
Abstract
Biofilm-degrading enzymes could be used for the gentle cleaning of industrial and medical devices and the manufacture of biofilm-resistant materials. We therefore investigated 20 species and strains of the bacterial genus Lysobacter for their ability to degrade experimental biofilms formed by Staphylococcus epidermidis, a common nosocomial pathogen typically associated with device-related infections. The highest biofilm-degradation activity was achieved by L. gummosus. The corresponding enzymes were identified by sequencing the L. gummosus genome. Partial purification of the biofilm-degrading activity from an extract of extracellular material followed by peptide mass fingerprinting resulted in the identification of two peptidases (α-lytic protease and β-lytic metalloendopeptidase) that were predicted to degrade bacterial cell walls. In addition, we identified two isoforms of a lysyl endopeptidase and an enzyme similar to metalloproteases from Vibrio spp. Potential peptidoglycan-binding C-terminal fragments of two OmpA-like proteins also co-purified with the biofilm-degrading activity. The L. gummosus genome was found to encode five isoenzymes of α-lytic protease and three isoenzymes of lysyl endopeptidase. These results indicated that the extracellular digestion of biofilms by L. gummosus depends on multiple bacteriolytic and proteolytic enzymes, which could now be exploited for biofilm control.
Introduction
Biofilms cause significant problems in industrial water facilities, and biofilms forming on indwelling medical devices are responsible for a large proportion of hospital-acquired infections. Environmental biofilms are reservoirs for pathogens, which may acquire increased virulence following their adaptation to the biofilm growth mode.Citation1 The prevention and removal of biofilms remains a largely unsolved challenge. Most current procedures rely on rigorous mechanical and chemical cleaning of colonized surfaces, whereas implant infections typically require surgical replacement.Citation2 Enzymatic procedures for killing microorganisms inside biofilms are also available. Some are based on the in situ production of inorganic antimicrobial compounds such as hypothiocyanite or hypoiodite, using a combination of glucose oxidase and lactoperoxidase.Citation3 The degradation of other biofilms can be achieved with commercially available proteases such as subtilisin, trypsin, proteinase K, and Antarctic krill shrimp proteases.Citation4-Citation6 Biofilms can also be removed using polysaccharide-degrading enzymes such as cellulaseCitation7 and Pectinex Ultra SP, a complex mixture commercialized for the clarification of fruit juices.Citation5 Further approaches employ cell wall-degrading enzymes such as phage-derived endolysins,Citation8 bacterial autolysins,Citation9 and lysostaphin.Citation10 DNase I, which is thought to degrade extracellular DNA, is also useful for biofilm inhibition and removal in some cases.Citation11 Probably the most specific biofilm-degrading agent known at present is dispersin B, which hydrolyzes poly-β-1,6-N-acetyl-d-glucosamine (PNAG).Citation12 This structurally-unique exopolysaccharide is the major constituent of biofilms produced by many nosocomial Staphylococcus epidermidis and S. aureus strains, and has also been detected in other gram-positive and gram-negative bacteria.Citation13 However, S. epidermidis strains are also known to form biofilms in a PNAG-independent manner, mediated by the accumulation-associated protein (Aap) or the extracellular matrix binding protein (Embp).Citation14 Enzymes have also been used for the production of biofilm-resistant materials by covalent attachment or physical entrapment within suitable matrices.Citation4,Citation7,Citation15
The bacterial genus Lysobacter comprises gram-negative rods with gliding motility, which typically produce exoenzymes that lyse and degrade other microorganisms.Citation16-Citation18 This type of facultative predatory lifestyle, often described as a microbial wolf pack, is also typical of the myxobacteria, with which Lysobacter species were confused for a long time. In contrast to myxobacteria, Lysobacter species do not form fruiting bodies. L. enzymogenes, which has been studied in the most detail, can lyse gram-positive and gram-negative bacteria, cyanobacteria, certain algae, yeasts and filamentous fungi, in addition to killing nematodes. Although the ability of different Lysobacter strains to lyse pathogenic bacteria such as S. aureus is well documented, the use of Lysobacter-derived enzymes to combat biofilms has not been investigated thus far. We therefore tested preparations of extracellular material secreted by several Lysobacter species for their ability to degrade experimental biofilms formed by a PNAG-producing strain of S. epidermidis. As a commensal resident of human skin and mucous membranes, S. epidermidis is thought to play a key role in the colonization of medical devices.Citation19 Among the species we tested, the highest biofilm-degrading activity was achieved by L. gummosus. After partial purification of the extracellular material, two types of lytic proteases, two isoforms of a lysyl endopeptidase, a metalloprotease, and putative peptidoglycan-binding fragments of two OmpA-like proteins were identified in fractions with biofilm-degrading activity. Further analysis of the L. gummosus genome also revealed the presence of genes encoding additional related enzymes. L. gummosus therefore appears to represent a rich source of enzymes for applications in biofilm control.
Results
Identification of biofilm-degrading Lysobacter species
A total of 20 Lysobacter species and strains (see Materials and Methods) were tested for biofilm-degrading activity. Supernatants from liquid cultures as well as extracts of extracellular material from cultures on solid medium were added to S. epidermidis biofilms established on 24-well plates. After incubation, the plates were rinsed and the remaining biofilm was stained with crystal violet. The most efficient reduction in staining was achieved using an extract from L. gummosus grown for 3 d on solid medium (). Less pronounced activity was also observed with samples from L. antibioticus, L. enzymogenes, Lysobacter sp. DSM 3655, and L. panaciterrae.
Figure 1. Detection and chromatographic characterization of biofilm-degrading activity from L. gummosus. (A) Crude extracts prepared from the extracellular material of different Lysobacter species grown for 3 d on solid medium were tested against S. epidermidis biofilms on 24-well plates. Lant, L. antibioticus; Lenz, L. enzymogenes (DSMZ 2043); Lgum, L. gummosus; Lsp, Lysobacter sp. (DSMZ 3655); Lbru, L. brunescens; C water control. (B) L. gummosus extract was loaded onto a Q Sepharose Fast Flow column, and eluted isocratically with 100 mM NaCl (fractions 1–5). The flow-through (FT), the wash fractions (W), and the elution fractions were tested for biofilm-degrading activity. (C) The active fraction from the Q Sepharose column was passed over a Mono S 5/50 GL column and subsequently loaded on a Mono Q 5/50 GL column. Fractions 1–18 were eluted with 0–225 mM NaCl and tested for biofilm-degrading activity.
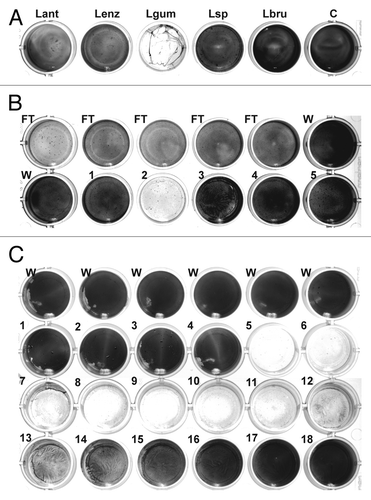
Chromatographic characterization of biofilm-degrading activity from L. gummosus
An aqueous extract of extracellular material was prepared from L. gummosus cultured on 20 145-mm Petri dishes. When bacteria scratched from the plates were stirred with water, a jelly-like mass was produced, which needed to be liquefied by homogenization in a blender followed by ultra-sonication. The extract was adjusted to pH 8.0 and loaded onto a strong anion-exchange column (Q Sepharose Fast Flow), and then eluted in a stepped gradient of NaCl. Biofilm-degrading activity was recovered at the onset of the 100 mM NaCl elution step and was also observed in the flow-through fraction (). In contrast to the starting material, the active elution fraction was not viscous and was therefore suitable for further analytical chromatography steps. Because the active molecules eluted from the anion-exchange column at a low salt concentration, they were likely to carry only a weak negative charge. We therefore reduced the pH to 6.0 to increase the positive charge of the molecules, and passed the sample through a strong cation-exchange column (Mono S 5/50 GL). Unexpectedly, the activity was almost completely recovered in the flow-through. Therefore, we re-adjusted the sample pH to 8.0 and repeated the separation using a small, high-capacity anion-exchange column (Mono Q 5/50 GL). In this experiment, a clear activity peak was observed, eluting at ~60 mM NaCl (, fractions 5 and 6). SDS-PAGE analysis of the active fractions produced a complex band pattern, so further purification was carried out by gel permeation chromatography (Superdex 75 10/30). However, this step resulted in the loss of most of the activity and only residual activity was observed in the fractions eluting at an apparent molecular mass <6.5 kDa (data not shown). This suggested that the active molecules were retained on the column material.
Purification of biofilm-degrading activity for peptide mass fingerprinting
In order to identify individual enzymes associated with the observed biofilm-degrading activity, we developed a purification procedure based on the experiments described above. An extract prepared from 100 L. gummosus culture plates was fractionated by Q Sepharose Fast Flow chromatography using a step gradient in 12 consecutive runs. The combined fractions from the 100 mM NaCl step were then fractionated in three consecutive runs on Source 15 Q resin (a strong anion-exchange material equivalent to Mono Q, but with a larger particle size for higher flow rates). The most active fractions eluting at ~50 mM NaCl were combined and fractionated on a ProPac SAX-10 strong anion-exchange column, featuring a nonporous pellicular solid-phase resin for high-resolution separation. The fractions eluting at a salt concentration of ~38 mM (fractions 12 and 13) showed biofilm degradation activity after incubation with the substrate for only after 2 h. After overnight incubation, biofilm degradation activity was also observed in additional fractions eluting over a much broader range (, 15–100 mM, fractions 6–31). Further activity was observed in the flow-through. SDS-PAGE analysis revealed three protein bands with apparent molecular masses of 33 kDa (band 1), 18 kDa (band 2), and 12 kDa (band 3), which were present at different intensities in fractions 9–15 (). In fractions 7 and 8, only the faint band 2 was visible, whereas only band 1 was detected in fractions 17–20. Remarkably, despite the absence of clear bands in some of the fractions (e.g. 6, 21 and 22), biofilm-degradation activity was still present in the corresponding assay. Our attempts to purify or concentrate the samples further were unsuccessful, possibly reflecting the nonspecific adsorption and/or aggregation of the active molecules.
Figure 2. Purification of biofilm-degrading activity for peptide mass fingerprinting. (A) The fractions obtained by chromatography of the L. gummosus extract on a ProPac-SAX10 column were tested for biofilm-degrading activity (ST, starting material; FT, flow-through; W, wash; C, water control). (B) The fractions were analyzed by SDS-PAGE followed by staining with Flamingo Fluorescent Gel Stain. The bands labeled 1, 2, and 3 were excised and digested in-gel with trypsin for peptide mass fingerprinting. Fractions 8 and 17 were digested in solution with trypsin for peptide mass fingerprinting.
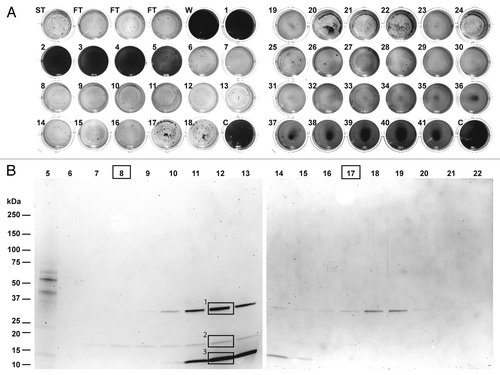
Identification of proteins co-purifying with biofilm-degrading activity
The three bands detected by SDS-PAGE in fractions 9–15 () were excised, digested in-gel with trypsin and analyzed by nano-LC-Orbitrap-MS/MS. Further relevant proteins present at concentrations below the detection limit of SDS-PAGE were identified by digesting fractions 8 and 17 in-solution with trypsin, and capturing the resulting peptides on a reversed-phase cartridge followed by nano-LC-Orbitrap-MS/MS. The MS/MS data were searched against the L. gummosus genome using Mascot software (). The protein correlating to band 1 was identified as an M4 family metalloprotease with similarity to an enzyme from Vibrio cholerae previously described as a hemagglutinin/proteinase (sequence alignments are presented in Figs. S1–S6). Band 2 was found to contain both a serine peptidase and a metallopeptidase with similarity to enzymes from other Lysobacter species known as α-lytic protease and β-lytic metalloendopeptidase, respectively. The proteins co-migrated in the gel because the predicted molecular masses of the mature enzymes were similar (20.0 and 19.1 kDa, respectively). Peptides matching the α‑lytic protease and β-lytic metalloendopeptidase were also detected in band 3, indicating the presence of proteolytic fragments of these enzymes. Further constituents of band 3 were found to originate from two OmpA-like outer membrane proteins, both with a calculated molecular mass of 36.6 kDa. However, the corresponding bands had apparent molecular masses of ~12 kDa suggesting they were proteolytic fragments rather than full-length proteins. Accordingly, only peptides matching the C-terminal parts of these proteins were detected (Fig. S7). Peptides from fraction 8 (digested in solution), in which only band 2 was detectable by SDS-PAGE, also matched the α-lytic protease and β-lytic metalloendopeptidase. Fraction 17 (digested in solution) yielded peptides matching the hemagglutinin/proteinase homolog, the α-lytic protease and the β-lytic metalloendopeptidase, even though only band 1 was detected weakly by SDS-PAGE. Both fractions digested in solution also yielded peptides derived from two isoforms of a protein resembling a lysyl endopeptidase known from other Lysobacter species and from Achromobacter lyticus. No SDS-PAGE bands were assigned to these proteins. The mature enzymes had predicted molecular masses of 27.4 and 28.7 kDa, respectively.
Table 1.L. gummosus proteins that co-purified with biofilm-degrading activity
Identification of additional potential biofilm-degrading enzymes in the L. gummosus genome
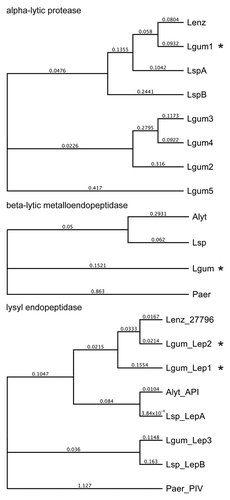
A L. gummosus shotgun library and 8-kbp paired-end library were sequenced using the GS FLX+ system followed by PCR and conventional sequencing to close gaps, yielding a 5 901 415-bp contig with a GC content of 66.7%. Coding sequences predicted with GeneMarkS were screened against the enzyme sequences derived by peptide mass fingerprinting. The L. gummosus α-lytic protease matched four additional α-lytic proteases with more distantly-related amino acid sequences, which showed 51%, 44%, 44%, and 48% identity to the prototypical L. enzymogenes preproenzyme (; Fig. S1). In addition to the two L. gummosus lysyl endopeptidases (named Lep1 and Lep2) identified by peptide mass fingerprinting, we also found a L. gummosus gene encoding a third lysyl endopeptidase (Lep3) (, Fig. S3). Lep2 and Lep3 shared a C-terminal extension domain similar to the homologous enzymes from other Lysobacter species, whereas Lep1 did not contain such a domain.
Figure 3. Phylogeny of L. gummosus enzymes that co-purified with biofilm-degrading activity. The phylogenetic trees were constructed on the basis of the amino acid sequences of the predicted preproenzymes using Geneious Tree Builder software (Blosum62; gap open penalty, 3; gap extension penalty 3). Alyt, Achromobacter lyticus; Lenz, L. enzymogenes; Lgum, L. gummosus; Lsp, Lysobacter sp.; Paer, Pseudomonas aeruginosa. The proteins in the fractions with biofilm-degrading activity are marked with an asterisk. Accession numbers are listed in the supplemental material.
Discussion
Identification of L. gummosus proteins co-purifying with biofilm-degrading activity
We have identified L. gummosus as a potent candidate for the production of novel, biofilm-degrading enzymes. The ability to secrete a cocktail of such enzymes may enable L. gummosus to compete with and to feed on other microorganisms not only in soil and fresh water habitats but also those associated with animals including humans. For example, L. gummosus has been isolated from the skin of redback salamanders and was identified by culture-independent methods as the most abundant bacterial species on the surface of failed prosthetic hip joints removed during arthroplasty.Citation20,Citation21 After purifying the biofilm-degrading activity using three different types of strong anion-exchange columns we identified by peptide mass fingerprinting an α-lytic protease, a β-lytic metalloendopeptidase, two isoforms of a lysyl endopeptidase (Lep1 and Lep2), a metalloprotease homologous to the hemagglutinin/proteinase from V. cholera, and the C-terminal fragments of two OmpA-like proteins.
α-Lytic protease
The prototype α-lytic protease was purified from the culture medium of L. enzymogenes strain ATCC 29487 (formerly known as Myxobacter or Sorangium strain 495).Citation22,Citation23 Its bacteriolytic activity was demonstrated against several soil bacteria, and it was also shown to hydrolyze proteinaceous substrates such as casein, elastin and insulin. The enzyme is synthesized as a preproenzyme that is transported across the inner membrane to the periplasmic space, where the signal sequence is removed.Citation24-Citation26 The prodomain is a folding catalyst, allowing the protease domain to adopt the correct conformation. Following autocatalytic cleavage at the junction between the prodomain and the protease domain, the two regions remain associated while they are conveyed across the outer membrane. The prodomain not only stabilizes the folded protease domain but also functions as a strong inhibitor of its protease activity. Proteolytic degradation of the prodomain, either by the α-lytic protease itself or other enzymes, finally releases the mature active protease.Citation27 Two enzymes named α-lytic protease A (AlpA) and B (AlpB) are produced by Lysobacter sp. XL1, with 78% and 58% sequence identity to the L. enzymogenes preproenzyme.Citation28,Citation29 Another α-lytic protease was purified from the culture medium of Achromobacter lyticus, and N-terminal sequencing/amino acid compositional analysis of the mature enzyme suggested that large segments are identical to the L. enzymogenes prototype.Citation30 The A. lyticus α-lytic protease was shown to cleave the N-acetylmuramoyl-l-alanine amide bond and the d-Ala-Gly and Gly-Gly bonds of S. aureus peptidoglycan.
We found that the L. gummosus α-lytic protease that co-purified with the biofilm-degrading activity (L. gummosus α-lytic protease isoform 1) is closely related to the corresponding enzyme from L. enzymogenes (; Fig. S1). In contrast, the four additional α-lytic proteases (isoforms 2, 3, 4, and 5) encoded by the L. enzymogenes genome cluster into a separate phylogenetic clade, possibly indicating unusual physicochemical and/or catalytic properties (). Indeed, the predicted isoelectric points of the L. gummosus α-lytic protease isoforms 2, 3, 4, and 5 are acidic-to-neutral (pI values of 5.9, 6.3, 7.0, and 5.5, respectively), whereas the previously known α-lytic proteases are characterized by basic pI values (9.7, 9.6, and 9.4) for the α-lytic proteases from L. enzymoges and Lysobacter sp. XL1 isoforms A and B, respectively).
β-Lytic metalloendopeptidase
The prototype β-lytic metalloendopeptidase (formerly β-lytic protease) was originally discovered together with the α-lytic protease in the culture medium of L. enzymogenes.Citation22 Its primary structure has been determined only by direct protein sequencing of the mature enzymeCitation31 but the molecular cloning of an A. lyticus homolog confirmed that the enzyme is produced as a preproprotein.Citation32 The mature peptidase domain of the A. lyticus enzyme is highly similar to its counterpart from L. enzymogenes (96% sequence identity). A full-length gene encoding the Lysobacter sp. IB-9374 homolog has also been sequenced and the mature peptidase domain was found to be completely identical to that of the A. lyticus enzyme, whereas the preprodomains display only 41% sequence identity.Citation33 The L. enzymogenes enzyme can degrade casein and elastin-orcein.Citation32,Citation34 The A. lyticusCitation35 and Lysobacter sp. IB-9374Citation33 homologs cleave synthetic peptides downstream of glycine residues. The Lysobacter sp. IB-9374 enzyme cleaves Gly5 and Gly4 into Gly3 and Gly2, and can also degrade elastin-Congo red. The A. lyticus enzyme is specific for d-Ala-Gly and Gly-Gly bonds in S. aureus peptidoglycan, and for d-Ala-Ala bonds in Micrococcus luteus peptidoglycan.Citation35 The Lysobacter sp. IB-9374 enzymes showed pronounced bacteriolytic activity against M. luteus, S. aureus, S. caseolyticus, Microbacterium arborescens, Bacillus subtilis, Arthrobacter globiformis, and Enterococcus faecalis.Citation33 Significant but lower activity was also observed against Corynebacterium aquaticum, five species of lactic acid bacteria and four species of gram-negative bacteria.
Staphylolysin (also known as LasA or staphylolytic protease) from Pseudomonas aeruginosa is another bacteriolytic metalloendopeptidase which is homologous to β-lytic metalloendopeptidase albeit with a lower sequence identity to the proteins discussed above. In contrast to α-lytic protease and most other bacterial proteases produced as preproproteins, the cleavage of staphylolysin requires exogenous enzymes such as P. aeruginosa-derived elastase, lysine-specific protease, alkaline proteininase or, under experimental conditions, trypsin.Citation36 It is not known whether the A. lyticus and Lysobacter spp. β-lytic metalloendopeptidase proproteins are processed using a similar mechanism.
The L. gummosus β-lytic metalloendopeptidase that co-purified with the biofilm-degrading activity was similar to the L. enzymogenes, A. lyticus, and Lysobacter sp. IB-9374 homologs in the mature peptidase domains (84%, 88%, and 88% sequence identity, respectively). However, the predicted preprodomain of the L. gummosus enzyme is more closely related to the corresponding domain of the Lysobacter sp. IB-9374 homolog (74% identity) and shows little resemblance to the corresponding domain of the A. lyticus enzyme (26% identity) (; Fig. S2).
Lysyl endopeptidase
A lysyl endopeptidase that specifically cleaves peptide bonds downstream of lysine residues was originally purified from A. lyticus culture medium and named Achromobacter protease I (API).Citation37 A similar enzyme named endoproteinase Lys-C was purified from L. enzymogenes culture supernatant.Citation38,Citation39 The A. lyticus enzyme is derived from a preproprotein comprising a signal peptide, a prodomain, the peptidase domain and a C-terminal extension domain (Fig. S3).Citation40 A lysyl endopeptidase gene from L. enzymogenes strain ATCC 29487 was found to encode a preproenzyme nearly identical to its A. lyticus counterpart, except for three residues in the prodomain and one in the C-terminal extension (not included in the alignment in Fig. S3).Citation41 The corresponding sequence from L. enzymogenes strain ATCC 27796 has recently been published.Citation39 Lysobacter sp. strain IB-9374 contains two genes encoding different lysyl endopeptidases named LepA and LepB.Citation42 The predicted mature domain of LepA is 100% identical to the A. lyticus lysyl endopeptidase, although there are differences in the C-terminal extension domain, whereas the predicted mature domain of LepB is only 72% identical to the A. lyticus enzyme. Cleavage of the LepB prodomain was shown to involve an autocatalytic mechanism.Citation42 Little is known about the function or cleavage of the C-terminal extension. The naturally-produced lysyl endopeptidases that have been isolated thus far are devoid of the C-terminal extension, which potentially mediates transport through the outer membrane, but apparently does not influence catalytic activity.Citation40-Citation42
In Pseudomonas aeruginosa, a secreted protease named protease IV has been identified which is homologous to the A. lyticus and Lysobacter spp. lysyl endopeptidases (albeit with limited sequence identity) and has similar substrate specificity.Citation43 Protease IV is produced from a precursor comprising a signal peptide, a prodomain and the mature peptidase domain, but lacking a C-terminal extension. The prodomain is likely to function as a folding catalyst and is removed autocatalytically.Citation44
Our analysis revealed the presence of two L. gummosus lysyl endopeptidases (named Lep1 and Lep2) in the biofilm-degrading fractions. In addition, we found a gene encoding a third lysyl endopeptidase homolog (Lep3) in the L. gummosus genome (; Fig. S3). As in the other Lysobacter species, Lep2 and Lep3 possess the C-terminal extension. In contrast, Lep1 lacks this domain making its organization more similar to that of P. aeruginosa protease IV. Remarkably, we also detected peptides matching the C-terminal extension domain of Lep2, in addition to those derived from the mature peptidase domain (Fig. S7).
Hemagglutinin/protease
An enzyme described as hemagglutinin/protease was first recognized in V. cholerae culture supernatants on the basis of its hemagglutinating activity and was subsequently shown to be similar to the P. aeruginosa elastase (LasB), which is an M4 family metallopeptidase.Citation45 Hemagglutination was only observed with certain types of chicken erythrocytes and was dependent on a proteolytic event.Citation46 Depending on its proteolytic activity, hemagglutinin/protease can activate cholera toxin, El Tor cytolysin/hemolysin and can hydrolyze mucin, fibronectin, and lactoferrin. The V. cholerae hemagglutinin/protease is produced as a preproenzyme comprising a signal peptide, an N-terminal prodomain, the protease domain, and a C-terminal extension domain.Citation47 The same domain architecture is found in the closely-related metalloprotease from V. vulnificus.Citation48 The C-terminal domains of both proteins remain covalently connected to the protease domain while it is secreted into the medium, at which point the C-terminal domain is autocatalytically removed. The exact cleavage sites are unknown, but have been tentatively predicted based on the molecular masses of the precursor and mature enzymes.Citation47,Citation49 The V. vulnificus metalloprotease was more active against insoluble protein substrates when it was still connected to the C-terminal extension, whereas the variants with and without this domain showed comparable activity against soluble substrates.Citation47,Citation49
Analysis of the deduced amino acid sequence of the L. gummosus hemagglutinin/protease (the major component of band 1) revealed that the enzyme is produced from a precursor with a domain architecture similar to that of the V. cholerae and V. vulnificus counterparts (Fig. S4). However, the sequence of the L. gummosus C-terminal extension domain differs substantially from the corresponding parts of the Vibrio spp. enzymes, and indeed we found that the L. gummosus hemagglutinin/protease C-terminal domain is remarkably similar to the last 99 residues of the C-terminal domain of the L. gummosus lysyl endopeptidases (Fig. S5).
OmpA-like proteins
Proteins with a conserved C-terminal peptidoglycan-binding domain are widespread among gram-negative bacteria and can be assigned either as members of the OmpA family or the peptidoglycan-associated lipoproteins (PALs), in addition to further less well characterized proteins.Citation50 In both gram-negative and gram-positive bacteria, this structural motif is also part of the MotB protein, which links the rotational machinery of the flagellar motor to the cell wall. OmpA proteins are characterized by an N-terminal eight-stranded β-barrel domain forming an integral component of the outer membrane.Citation51 Thus the complete OmpA molecule physically links the outer membrane to the peptidoglycan layer. In contrast to OmpA proteins, PALs become covalently modified by the addition of a phosphatidylglyceryl moiety.Citation52
The in-gel digestion of band 3 identified peptides derived from two L. gummosus proteins with 92% sequence identity to each other, both possessing the C-terminal peptidoglycan-binding domain, as predicted by sequence comparison (Fig. S6). The N-terminal parts displayed only moderate sequence similarity to known proteins. However, based on the analysis of individual conserved amino acid residues combined with secondary structure predictions, we suggest that the two proteins belong to the OmpA family. The absence of a cysteine-containing sequence motif defining the lipid attachment site (the so-called lipobox) clearly distinguished the two proteins from PALs.Citation52 Remarkably, the two L. gummosus OmpA-like proteins possess an extended linker region of 33 amino acid residues between the putative N-terminal integral membrane domain and the C-terminal peptidoglycan-binding domain. The ~12-kDa polypeptides detected by SDS-PAGE are probably derived by proteolytic cleavage in this region. Accordingly, all peptides derived from band 3 were matched to the C-terminal domain of the OmpA-like proteins (Fig. S7).
Possible interaction of different proteins in biofilm degradation
Among the L. gummosus proteins we identified, the α-lytic protease and the β-lytic metalloendopeptidase appear to play a key role in biofilm degradation reflecting their predicted ability to degrade bacterial cell walls. The lysyl endopeptidases and the hemagglutinin/protease may contribute to biofilm degradation by hydrolyzing proteinaceous components. The C-terminal fragments of the OmpA-like proteins could also participate in biofilm degradation, possibly by mediating the attachment of the peptidases to bacterial surfaces based on their predicted peptidoglycan-binding ability. The assembly of these individual proteins into supramolecular structures could improve their ability to bind bacterial target structures thus allowing the enzymes to operate cooperatively. Indeed, the proteins that co-purified with the biofilm-degrading activity could not be fully separated by chromatography under native conditions. Furthermore, when we calculated the theoretical isoelectric points, we recognized that the α-lytic protease, the β-lytic metalloendopeptidase, lysyl endopeptidase Lep1, and the C-terminal fragments of the OmpA-like proteins are positively charged (pI > 7), whereas lysyl endopeptidase Lep2 and the hemagglutinin/protease homolog are negatively charged (pI < 7) (). Because positively charged molecules were not expected to bind to the anion-exchange columns we used during purification, it is likely these proteins form complexes with an overall negative net charge. As well as forming aggregates by electrostatic attraction, such structures may include outer membrane vesicles, in which the different proteins are enclosed. A previous study showed that one of the two isoforms of α-lytic protease (AlpA) produced by Lysobacter sp. XL1 is released by forming outer membrane vesicles, whereas the other isoform (AlpB) is secreted directly into the medium.Citation53,Citation54 Although the S. epidermidis strain used in our biofilm degradation assay forms a biofilm mainly comprising PNAG, we did not detect L. gummosus enzymes that would be expected to degrade this polymer. Instead, we found that the L. gummosus genome contains genes encoding all enzymes required to synthesize, secrete, and modify PNAG, suggesting that PNAG represents a component of the gummy extracellular material that characterizes this bacterium, but has not yet been chemically analyzed. This finding does not exclude the possibility that L. gummosus may be capable of producing PNAG-degrading enzymes in small amounts, similar to the PNAG-producing bacterium Aggregatibacter actinomycetemcomitans, which uses such an enzyme to allow cells to spread from the extracellular matrix.Citation12 The unambiguous assignment of the biofilm-degrading activity of L. gummosus to individual proteins or combinations of proteins will ultimately depend on their production in heterologous host cells. Work is currently underway to identify appropriate E. coli expression systems that allow the secretion of the correctly processed recombinant proteins into the periplasmic spaced or culture medium.
Materials and Methods
Biofilm degradation assay
S. epidermidis RP62A (ATCC 35984) was cultured in Tryptic Soy Broth (TSB) medium (Fluka, T8907) at 37 °C. Cryopreserved cells were prepared using Roti-Store Cryotubes (Carl Roth, P730.1). A TSB plate containing 1.5% agar–agar was inoculated by streaking the plate with a glass pellet carrying cryopreserved cells. After incubation at 37 °C for 72 h, bacteria scratched from the plate with a 1-mL serological plastic pipette were used to inoculate 25 mL TSB medium in a 50-mL polypropylene conical tube. The tube was incubated at 37 °C for 5 h without shaking and without aeration. We then transferred 1 mL of the bacterial suspension to each well of a 24-well tissue culture plate, which was incubated at 37 °C for 24 h. The medium was aspirated from the wells, and the biofilm that had formed on the bottom was washed with 1 mL water. We added 0.3 mL of the test sample to each well. The plate was then incubated at 28 °C with gentle shaking (50 rpm) for 15 h. The supernatant was aspirated, the wells washed with 1 mL water, and the remaining biofilm stained with 0.4 mL 0.1% crystal violet solution in water for 5 min. The dye solution was removed and the wells were washed with 1 mL water.
Cultivation of Lysobacter spp. and sample preparation
The following Lysobacter strains were cultivated in liquid medium under the conditions recommended by the German Collection of Microorganisms and Cell Cultures (DSMZ): L. antibioticus (DSMZ 2044), L. antibioticus (DSMZ 2045), L. brunescens (DSMZ 6979), L. capsici (DSMZ 23109), L. capsici (DSMZ 19286), L. concretionis (DSMZ 16239), L. daejeonensis (DSMZ 17634), L. defluvii (DSMZ 18482), L. enzymogenes (DSMZ 1895), L. enzymogenes (DSMZ 2043), L. gummosus (DSMZ 6980), L. niabensis (DSMZ 18244), L. niastensis (DSMZ 18481), L. oryzae (DSMZ 21044), L. panaciterrae (DSMZ 17927), Lysobacter sp. (DSMZ 3655), Lysobacter sp. (DSMZ 30821), L. spongiicola (DSMZ 21749), L. ximonensis (DSMZ 23410), and L. yangpyeongensis (DSMZ 17635). With the exception of L. antibioticus (DSMZ 2045), L. brunescens, and L. defluvii, all strains were also cultivated on solid medium comprising 10 g skimmed milk, 1 g yeast extract and 15 g agar-agar in 1 L water. Samples were taken for biofilm activity tests after 3, 7, and 10 days. The liquid cultures were centrifuged and the supernatant was tested. For cultures on solid medium, the grown layer comprising the bacteria and extracellular material was scraped off the culture plates and transferred to a beaker. The material was mixed with water (4 mL per gram wet weight) and stirred at room temperature for 15 min. After centrifugation, the supernatant was tested as above.
Characterization of biofilm-degrading activity by chromatography
L. gummosus was cultured in 20 large petri dishes (145 mm diameter) on skimmed milk medium at 28 °C for 3 days. The harvested bacterial layer (125 g wet weight) was stirred with 500 mL water, resulting in a highly viscous mass, which was comminuted in a blender. After centrifugation (75 000 × g, 30 min, 4 °C), the supernatant was treated with ultrasound until a less-viscous, liquid was obtained suitable for pipetting. Sonication (50% pulse time, 80% amplitude) was carried out in a Rosett Cell (RZ 5, Bandelin) packed in an ice-water bath, using an ultrasonic homogenizer fitted with a GM 2200 generator, UW 2200 ultrasonic convertor, SH 213 G booster horn, and a 13-mm diameter extended probe VS 70 T (Bandelin). The material was centrifuged as above and the pH of the supernatant adjusted to 8.0 by adding tris(hydroxymethyl)aminomethane (Tris) base.
The supernatant was loaded onto an ÄKTAprime plus (GE Healthcare) chromatography system at a flow rate of 5 mL/min on a 50 × 50 mm Q Sepharose Fast Flow (GE Healthcare) column equilibrated with 20 mM Tris-HCl (pH 8.0). The column was washed with 20 mM Tris-HCl (pH 8.0) and eluted with a step gradient of 100, 300, 500, and 1000 mM NaCl in the same buffer. Further chromatography was carried out using a Dionex HPLC system consisting of an ICS-3000 low-pressure mixing quaternary gradient pump, an AXP auxiliary single-piston pump for sample loading, an UltiMate 3000 diode array detector, fluorescence detector and fraction collector. The samples were loaded at a flow rate of 2 mL/min and eluted at 1 mL/min.
The most active fraction (45 mL), which eluted from the Q Sepharose column at the beginning of the 100 mM NaCl step, was diluted with 180 mL 10 mM sodium phosphate buffer (pH 6.0), adjusted to pH 6.0 with HCl, and applied to a Mono S 5/50 GL column (GE Healthcare) equilibrated with 10 mM sodium phosphate buffer (pH 6.0). Samples were eluted with a linear gradient from 0 to 500 mM NaCl in the same buffer in 20 min. The flow-through fraction (250 mL) containing the main activity was diluted with 70 mL 5 mM Tris-HCl (pH 8.0) and 210 mL water, and applied to a Mono Q 5/50 GL column (GE Healthcare) equilibrated with 5 mM Tris-HCl (pH 8.0). Samples were eluted with a linear gradient from 0 to 500 mM NaCl in the same buffer in 20 min. The two most active fractions (0.5 mL each) were combined and concentrated to 0.2 mL by ultrafiltration through a 10-kDa filter, and applied to a Superdex 75 10/30 column (GE Healthcare) equilibrated with 100 mM NaCl, 20 mM Tris-HCl (pH 8.0). Isocratic elution was carried out using the same buffer.
Purification of biofilm-degrading activity
L. gummosus was cultured in 100 petri dishes, and an extract was prepared from the harvested bacterial layer (650 g wet weight) as described above. After adjusting the pH to 8.0 with Tris base, 200 mL of the extract was loaded on a 50 × 150 mm Q Sepharose Fast Flow column equilibrated with 20 mM Tris-HCl (pH 8.0), and eluted with 100 mM NaCl in the same buffer. The chromatography was carried out 12 times, with the column material replaced after every three runs. The active fractions were combined (600 mL). Three 200-mL aliquots were diluted with 1 L water each, and loaded in three consecutive runs at a flow rate of 10 mL/min on a 26 × 100 mm Source 15 Q column equilibrated with 5 mM Tris-HCl (pH 8.0). Samples were eluted with a linear gradient from 0 to 300 mM NaCl in the same buffer at a flow rate of 10 mL/min in 30 min. The main fractions were combined (30 mL), diluted with 100 mL water, re-adjusted to pH 8.0 with Tris base, and loaded at a flow rate of 1 mL/min on a 4 × 250 mm ProPac SAX-10 (Dionex) column equilibrated with 5 mM Tris-HCl (pH 8.0). Samples were eluted with a linear gradient from 0 to 200 mM NaCl in the same buffer in 60 min. Fractions (1 mL each) were analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) using Mini-PROTEAN TGX 4–20% gradient gels (Bio-Rad). Proteins were stained with Flamingo Fluorescent Gel Stain (Bio-Rad) according to the manufacturer’s instructions and visualized with a VersaDoc MP 4000 imaging system (Bio-Rad).
Peptide mass fingerprinting
Peptide mass fingerprinting was carried out by Toplab, Martinsried. Gel slices were washed and destained in several 100-µL aliquots of 50 mM NH4HCO3 containing 30% acetonitrile, and were then submerged in 100 µL of the same buffer. Disulfide bonds were reduced by adding 10 µL 45 mM dithiothreitol followed by incubation at 55 °C for 30 min. Thiol groups were alkylated by adding 10 µL 100 mM iodoacetamide followed by incubation for 15 min at room temperature in the dark. After washing in the buffer described above, the slices were submerged in acetonitrile before the acetonitrile was removed and the slices were allowed to air dry. The slices were rehydrated in 10 mM NH4HCO3 containing ~50 ng porcine trypsin (Serva) and incubated at 37 °C overnight. Liquid samples (~1 mL) were supplemented with 250 µL 8 M urea in 400 mM NH4HCO3 followed by 100 µL 45 mM dithiothreitol and incubation at 55 °C for 30 min. Samples were then digested by adding 2.5 µg trypsin and incubating at 37 °C overnight.
The resulting peptides were adsorbed to a SepPac C18 cartridge (Millipore) and eluted with 80% acetonitrile containing 0.1% trifluoroacetic acid. The volume was reduced to 10 µL under vacuum in a centrifugal evaporator. LC-ESI-MS/MS analysis was performed using an Ettan MDLC nano-LC system (GE Healthcare) coupled to an LTQ Orbitrap mass spectrometer (Thermo Scientific). The peptides were applied in 0.1% formic acid to a 0.3 × 5 mm C18 PepMap 100 trapping column (5 µm particle size, LC Packings) at a flow rate of 6 µL/min, and subsequently separated on a 0.075 × 150 mm C18 PepMap column (3 µm particle size). Samples were eluted in mixtures of buffer A (0.1% formic acid) and B (84% acetonitrile containing 0.1% formic acid). A gradient of 0 to 30% A in 80 min, 30 to 60% B in 30 min, and 100% B for 10 min was applied. The LTQ Orbitrap was operated in parallel mode, with a precursor ion scan in the Orbitrap (300–2000 m/z, 60 000 full-width at half-maximum resolution at m/z 400) and concurrent acquisition of three data-dependent collision-induced dissociation MS/MS scans in the linear ion trap. Mascot software was used to search the acquired MS/MS data against the L. gummosus genome sequence.
DNA isolation and genome sequencing
L. gummosus genomic DNA was isolated using a modified hexadecyltrimethyl ammonium bromide (CTAB) method.Citation55 The CTAB/NaCl solution was prepared by dissolving 4.1 g NaCl in 80 mL water, and 10 g CTAB was stirred in slowly while heating to 65 °C. The solution was filter-sterilized before use. The bacterial layers from eight L. gummosus culture plates (50 g wet weight) were harvested, mixed with 200 mL water and homogenized in a blender. The cells were collected by centrifugation (75 000 × g, 30 min, 4 °C), resuspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and adjusted to an OD600 of 1.0. The suspension (14.8 mL) was transferred to a 50-mL polypropylene conical tube, supplemented with 400 µL lysozyme (100 mg/mL) and incubated at room temperature for 5 min. The mixture was then supplemented with 800 µL 10% SDS and 160 µL proteinase K (10 mg/mL) and incubated at 37 °C for 1 h. We then added 2 mL 5 M NaCl and 2 mL CTAB/NaCl solution, incubated at 65 °C for 10 min, and extracted the preparation with 24:1 (v/v) chloroform:isoamyl alcohol, twice with 25:24:1 phenol:chloroform:isoamyl alcohol and once more with 24:1 chloroform:isoamyl alcohol. The DNA in the aqueous phase was precipitated with 0.6 volumes of isopropanol at room temperature for 30 min, washed with 70% ethanol and dissolved in 100 µL TE buffer containing 0.1 mg RNase A. Genome sequencing was carried out by Eurofins MWG, Ebersberg, who prepared a non-cloned shotgun library and an 8-kbp paired-end library according to standard protocols (Roche). The libraries were sequenced using GS FLX Titanium-series chemistry. Gap closure by PCR and Sanger sequencing resulted in a 5 901 415-bp single contig with a G/C content of 66.7%. Coding sequences were predicted using GeneMarkS software and annotated by homology searching against the Swiss-Prot and TrEMBL databases using NCBI BLAST 2.2.25+.
Additional material
Download Zip (184.3 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We wish to thank Rüdiger Lehmann for help with the bioinformatics analysis and Wilma Ziebuhr for helpful discussion and for providing the S. epidermidis strain. The work was supported by the German federal state of Hessen as part of the LOEWE-Schwerpunkt Insektenbiotechnologie.
References
- Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med 2013; 3:a010306; http://dx.doi.org/10.1101/cshperspect.a010306; PMID: 23545571
- Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 2012; 33:5967 - 82; http://dx.doi.org/10.1016/j.biomaterials.2012.05.031; PMID: 22695065
- Cooper RA. Inhibition of biofilms by glucose oxidase, lactoperoxidase and guaiacol: the active antibacterial component in an enzyme alginogel. Int Wound J 2013; Forthcoming http://dx.doi.org/10.1111/iwj.12083; PMID: 23672196
- Regina VR, Søhoel H, Lokanathan AR, Bischoff C, Kingshott P, Revsbech NP, Meyer RL. Entrapment of subtilisin in ceramic sol-gel coating for antifouling applications. ACS Appl Mater Interfaces 2012; 4:5915 - 21; http://dx.doi.org/10.1021/am301554m; PMID: 23020255
- Chaignon P, Sadovskaya I, Ragunah Ch, Ramasubbu N, Kaplan JB, Jabbouri S. Susceptibility of staphylococcal biofilms to enzymatic treatments depends on their chemical composition. Appl Microbiol Biotechnol 2007; 75:125 - 32; http://dx.doi.org/10.1007/s00253-006-0790-y; PMID: 17221196
- Berg CH, Kalfas S, Malmsten M, Arnebrant T. Proteolytic degradation of oral biofilms in vitro and in vivo: potential of proteases originating from Euphausia superba for plaque control. Eur J Oral Sci 2001; 109:316 - 24; http://dx.doi.org/10.1034/j.1600-0722.2001.00099.x; PMID: 11695752
- Cordeiro AL, Hippius C, Werner C. Immobilized enzymes affect biofilm formation. Biotechnol Lett 2011; 33:1897 - 904; http://dx.doi.org/10.1007/s10529-011-0643-3; PMID: 21618024
- Sass P, Bierbaum G. Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus.. Appl Environ Microbiol 2007; 73:347 - 52; http://dx.doi.org/10.1128/AEM.01616-06; PMID: 17085695
- Domenech M, García E, Moscoso M. In vitro destruction of Streptococcus pneumoniae biofilms with bacterial and phage peptidoglycan hydrolases. Antimicrob Agents Chemother 2011; 55:4144 - 8; http://dx.doi.org/10.1128/AAC.00492-11; PMID: 21746941
- Wu JA, Kusuma C, Mond JJ, Kokai-Kun JF. Lysostaphin disrupts Staphylococcus aureus and Staphylococcus epidermidis biofilms on artificial surfaces. Antimicrob Agents Chemother 2003; 47:3407 - 14; http://dx.doi.org/10.1128/AAC.47.11.3407-3414.2003; PMID: 14576095
- Izano EA, Amarante MA, Kher WB, Kaplan JB. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol 2008; 74:470 - 6; http://dx.doi.org/10.1128/AEM.02073-07; PMID: 18039822
- Kaplan JB, Ragunath C, Ramasubbu N, Fine DH. Detachment of Actinobacillus actinomycetemcomitans biofilm cells by an endogenous beta-hexosaminidase activity. J Bacteriol 2003; 185:4693 - 8; http://dx.doi.org/10.1128/JB.185.16.4693-4698.2003; PMID: 12896987
- Gökçen A, Vilcinskas A, Wiesner J. Methods to identify enzymes that degrade the main extracellular polysaccharide component of Staphylococcus epidermidis biofilms. Virulence 2013; 4:260 - 70; http://dx.doi.org/10.4161/viru.23560; PMID: 23357872
- Schommer NN, Christner M, Hentschke M, Ruckdeschel K, Aepfelbacher M, Rohde H. Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A.1. Infect Immun 2011; 79:2267 - 76; http://dx.doi.org/10.1128/IAI.01142-10; PMID: 21402760
- Pavlukhina SV, Kaplan JB, Xu L, Chang W, Yu X, Madhyastha S, Yakandawala N, Mentbayeva A, Khan B, Sukhishvili SA. Noneluting enzymatic antibiofilm coatings. ACS Appl Mater Interfaces 2012; 4:4708 - 16; http://dx.doi.org/10.1021/am3010847; PMID: 22909396
- Christensen P, Cook FD. Lysobacter, a new genus of nonfruiting, gliding bacteria with a high base ratio. Int J Syst Bacteriol 1978; 28:367 - 93; http://dx.doi.org/10.1099/00207713-28-3-367
- Reichenbach H. The genus Lysobacter, p 939–957. In Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (ed), Prokaryotes 2006; Springer, New York.
- Hayward AC, Fegan N, Fegan M, Stirling GR. Stenotrophomonas and Lysobacter: ubiquitous plant-associated gamma-proteobacteria of developing significance in applied microbiology. J Appl Microbiol 2010; 108:756 - 70; http://dx.doi.org/10.1111/j.1365-2672.2009.04471.x; PMID: 19702860
- Schoenfelder SM, Lange C, Eckart M, Hennig S, Kozytska S, Ziebuhr W. Success through diversity - how Staphylococcus epidermidis establishes as a nosocomial pathogen. Int J Med Microbiol 2010; 300:380 - 6; http://dx.doi.org/10.1016/j.ijmm.2010.04.011; PMID: 20451447
- Brucker RM, Baylor CM, Walters RL, Lauer A, Harris RN, Minbiole KP. The identification of 2,4-diacetylphloroglucinol as an antifungal metabolite produced by cutaneous bacteria of the salamander Plethodon cinereus.. J Chem Ecol 2008; 34:39 - 43; http://dx.doi.org/10.1007/s10886-007-9352-8; PMID: 18058176
- Riggio MP, Dempsey KE, Lennon A, Allan D, Ramage G, Bagg J. Molecular detection of transcriptionally active bacteria from failed prosthetic hip joints removed during revision arthroplasty. Eur J Clin Microbiol Infect Dis 2010; 29:823 - 34; http://dx.doi.org/10.1007/s10096-010-0934-y; PMID: 20449620
- Whitaker DR. Lytic enzymes of Sorangium sp. Isolation and enzymatic properties of the alpha- and beta-lytic proteases. Can J Biochem 1965; 43:1935 - 54; http://dx.doi.org/10.1139/o65-217; PMID: 5326246
- Olson MO, Nagabhushan N, Dzwiniel M, Smillie LB, Whitaker DR. Priaary structure of alpha-lytic protease: a bacterial homologue of the pancreatic serine proteases. Nature 1970; 228:438 - 42; http://dx.doi.org/10.1038/228438a0; PMID: 5482494
- Silen JL, McGrath CN, Smith KR, Agard DA. Molecular analysis of the gene encoding alpha-lytic protease: evidence for a preproenzyme. Gene 1988; 69:237 - 44; http://dx.doi.org/10.1016/0378-1119(88)90434-9; PMID: 3234766
- Silen JL, Frank D, Fujishige A, Bone R, Agard DA. Analysis of prepro-alpha-lytic protease expression in Escherichia coli reveals that the pro region is required for activity. J Bacteriol 1989; 171:1320 - 5; PMID: 2646278
- Fujishige A, Smith KR, Silen JL, Agard DA. Correct folding of alpha-lytic protease is required for its extracellular secretion from Escherichia coli.. J Cell Biol 1992; 118:33 - 42; http://dx.doi.org/10.1083/jcb.118.1.33; PMID: 1618906
- Cunningham EL, Agard DA. Disabling the folding catalyst is the last critical step in alpha-lytic protease folding. Protein Sci 2004; 13:325 - 31; http://dx.doi.org/10.1110/ps.03389704; PMID: 14739318
- Muranova TA, Krasovskaya LA, Tsfasman IM, Stepnaya OA, Kulaev IS. Structural investigations and identification of the extracellular bacteriolytic endopeptidase L1 from Lysobacter sp. XL1. Biochemistry (Mosc) 2004; 69:501 - 5; http://dx.doi.org/10.1023/B:BIRY.0000029847.40511.26; PMID: 15193123
- Lapteva YS, Zolova OE, Shlyapnikov MG, Tsfasman IM, Muranova TA, Stepnaya OA, Kulaev IS, Granovsky IE. Cloning and expression analysis of genes encoding lytic endopeptidases L1 and L5 from Lysobacter sp. strain XL1. Appl Environ Microbiol 2012; 78:7082 - 9; http://dx.doi.org/10.1128/AEM.01621-12; PMID: 22865082
- Li S, Norioka S, Sakiyama F. Purification, staphylolytic activity, and cleavage sites of alpha-lytic protease from Achromobacter lyticus.. J Biochem 1997; 122:772 - 8; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a021822; PMID: 9399581
- Damaglou AP, Allen LC, Whitaker DR. Beta-lytic protease – Myxobacter 495, p 198. In Dayhoff MO (ed), Atlas of protein sequence and structure, vol 5. National Biomedical Research Foundation 1976; Washington DC.
- Li SL, Norioka S, Sakiyama F. Molecular cloning and nucleotide sequence of the beta-lytic protease gene from Achromobacter lyticus.. J Bacteriol 1990; 172:6506 - 11; PMID: 2228973
- Ahmed K, Chohnan S, Ohashi H, Hirata T, Masaki T, Sakiyama F. Purification, bacteriolytic activity, and specificity of beta-lytic protease from Lysobacter sp. IB-9374. J Biosci Bioeng 2003; 95:27 - 34; PMID: 16233362
- Oza NB. Beta-lytic protease, a neutral sorangiopeptidase. Int J Pept Protein Res 1973; 5:365 - 9; http://dx.doi.org/10.1111/j.1399-3011.1973.tb02341.x; PMID: 4782638
- Li S, Norioka S, Sakiyama F. Bacteriolytic activity and specificity of Achromobacter beta-lytic protease. J Biochem 1998; 124:332 - 9; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a022116; PMID: 9685723
- Kessler E, Safrin M, Gustin JK, Ohman DE. Elastase and the LasA protease of Pseudomonas aeruginosa are secreted with their propeptides. J Biol Chem 1998; 273:30225 - 31; http://dx.doi.org/10.1074/jbc.273.46.30225; PMID: 9804780
- Masaki T, Tanabe M, Nakamura K, Soejima M. Studies on a new proteolytic enzyme from A chromobacter lyticus M497-1. I. Purification and some enzymatic properties. Biochim Biophys Acta 1981; 660:44 - 50; http://dx.doi.org/10.1016/0005-2744(81)90106-6; PMID: 6791693
- Sakiyama F, Masaki T. Lysyl endopeptidase of Achromobacter lyticus.. Methods Enzymol 1994; 244:126 - 37; http://dx.doi.org/10.1016/0076-6879(94)44011-5; PMID: 7845202
- Kuhlman PA, Chen R, Alcantara J, Szarka S. Rapid purification of Lys-C from Lysobacter enzymogenes cultures: A sequential chromatography technique. BioProcess Int 2009; 7:28 - 38
- Ohara T, Makino K, Shinagawa H, Nakata A, Norioka S, Sakiyama F. Cloning, nucleotide sequence, and expression of Achromobacter protease I gene. J Biol Chem 1989; 264:20625 - 31; PMID: 2684982
- Chohnan S, Nonaka J, Teramoto K, Taniguchi K, Kameda Y, Tamura H, Kurusu Y, Norioka S, Masaki T, Sakiyama F. Lysobacter strain with high lysyl endopeptidase production. FEMS Microbiol Lett 2002; 213:13 - 20; http://dx.doi.org/10.1111/j.1574-6968.2002.tb11279.x; PMID: 12127482
- Chohnan S, Shiraki K, Yokota K, Ohshima M, Kuroiwa N, Ahmed K, Masaki T, Sakiyama F. A second lysine-specific serine protease from Lysobacter sp. strain IB-9374. J Bacteriol 2004; 186:5093 - 100; http://dx.doi.org/10.1128/JB.186.15.5093-5100.2004; PMID: 15262946
- Elliott BW Jr., Cohen C. Isolation and characterization of a lysine-specific protease from Pseudomonas aeruginosa.. J Biol Chem 1986; 261:11259 - 65; PMID: 3090046
- Traidej M, Marquart ME, Caballero AR, Thibodeaux BA, O’Callaghan RJ. Identification of the active site residues of Pseudomonas aeruginosa protease IV. Importance of enzyme activity in autoprocessing and activation. J Biol Chem 2003; 278:2549 - 53; http://dx.doi.org/10.1074/jbc.M208973200; PMID: 12419815
- Lutfullah G, Amin F, Khan Z, Azhar N, Azim MK, Noor S, Shoukat K. Homology modeling of hemagglutinin/protease [HA/P (vibriolysin)] from Vibrio cholerae: sequence comparision, residue interactions and molecular mechanism. Protein J 2008; 27:105 - 14; http://dx.doi.org/10.1007/s10930-007-9113-0; PMID: 18074211
- Booth BA, Boesman-Finkelstein M, Finkelstein RA. Vibrio cholerae soluble hemagglutinin/protease is a metalloenzyme. Infect Immun 1983; 42:639 - 44; PMID: 6417020
- Häse CC, Finkelstein RA. Cloning and nucleotide sequence of the Vibrio cholerae hemagglutinin/protease (HA/protease) gene and construction of an HA/protease-negative strain. J Bacteriol 1991; 173:3311 - 7; PMID: 2045361
- Miyoshi S, Wakae H, Tomochika K, Shinoda S. Functional domains of a zinc metalloprotease from Vibrio vulnificus.. J Bacteriol 1997; 179:7606 - 9; PMID: 9393733
- Chang AK, Park JW, Lee EH, Lee JS. The N-terminal propeptide of Vibrio vulnificus extracellular metalloprotease is both an inhibitor of and a substrate for the enzyme. J Bacteriol 2007; 189:6832 - 8; http://dx.doi.org/10.1128/JB.00396-07; PMID: 17644589
- Parsons LM, Lin F, Orban J. Peptidoglycan recognition by Pal, an outer membrane lipoprotein. Biochemistry 2006; 45:2122 - 8; http://dx.doi.org/10.1021/bi052227i; PMID: 16475801
- Pautsch A, Schulz GE. Structure of the outer membrane protein A transmembrane domain. Nat Struct Biol 1998; 5:1013 - 7; http://dx.doi.org/10.1038/2983; PMID: 9808047
- Kovacs-Simon A, Titball RW, Michell SL. Lipoproteins of bacterial pathogens. Infect Immun 2011; 79:548 - 61; http://dx.doi.org/10.1128/IAI.00682-10; PMID: 20974828
- Vasilyeva NV, Tsfasman IM, Suzina NE, Stepnaya OA, Kulaev IS. Secretion of bacteriolytic endopeptidase L5 of Lysobacter sp. XL1 into the medium by means of outer membrane vesicles. FEBS J 2008; 275:3827 - 35; http://dx.doi.org/10.1111/j.1742-4658.2008.06530.x; PMID: 18573103
- Vasilyeva NV, Tsfasman IM, Kudryakova IV, Suzina NE, Shishkova NA, Kulaev IS, Stepnaya OA. The role of membrane vesicles in secretion of Lysobacter sp. bacteriolytic enzymes. J Mol Microbiol Biotechnol 2013; 23:142 - 51; http://dx.doi.org/10.1159/000346550; PMID: 23615202
- Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol 2001; Chapter 2:Unit 2.4. doi: http://dx.doi.org/10.1002/0471142727.mb0204s56.