellular antiviral mechanisms.Citation65 For example, West Nile virus NS1 prevents IRF-7 activation; Kunjin virus (a West Nile virus variant) NS2A prevents IFN-β transcription; and Japanese encephalitis virus NS5 prevents Tyk2 phosphorylation. Flavivirus-derived proteins have also been shown to interact with host PRRs and the type-1 interferon receptor. Flaviviruses have also evolved mechanisms to evade the adaptive immune response.Citation65
In a CD4+ T-cell model, it has been shown that HIV-derived viral proteases interfere with RIG-1 signaling. Specifically, the vif and vpr proteins lead to IRF-3 degradation.Citation66 In macrophages and lymphocytes, it has been shown that HIV exploits the cell-intrinsic 3′ repair exonuclease to digest viral DNA, thereby preventing its recognition by host PRRs.
HSV-1-derived proteins have been shown to interfere with both type-1 and type-2 interferon responses.Citation67 Specifically, HSV-1-derived ICP0 prevents IRF and STAT activation; ICP27 shuts down the splicing and translation machineries in host cells; ICP34.5 and vhs inhibit protein synthesis leading to interferon production; Us11 prevents PKR activation; and Us3 interferes with TLR3 signaling and induces posttranslational modifications on the type-2 interferon receptor.Citation67
Osterlund et al. have compared the mechanisms through with the interferon pathway is activated by influenza A vs. influenza B viruses in host dendritic cells. The investigators find that the host interferon response is activated immediately following the entry of influenza B. This was observed to occur via the activation of IRF3. On the contrary, interferon induction by influenza A was only observed after viral RNA synthesis—an event that occurs after viral entry.Citation68 These findings demonstrate the differences in early vs. late induction of the interferon response. In addition, the findings also highlight the mechanism through which influenza A evades early recognition by host cells.
Conclusion and Perspective
This review discusses the mechanisms viruses use to evade the host interferon system. There are however, other maneuvers viruses use to persist in the human host that do not directly impact interferon signaling or production. Examples of these mechanisms include (1) protected regions of the HIV-1 viral genome integrating into the host DNA without evoking cell intrinsic viral sensorsCitation69 and (2) hepatitis C virus using microRNA-122, which is endogenous to host hepatic cells, to facilitate its replication.Citation70 These findings add to the growing body of knowledge in our understanding of virus–host interactions. Viruses have played a pivotal role in the discovery of interferons. Studying how viruses evade the interferon system could lead to new discoveries in the fields of virology and innate immunity.
Although many developed nations have adopted strategies to significantly reduce the incidence of viral infections resulting from rabies and polio, statistical data from the World Health Organization show that these diseases still affect many people in developing countries. A survey conducted by the Centers for Disease Control shows that there are over 5 million individuals in the United States who suffer from liver disease associated with hepatitis B.Citation71 When faced with the challenge of combating viral infections of endemic or epidemic proportions, new knowledge of the mechanisms used by viruses to evade the host antiviral response may prove to be indispensable in more efficient vaccine design and drug development.
| Abbreviations: | ||
| ADAR1 | = | adenosine deaminase acting on RNA 1 |
| AP-1 | = | activator protein 1 |
| CARD | = | caspase activation and recruitment domain |
| CBP | = | CREB-binding protein |
| eIF2α | = | eukaryotic initiation factor 2α |
| HAT | = | histone acetyltransfarase |
| HDAC | = | histone deacetylase |
| IFN | = | interferon |
| IKK | = | inhibitor of kappa kinase |
| IRF | = | interferon regulatory factor |
| JAK | = | Janus kinase |
| JNK | = | c-Jun N terminal kinase |
| MAVS | = | mitochondrial antiviral signaling |
| MDA-5 | = | melanoma differentiation-associated protein 5 |
| NFκB | = | nuclear factor kappa B |
| PAMP | = | pathogen associated molecular pattern |
| PKR | = | protein kinase R |
| RANTES | = | regulated on activation, normal T-cell expressed and secreted |
| RIG-1 | = | retinoic acid-inducible gene 1 |
| SOCS | = | suppressor of cytokine signaling |
| STAT | = | signal transducer and activator of transcription |
| TLR | = | Toll-like receptor |
| TNF | = | tumor necrosis factor |
| TRIM25 | = | tripartite motif containing 25 |
| TYK2 | = | tyrosine kinase 2 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
A.S.D. would like to thank Dr Thomas B Tomasi for his support and encouragement in completing this work.
References
- Hengel H, Koszinowski UH, Conzelmann KK. Viruses know it all: new insights into IFN networks. Trends Immunol 2005; 26:396 - 401; http://dx.doi.org/10.1016/j.it.2005.05.004; PMID: 15922665
- Wilfert L, Jiggins FM. Host-parasite coevolution: genetic variation in a virus population and the interaction with a host gene. J Evol Biol 2010; 23:1447 - 55; http://dx.doi.org/10.1111/j.1420-9101.2010.02002.x; PMID: 20456575
- Lobo FP, Mota BE, Pena SD, Azevedo V, Macedo AM, Tauch A, Machado CR, Franco GR. Virus-host coevolution: common patterns of nucleotide motif usage in Flaviviridae and their hosts. PLoS One 2009; 4:e6282; http://dx.doi.org/10.1371/journal.pone.0006282; PMID: 19617912
- Vink C, Smit MJ, Leurs R, Bruggeman CA. The role of cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines in manipulation of and evasion from the immune system. J Clin Virol 2001; 23:43 - 55; http://dx.doi.org/10.1016/S1386-6532(01)00184-6; PMID: 11595583
- Taylor DJ, Ballinger MJ, Bowman SM, Bruenn JA. Virus-host co-evolution under a modified nuclear genetic code. PeerJ 2013; 1:e50; http://dx.doi.org/10.7717/peerj.50; PMID: 23638388
- Weber F, Kochs G, Haller O. Inverse interference: how viruses fight the interferon system. Viral Immunol 2004; 17:498 - 515; http://dx.doi.org/10.1089/vim.2004.17.498; PMID: 15671747
- McInerney GM, Karlsson Hedestam GB. Direct cleavage, proteasomal degradation and sequestration: three mechanisms of viral subversion of type I interferon responses. J Innate Immun 2009; 1:599 - 606; http://dx.doi.org/10.1159/000235861; PMID: 20375615
- Schlender J, Hornung V, Finke S, Günthner-Biller M, Marozin S, Brzózka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol 2005; 79:5507 - 15; http://dx.doi.org/10.1128/JVI.79.9.5507-5515.2005; PMID: 15827165
- Brzózka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol 2005; 79:7673 - 81; http://dx.doi.org/10.1128/JVI.79.12.7673-7681.2005; PMID: 15919920
- Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J Virol 2008; 82:8296 - 306; http://dx.doi.org/10.1128/JVI.00108-08; PMID: 18562542
- Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog 2009; 5:e1000287; http://dx.doi.org/10.1371/journal.ppat.1000287; PMID: 19197350
- Oshansky CM, Krunkosky TM, Barber J, Jones LP, Tripp RA. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a toll-like receptor pathway. Viral Immunol 2009; 22:147 - 61; http://dx.doi.org/10.1089/vim.2008.0098; PMID: 19435411
- Heise MT, Connick M, Virgin HW 4th. Murine cytomegalovirus inhibits interferon gamma-induced antigen presentation to CD4 T cells by macrophages via regulation of expression of major histocompatibility complex class II-associated genes. J Exp Med 1998; 187:1037 - 46; http://dx.doi.org/10.1084/jem.187.7.1037; PMID: 9529320
- Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009; 5:439 - 49; http://dx.doi.org/10.1016/j.chom.2009.04.006; PMID: 19454348
- Zhou S, Cerny AM, Zacharia A, Fitzgerald KA, Kurt-Jones EA, Finberg RW. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J Virol 2010; 84:9452 - 62; http://dx.doi.org/10.1128/JVI.00155-10; PMID: 20592086
- Borrow P, Martínez-Sobrido L, de la Torre JC. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2010; 2:2443 - 80; http://dx.doi.org/10.3390/v2112443; PMID: 21994626
- Gori-Savellini G, Valentini M, Cusi MG. Toscana virus NSs protein inhibits the induction of type I interferon by interacting with RIG-I. J Virol 2013; 87:6660 - 7; http://dx.doi.org/10.1128/JVI.03129-12; PMID: 23552410
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 2004; 101:17264 - 9; http://dx.doi.org/10.1073/pnas.0407639101; PMID: 15563593
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675 - 80; http://dx.doi.org/10.1038/90609; PMID: 11477402
- Meylan E, Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell 2006; 22:561 - 9; http://dx.doi.org/10.1016/j.molcel.2006.05.012; PMID: 16762830
- Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev 1998; 12:2061 - 72; http://dx.doi.org/10.1101/gad.12.13.2061; PMID: 9649509
- Grimm S, Baeuerle PA. The inducible transcription factor NF-kappa B: structure-function relationship of its protein subunits. Biochem J 1993; 290:297 - 308; PMID: 8452515
- Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, García-Sastre A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 2000; 74:7989 - 96; http://dx.doi.org/10.1128/JVI.74.17.7989-7996.2000; PMID: 10933707
- Neznanov N, Chumakov KM, Neznanova L, Almasan A, Banerjee AK, Gudkov AV. Proteolytic cleavage of the p65-RelA subunit of NF-kappaB during poliovirus infection. J Biol Chem 2005; 280:24153 - 8; http://dx.doi.org/10.1074/jbc.M502303200; PMID: 15845545
- Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, García-Sastre A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 2000; 74:11566 - 73; http://dx.doi.org/10.1128/JVI.74.24.11566-11573.2000; PMID: 11090154
- Ludwig S, Wang X, Ehrhardt C, Zheng H, Donelan N, Planz O, Pleschka S, García-Sastre A, Heins G, Wolff T. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J Virol 2002; 76:11166 - 71; http://dx.doi.org/10.1128/JVI.76.21.11166-11171.2002; PMID: 12368362
- Chen Z, Rijnbrand R, Jangra RK, Devaraj SG, Qu L, Ma Y, Lemon SM, Li K. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 2007; 366:277 - 92; http://dx.doi.org/10.1016/j.virol.2007.04.023; PMID: 17531282
- de Oliveira VL, Almeida SC, Soares HR, Crespo A, Marshall-Clarke S, Parkhouse RM. A novel TLR3 inhibitor encoded by African swine fever virus (ASFV). Arch Virol 2011; 156:597 - 609; http://dx.doi.org/10.1007/s00705-010-0894-7; PMID: 21203785
- Vitour D, Doceul V, Ruscanu S, Chauveau E, Schwartz-Cornil I, Zientara S. Induction and control of the type I interferon pathway by Bluetongue virus. Virus Res 2013; Forthcoming http://dx.doi.org/10.1016/j.virusres.2013.10.027; PMID: 24211608
- Bao D, Wang R, Qiao S, Wan B, Wang Y, Liu M, Shi X, Guo J, Zhang G. Antibody-dependent enhancement of PRRSV infection down-modulates TNF-α and IFN-β transcription in macrophages. Vet Immunol Immunopathol 2013; 156:128 - 34; http://dx.doi.org/10.1016/j.vetimm.2013.09.006; PMID: 24099951
- Peterhans E, Schweizer M. Pestiviruses: how to outmaneuver your hosts. Vet Microbiol 2010; 142:18 - 25; http://dx.doi.org/10.1016/j.vetmic.2009.09.038; PMID: 19846261
- Paulmann D, Magulski T, Schwarz R, Heitmann L, Flehmig B, Vallbracht A, Dotzauer A. Hepatitis A virus protein 2B suppresses beta interferon (IFN) gene transcription by interfering with IFN regulatory factor 3 activation. J Gen Virol 2008; 89:1593 - 604; http://dx.doi.org/10.1099/vir.0.83521-0; PMID: 18559929
- Lin L, Li Y, Pyo HM, Lu X, Raman SN, Liu Q, Brown EG, Zhou Y. Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J Virol 2012; 86:1942 - 54; http://dx.doi.org/10.1128/JVI.06362-11; PMID: 22171255
- Horvath CM. The Jak-STAT pathway stimulated by interferon gamma. Sci STKE 2004; 2004:tr8; PMID: 15561980
- Stark GR, Kerr IM. Interferon-dependent signaling pathways: DNA elements, transcription factors, mutations, and effects of viral proteins. J Interferon Res 1992; 12:147 - 51; http://dx.doi.org/10.1089/jir.1992.12.147; PMID: 1379292
- Neznanov N, Chumakov KM, Neznanova L, Almasan A, Banerjee AK, Gudkov AV. Proteolytic cleavage of the p65-RelA subunit of NF-kappaB during poliovirus infection. J Biol Chem 2005; 280:24153 - 8; http://dx.doi.org/10.1074/jbc.M502303200; PMID: 15845545
- de Los Santos T, Diaz-San Segundo F, Grubman MJ. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J Virol 2007; 81:12803 - 15; http://dx.doi.org/10.1128/JVI.01467-07; PMID: 17881445
- Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J Biol Chem 2003; 278:17775 - 84; http://dx.doi.org/10.1074/jbc.M210900200; PMID: 12621033
- Lin RJ, Chang BL, Yu HP, Liao CL, Lin YL. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol 2006; 80:5908 - 18; http://dx.doi.org/10.1128/JVI.02714-05; PMID: 16731929
- Mazzon M, Jones M, Davidson A, Chain B, Jacobs M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis 2009; 200:1261 - 70; http://dx.doi.org/10.1086/605847; PMID: 19754307
- Horvath CM. The Jak-STAT pathway stimulated by interferon alpha or interferon beta. Sci STKE 2004; 2004:tr10; PMID: 15561979
- Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, Lamb RA, Horvath CM. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology 2001; 283:230 - 9; http://dx.doi.org/10.1006/viro.2001.0856; PMID: 11336548
- Ulane CM, Horvath CM. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002; 304:160 - 6; http://dx.doi.org/10.1006/viro.2002.1773; PMID: 12504558
- Rodriguez JJ, Wang LF, Horvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol 2003; 77:11842 - 5; http://dx.doi.org/10.1128/JVI.77.21.11842-11845.2003; PMID: 14557668
- Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol 2004; 78:5358 - 67; http://dx.doi.org/10.1128/JVI.78.10.5358-5367.2004; PMID: 15113915
- Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology 2003; 306:135 - 46; http://dx.doi.org/10.1016/S0042-6822(02)00026-0; PMID: 12620806
- Garcin D, Marq JB, Goodbourn S, Kolakofsky D. The amino-terminal extensions of the longer Sendai virus C proteins modulate pY701-Stat1 and bulk Stat1 levels independently of interferon signaling. J Virol 2003; 77:2321 - 9; http://dx.doi.org/10.1128/JVI.77.4.2321-2329.2003; PMID: 12551969
- Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol 2002; 76:4190 - 8; http://dx.doi.org/10.1128/JVI.76.9.4190-4198.2002; PMID: 11932384
- Gotoh B, Takeuchi K, Komatsu T, Yokoo J. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J Virol 2003; 77:3360 - 70; http://dx.doi.org/10.1128/JVI.77.6.3360-3370.2003; PMID: 12610111
- Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, Heinrich PC, Häussinger D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J 2003; 17:488 - 90; PMID: 12551851
- Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Miura S, Jimbow K, Fujii N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J Virol 2004; 78:6282 - 6; http://dx.doi.org/10.1128/JVI.78.12.6282-6286.2004; PMID: 15163721
- Fan L, Briese T, Lipkin WI. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J Virol 2010; 84:1785 - 91; http://dx.doi.org/10.1128/JVI.01362-09; PMID: 20007272
- Pothlichet J, Chignard M, Si-Tahar M. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol 2008; 180:2034 - 8; PMID: 18250407
- Pfaller CK, Li Z, George CX, Samuel CE. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol 2011; 23:573 - 82; http://dx.doi.org/10.1016/j.coi.2011.08.009; PMID: 21924887
- Li Z, Okonski KM, Samuel CE. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J Virol 2012; 86:3787 - 94; http://dx.doi.org/10.1128/JVI.06307-11; PMID: 22278222
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007; 315:1137 - 40; http://dx.doi.org/10.1126/science.1138050; PMID: 17322061
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 2006; 13:13 - 21; http://dx.doi.org/10.1038/nsmb1041; PMID: 16369484
- Upton C, Mossman K, McFadden G. Encoding of a homolog of the IFN-gamma receptor by myxoma virus. Science 1992; 258:1369 - 72; http://dx.doi.org/10.1126/science.1455233; PMID: 1455233
- Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 2013; 94:2367 - 92; http://dx.doi.org/10.1099/vir.0.055921-0; PMID: 23999164
- Amsler L, Verweij MC, DeFilippis VR. The tiers and dimensions of evasion of the type I interferon response by human cytomegalovirus. J Mol Biol 2013; 425:4857 - 71; http://dx.doi.org/10.1016/j.jmb.2013.08.023; PMID: 24013068
- Zinzula L, Tramontano E. Strategies of highly pathogenic RNA viruses to block dsRNA detection by RIG-I-like receptors: hide, mask, hit. Antiviral Res 2013; 100:615 - 35; http://dx.doi.org/10.1016/j.antiviral.2013.10.002; PMID: 24129118
- Karim R, Tummers B, Meyers C, Biryukov JL, Alam S, Backendorf C, Jha V, Offringa R, van Ommen GJ, Melief CJ, et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog 2013; 9:e1003384; http://dx.doi.org/10.1371/journal.ppat.1003384; PMID: 23717208
- Kolli D, Bao X, Casola A. Human metapneumovirus antagonism of innate immune responses. Viruses 2012; 4:3551 - 71; http://dx.doi.org/10.3390/v4123551; PMID: 23223197
- Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, Otwinowski Z, Liu G, Huh J, Basler CF, et al. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A 2012; 109:20661 - 6; http://dx.doi.org/10.1073/pnas.1213559109; PMID: 23185024
- Ye J, Zhu B, Fu ZF, Chen H, Cao S. Immune evasion strategies of flaviviruses. Vaccine 2013; 31:461 - 71; http://dx.doi.org/10.1016/j.vaccine.2012.11.015; PMID: 23153447
- Marsili G, Remoli AL, Sgarbanti M, Perrotti E, Fragale A, Battistini A. HIV-1, interferon and the interferon regulatory factor system: an interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev 2012; 23:255 - 70; http://dx.doi.org/10.1016/j.cytogfr.2012.06.001; PMID: 22748237
- Paladino P, Mossman KL. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interferon Cytokine Res 2009; 29:599 - 607; http://dx.doi.org/10.1089/jir.2009.0074; PMID: 19694546
- Österlund P, Strengell M, Sarin LP, Poranen MM, Fagerlund R, Melén K, Julkunen I. Incoming influenza A virus evades early host recognition, while influenza B virus induces interferon expression directly upon entry. J Virol 2012; 86:11183 - 93; http://dx.doi.org/10.1128/JVI.01050-12; PMID: 22855501
- Kuehn BM. Silent epidemic of viral hepatitis may lead to boom in serious liver disease. J Am Med Assoc 1949-50; 2009:54
- Manel N, Littman DR. Hiding in plain sight: how HIV evades innate immune responses. Cell 2011; 147:271 - 4; http://dx.doi.org/10.1016/j.cell.2011.09.010; PMID: 22000008
- Lee HR, Kim MH, Lee JS, Liang C, Jung JU. Viral interferon regulatory factors. J Interferon Cytokine Res 2009; 29:621 - 7; http://dx.doi.org/10.1089/jir.2009.0067; PMID: 19715458