
Abstract
An estimated 30–40% of patients with chronic hepatitis C have elevated serum iron, transferrin saturation, and ferritin levels. Clinical data suggest that iron is a co-morbidity factor for disease progression following HCV infection. Iron is essential for a number of fundamental metabolic processes in cells and organisms. Mammalian iron homeostasis is tightly regulated and this is maintained through the coordinated action of sensory and regulatory networks that modulate the expression of iron-related proteins at the transcriptional and/or posttranscriptional levels. Disturbances of iron homeostasis have been implicated in infectious disease pathogenesis. Viruses, similarly to other pathogens, can escape recognition by the immune system, but they need iron from their host to grow and spread. Hepcidin is a 25-aa peptide, present in human serum and urine and represents the key peptide hormone, which modulates iron homeostasis in the body. It is synthesized predominantly by hepatocytes and its mature form is released in circulation. In this review, we discuss recent advances in the exciting crosstalk of molecular mechanisms and cell signaling pathways by which iron and hepcidin production influences HCV-induced liver disease.
Introduction
Iron is essential for several substantial metabolic processes in cells and organisms. It is incorporated in heme, proteins with iron–sulfur clusters and enzymes that participate in DNA synthesis and mitochondrial respiration.Citation1 However, iron’s reaction with oxygen may lead to the generation of reactive oxygen species (ROS) and ultimately induce cellular toxicity and damage. Therefore, mammalian iron homeostasis is tightly regulated and maintained through the coordinated action of metabolic and signaling pathways that alter the expression of iron-related proteins at the transcriptional and/or posttranscriptional levels. There are mainly three regulatory systems: (1) the hepcidin/ferroportin axis, which acts systemically, (2) the IRP/IRE system that acts on cellular iron, and (3) the hypoxia inducible factors (HIF) factors that regulate several genes of iron homeostasis.Citation1
It was previously demonstrated in animal models that a host’s iron status can affect infection, with corresponding evidence in humans. Disturbances of iron homeostasis have been implicated in infectious disease pathogenesis. Viruses, similarly to other pathogens, can escape from immune recognition, but they require iron to grow and spread. If iron availability is high, infection can progress efficiently.Citation2 Deflecting iron away from invading pathogens retards their growth, providing a chance to immune mechanisms to clear the infection.
Approximately 65% of body iron is incorporated in heme, which is found in hemoglobin, myoglobin, and respiratory enzymes (cytochromes, oxidases, peroxidases, etc.). Non-hemin iron of the body (35%) is stored in a complex with ferritin, which is the major cytoplasmic iron-storage protein, mostly in hepatocytes and splenic and liver macrophages.Citation3 A minor fraction of iron exists in association with plasma protein transferrin (Tf), which transfers iron in cells and another one in proteins with iron–sulfur clusters. Erythropoiesis represents the largest physiological requirement for iron, where 90% of erythropoietic need is provided by iron recycling of senescent red blood cells through macrophages. It is known that iron excretion is not a regulated process.Citation3 A small amount of iron is lost by bleeding, skin desquamation, sweating, or urinary excretion. These losses are replenished by dietary iron absorption, so that the body maintains non-toxic amounts in stores.
Hepcidin
Hepcidin is a 25-aa cystein-rich peptide, found in human serum and urine, and represents the key peptide hormone, which modulates iron homeostasis in the body. The human hepcidin gene (Hamp) consists of three exons that encode an 84-aa preprohepcidin peptide that is cleaved at a characteristic furin cleavage site to produce the mature 25-aa hepcidin peptide.Citation4,Citation5 It is mainly produced by the liver and upon maturation, released in circulation.Citation6 However, hepcidin expression has also been recorded at different levels in monocytes, macrophages, adipocytes, and brain cells. Hepcidin is a type II acute phase proteinCitation7 whose upregulation systemically suspends iron absorption from intestinal enterocytes, as well as iron recycling from macrophages.Citation8,Citation9 Iron and inflammation elevate hepcidin expression, while anemia and hypoxia diminish it.Citation10 The interplay of these pathways and the relative prevalence of one upon another controls the expression of hepcidin.
Hepatitis C
Hepatitis C has emerged as a global health problem suffered by 170 million persons, which represents approximately 3% of the human population worldwide.Citation11 HCV is a positive-stranded RNA virus of the Flaviviridae family. Other known flaviviruses are the yellow fever and the dengue viruses. The HCV 9.6-kb genome contains 5′ and 3′ untranslated regions (UTRs) flanking a long open reading frame (ORF) that is translated via a highly structured mRNA stem-loop sequence, the internal ribosome entry site (IRES).Citation12 The resulting polyprotein is processed to yield structural (core, E1, and E2) and non-structural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins.Citation13 Abnormal iron levels have been recorded in about 50% of chronic hepatitis C (CHC)-infected patients.Citation14
In this review, we will focus on hepcidin and its role as a key player in iron homeostasis, outline the molecular mechanisms implicated in HCV-mediated hepcidin regulation and delineate the impact of iron on the pathogenesis and treatment of HCV infection.
Systemic and Cellular Iron Homeostasis
Dietary iron may be encountered either as heme or non-heme iron. Heme ferrous iron (Fe2+) is acquired through meat ingestion in the form of hemoglobin or myoglobin.Citation15 Heme is absorbed through heme carrier proteins into the enterocytes, where iron is released by heme oxygenase-1. The duodenal cytochrome b (DCYTB) enzyme reduces non-heme ferric iron (Fe3+). Subsequently, divalent metal transporter 1 (DMT1, also known as DCT1 or Nramp2) transports Fe2+ across the endosomal membrane of enterocytes into the cytoplasm. Within enterocytes, iron is stored in ferritin or, alternatively, exported into circulation via FPN1. Hephaestin oxidizes ferrous iron to Fe3+, which is then bound to transferrin (Tf) for transport within the plasma.Citation15,Citation16 Circulating Tf is subsequently endocytosed by transferring receptor 1 (TfR1) into endosomes through clathrin-coated pits, thereby providing iron to body cells ().
Figure 1. Hepcidin is primarily synthesized in hepatocytes and then released into the circulation. When hepcidin reaches circulation, it regulates iron metabolism by controlling iron transport to duodenal enterocytes and iron export from macrophages.
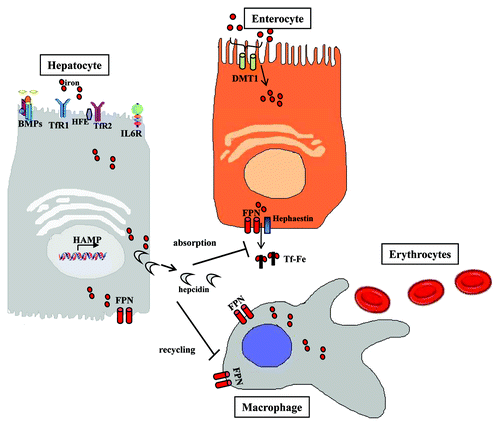
Systemic iron flux in the body is controlled by the liver secreted peptide hormone hepcidin.Citation1 Hepatic hepcidin binds to ferroportin (FPN1, also known as Ireg1),Citation17-Citation19 the only known cell iron exporter in the surface of enterocytes, hepatocytes, and macrophagesCitation18 (). It induces its ubiquitination, subsequent internalization into the cytoplasm and degradation,Citation9 thus preventing iron release from the cells. FPN1 expression is controlled by hepcidin and by the transcription factor nuclear factor erythroid 2-like 2 (NRF2).Citation20 Furthermore, hepcidin in enterocytes mainly inhibits DMT1 transcription, thus blocking apical intestinal iron absorption.Citation21,Citation22 Elevated iron stores or Tf-Fe3+ levels upregulate hepcidin expression through the BMP/HJV/SMAD and HFE/TfR1/TfR2 signaling pathways, respectively.Citation23,Citation24 During inflammation, hepcidin is also upregulated by proinflammatory cytokines, such as IL-6, IL-1, IL-22, and others, via the JAK-STAT signaling pathway. Hepcidin expression is reduced by erythropoiesis, hypoxia, and ROS via modulation of CCAAT enhancer binding protein α (C/EBPα) expression.Citation25 Recently, it has been demonstrated that hepcidin expression is regulated positively or negatively by microRNAs (miRNA).Citation26,Citation27
Monocyte and macrophage hepcidin is likely to be more important on a local rather than systemic level, contributing to host defense and may modulate inflammatory processes.Citation28
Iron is crucial for life. Because of its highly reactive nature that may cause cell damage through ROS formation, the whole homeostasis network must be tightly controlled so that iron toxicity is kept to a minimum.Citation29 This tricky task is mainly accomplished posttranscriptionally by the iron-regulatory protein (IRP)/iron-responsive element (IRE) regulatory network. IRP1 and IRP2 are cytoplasmic proteins, which belong to the aconitase superfamily. They adjust intracellular iron homeostasis by binding specifically and with high affinity to conserved IREs in mRNA UTRs.Citation30,Citation31 Depending on the location of the IREs on the 5′- or 3′-UTR, IRP binding regulates gene expression differentially. Intracellular iron levels are essentially modulated through coordinated alterations in ferritin and TfR1. In a state of iron depletion, IRPs bind to IREs and stabilize TfR1 mRNA (IRE in 3′-UTR), while decreasing translation of ferritin mRNA (IRE in 5′-UTR). Eventually, the uptake and availability of iron within the cell is increased. Equally, elevated iron stores lessen IRE binding activity, thereby improving ferritin translation, reducing stability of TfR1 mRNA and eventually favoring iron sequestration over uptake. IRP2, which is homologous to IRP1 is regulated in response to stimuli other than iron, since it lacks aconitase activity. These stimuli include hypoxia and oxidative stress characterized by increased ROS production or reactive nitrogen intermediates. Apart from adjusting ferritin and TfR1 levels, IRPs can also affect the stability of mRNAs of other proteins involved in iron acquisition, such as DMT1, Alas2, Aco2, and FPN1.Citation32
Approximately one-third of the body’s total iron is stored in hepatocytes and liver macrophages. Conglomeration of iron produces oxidative stress resulting in increased lipid peroxidation. This induces the destruction of organelle membranes and, as a result, cell death via hepatocyte necrosis or/and apoptosis. Products of oxidative stress cause the release of profibrogenic cytokines and promote an inflammatory reaction that acts as a stimulus for liver macrophages. Those mechanisms activate hepatic stellate cells (HSC)—sources of collagen and other extracellular matrix elements.Citation33,Citation34 On the contrary, when hepatoma cells were directly exposed to hypoxia, hepcidin production was reduced, implying that low oxygen intracellularly is linked to hepcidin regulation.Citation35,Citation36
Increased hepatic iron stores are associated with a higher risk of hepatocellular carcinoma (HCC) development, failure of antiviral treatment, and significantly worse clinical outcomes in patients suffering from CHC.Citation37-Citation40 Hereditary defects in iron homeostasis-related host genes have been deemed responsible for iron overload; nevertheless, it may be the product of viral propagation, in which diminished hepcidin expression may play a key role.Citation41
In summary, the three major molecular pathways implicated in the regulation of hepcidin are: JAK/STAT, BMP/SMAD, and HFE/TfR2 followed by ERK1/2. Formation of the hepcidin-ferroportin complex triggers phosphorylation, internalization and degradation of ferroportin through activation of JAK2 kinase. This, in turn, signals the phosphorylation of STAT3 and initiates changes in the expression of STAT3-responsive genes. Among the latter, SOCS3 reduces IL-6 and TNFα expression, thus diminishing hepcidin production further and completing a negative feedback loop.Citation42 Alternatively, hepcidin may be regulated through the bone morphogenetic protein/SMAD (BMP/SMAD) pathway. The TGF-β superfamily members BMPs bind to and activate their respective receptors, thereby promoting phosphorylation of the regulatory SMADS (R-SMADs), which heterodimerize with SMAD4. These complexes then translocates into the nucleus, where they increase Hamp gene transcription.Citation43 Finally, induction of phosphorylated ERK1/2 seems to be linked to TfR2 and to its binding to holotransferrin. Interestingly, phosphorylated ERK1/2 have been shown to regulate furin mRNA and protein expression and ultimately, through furin, MAPKs may control hepcidin expression.Citation44
HCV Life Cycle is Affected by Iron
It has been demonstrated that about 30–40% of CHC patients have elevated serum iron and ferritin, as well as transferrin saturation.Citation45-Citation47 A correlation between increased cellular and systemic iron levels and HCV patients with poor prognosis and severe deterioration in liver metabolic processes has been previously reported.Citation46-Citation48 To date, HCV is known to activate several different signaling pathways that lead to ROS production and/or ER stress, both in hepatocytes and liver-residing macrophages.Citation49
The precise molecular mechanisms by which iron may influence HCV-induced liver disease are still obscure and hepcidin expression in HCV infection remains controversial.
Iron and HCV IRES-dependent translation
Successful production of HCV viral proteins depends on the host cell protein machinery. HCV expression is predominantly regulated translationally, through the interaction of cellular translation initiation factors and the HCV IRES within the 5′-untranslated region (5′-UTR)Citation50. It has been proposed that increased intracellular levels of iron may be associated with enhanced HCV IRES-dependent translation. Cellular factors involved in promoting HCV translation, bind specifically to HCV RNA in an iron-dependent manner. Notably, these factors also interact with the IRE.Citation51
Studies have shown that the translation initiation factor 3 (eIF3) is absolutely required for HCV RNA translation initiation.Citation50,Citation52-Citation54 HCV genotypes respond differently to iron perturbations and this may be a matter of iron-dependent control of eIF3. For example, HCV-1b-infected patients have higher hepatic iron concentrations than patients infected with HCV-2a or -2b.Citation55 Therefore, one could hypothesize that HCV-1b translation is considerably influenced by iron in vivo. In these patients, responsiveness to treatment was accompanied by significantly lower hepatic iron content than that in non-responders, whereas this was not true for HCV-2a or 2b-infected patients.Citation55 Interestingly, it was also reported that HCV mRNA levels were positively correlated with intrahepatic eIF3 mRNA levels. Thus, iron-mediated eIF3 activation may increase HCV translation, thereby inducing HCV propagation through infection progression. Moreover, Theurl and colleagues showed that in HCV-1b, iron-dependent translation was accomplished via eIF3 induction, whereas HCV-6a remained iron-unresponsive. This group proposed that iron-mediated regulation of HCV translation may require high binding affinity of eIF3 to its corresponding binding site on the HCV IRES. This may not be the case for all HCV genotypes, which are known to exhibit sequence alterations in this part of the IRES.Citation53
Inhibition of iron-induced eIFs found in HCV patients with iron overloadCitation56 using small peptides and chemical compounds, might potentially be an exceptionally good candidate therapeutic target. To this end, a recent study proposed that the efficiency of IFN-α against HCV multiplication may correspond, partly, to its ability to modulate the expression of vital HCV translation factors.Citation55
Finally, La protein was one of the first host elements identified to interact with the IRES and stimulate HCV IRES-mediated translation. Together with eIF3, La protein was significantly elevated in iron-treated cells and this increase was partly inhibited by iron chelator deferoxamine (DFO).Citation56 Taken together, the above discussed data suggest that iron mediates the recruitment of specific factors onto HCV IRES, and, as a result, regulates viral translation.
Iron and HCV replication
Contradictory data exist as to whether iron exerts promoting or inhibitory effects on HCV replication.Citation51,Citation53,Citation57-Citation59 Clinical data suggested that iron is a co-morbidity factor for disease progression following HCV infection.Citation46 Increased iron can favor HCV replication, but it is more likely to promote disease progression by enhancing oxidative stress, thereby leading to chronic inflammation. In CHC, mutations in the hemochromatosis protein (HFE) gene have been linked to hepatic iron overload,Citation60 since numerous studies have described an association between HFE genotypes and iron overload in CHC patients.Citation46,Citation61 The direct effect of HCV on hepcidin synthesis may constitute an alternative mechanism.Citation62
Transgenic mice expressing the HCV polyprotein have been shown to have reduced hepatic expression of hepcidin as a result of HCV-induced oxidative stress.Citation62 Nishina and colleagues performed reporter gene experiments to identify which hepcidin promoter region was affected by HCV proteins. They concluded that the reduced hepcidin transcriptional activity in the HCV full-length open reading frame (FL-N/35 lineage) transgenic mouse was due to decreased C/EBPα DNA binding activity, which has been previously shown to regulate hepcidin transcription. Furthermore, this was associated with increased expression of a C/EBP repressor called C/EBP homology protein (CHOP). It was proposed that HCV-induced iron loading mediated an increase in ROS, which resulted in CHOP upregulation and prevention of C/EBPα binding to the hepcidin promoter. This, in turn, upregulated ferroportin expression, increased iron export from the duodenum and macrophages, raised serum iron and transferrin saturation, and eventually resulted in elevated hepatic iron levels. However, the animal models disregarded the role of inflammation, which is a potent hepcidin inducer.
On the other hand, hepcidin mRNA levels were significantly higher in the liver of the core gene transgenic mice, as opposed to control mice.Citation63 These mice have been known to develop HCC after a certain period with increased oxidative stress and steatosis. Indeed, in this study, deregulation of hepcidin resulted in hepatic iron overload, which ultimately deteriorated oxidative stress, as expected.Citation39,Citation63,Citation64
HCV-mediated inhibition of hepcidin was demonstrated in several in vitro studies.Citation39,Citation62,Citation65,Citation66 Again, these were criticized for not taking into consideration the effects of inflammation, which may counteract ROS-induced hepcidin repression in CHC patients, via the well-studied upregulation of hepcidin by proinflammatory cytokines, mainly IL-6.Citation67,Citation68 Miura and colleaguesCitation65 reported evidence that HCV-induced ROS downregulated hepcidin expression in Huh7 cells, while it was restored by the use of anti-oxidants in HCV replicon cells. Furthermore, it was shown that HCV-induced ROS could activate histone deacetylases (HDACs) with subsequent modulation of Hamp gene transcription. Inversely, suspension of HDAC activity restored histone acetylation, resulting in enhanced hepcidin expression, in full-length genomic replicon cells.
On the other hand, Miyachi and colleaguesCitation69 reported that hepcidin expression was surprisingly augmented in replicon cells, as compared with cured cells. Cytoplasmic STAT3 expression was increased in both cell lines as a result of IL-6 stimulation, with higher levels observed in the cured cells. In contrast, the phosphorylated form of the protein was more abundant in the nucleus of the replicon cells, thereby suggesting that JAK/STAT3 activation was more pronounced in this system. Interestingly, inhibition of SOCS-1 and SOCS-3 gene expression upregulated STAT3 activation and hepcidin expression. Hepcidin returned to background levels following SOCS-1 overexpression, most likely due to suppression of STAT3 activation.Citation69
In addition, having established that iron suppresses subgenomic HCV replication in replicon cells by binding to and silencing the HCV RNA-dependent RNA polymerase, NS5B, Fillebeen and colleaguesCitation70 speculated that the virus may manipulate the cell into reducing its iron stores, as part of its replicating strategy, during the early stages of infection. Alternatively, iron-deficient cells may be more receptive into hosting subgenomic HCV replication. According to these hypotheses, maintaining a “physiological” iron poolCitation7 may depend on many variables, including the particular stage of systemic and/or hepatic inflammatory disease.
Cell signaling pathways involved in hepcidin modulation and affected by HCV infection
HCV proteins are well known to interact with host cellular factors implicated in cell signaling pathways, cell cycle regulation, transcriptional regulation, cell proliferation, inflammation, liver fibrosis, and apoptosis.Citation71 Functional screening of HCV proteins revealed that almost all of them, structural and non-structural, are modulators of the cellular signaling pathways (STAT3, MAPKs, and BMPs-SMADs) that are implicated in hepcidin regulation (see ), making the interplay between these pathways more complex.
Figure 2. Schematic illustration of the crosstalk between the three main cell signaling pathways involved in the regulation of hepcidin and their modulation by HCV structural and non-structural proteins.
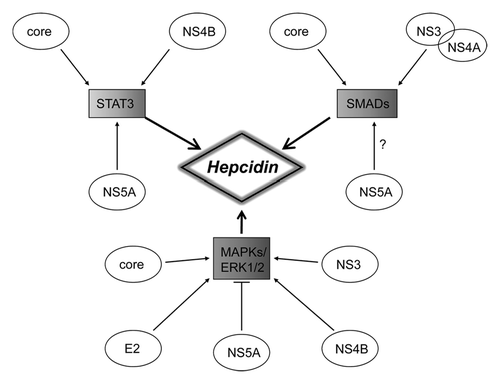
Core protein
Fukutomi and colleagues showed with microarray analysis that in HCV core-expressing Huh7 cells, 372 out of 12 500 known human genes were differentially regulated, with most of those involved in cell growth or oncogenic signaling.Citation72 Studies from our laboratory (Foka P, Dimitriadis A, et al., CMLS accepted) and others have implicated HCV core protein in the upregulation of IL-6Citation73,Citation74 and the subsequent transactivation of STAT3 through phosphorylation. In addition, it has been shown that in HCV core transgenic mice and HCV core transfected cell lines, STAT3 interacts with and is activated by HCV core through phosphorylation, while the latter increases Stat3 gene expressionCitation75,Citation76 (Foka P, Dimitriadis A, et al., CMLS accepted). The subsequent regulation of Hamp gene expression is very well documented. On the contrary, STAT1 and p-STAT1 expression are markedly reduced in HCV core transfected cells, since core binds STAT1 and causes its proteasome-dependent degradation.Citation77
Wild-type HCV core protein has been shown to activate the transforming growth factor-β1 (TGF-β1) promoter and upregulate its expression through the p38/MAPK pathway. As a result, the SMAD pathway is activated.Citation78 In contrast, HCV core variants that were isolated from a liver tumor preparation and exhibited specific mutations not found in adjacent non-tumor or serum isolates, were shown to be responsible for inhibiting the TGF-β cascade in stably transfected cell lines and primary human hepatocytes. Further investigation revealed that direct physical interaction between tumor-derived HCV core and SMAD3 inhibited the activated SMAD3/4 complex from binding to target gene promoters.Citation79,Citation80 This reduction in TGF-β levels was accompanied by an epithelial to mesenchymal transition. Finally, our research revealed that in HCV core expressing HepG2 cells, SMAD4 protein levels were upregulated. This upregulation was accompanied by increased SMAD4 translocation to the nucleus, while both were blocked by dorsomorphin, an inhibitor of the BMP type I receptor (TβR-I) that specifically inhibits the BMP/SMAD pathway. The BMP/SMAD pathway activation resulted in increased Hamp gene promoter activity, which was matched accordingly by an increase in hepcidin mRNA and protein levels (Foka P, Dimitriadis A, et al., CMLS accepted).
Recently submitted data by our team provide an insight for the link between the two aforementioned pathways. Specifically, HCV core transfection in hepatic cells led to an increase in the casein kinase 2 (CK2) levels and an augmentation in CK2 activity. The use of a specific CK2 inhibitor, dominant negative CK2 construct or RNAi interference inhibited the activation and the increase in STAT3 and SMAD4 expression, while it eradicated the HCV core-mediated hepcidin expression. Those data suggest the direct involvement of CK2 in HCV core-mediated hepcidin regulation by the BMP/SMAD and STAT3 pathways (Foka P, Dimitriadis A, et al., CMLS accepted).
Lastly, HCV core protein signals through the p38 MAPK/ERK cascade, since it activates the Raf-1 kinase, a central component of the pathway that phosphorylates MEK1.Citation81,Citation82 Subsequently, MEK1 phosphorylates ERK1 and ERK2.Citation83 We have shown that non-enveloped HCV nucleocapsids, which can be naturally found in the bloodstream of HCV infected patients,Citation84 were able to enter cells of hepatic origin, where they modulated the MAPK cascade by activating ERK1/2 pathway.Citation85
E1 and E2 glycoproteins
The impact of other structural proteins in cell signaling involved in hepcidin modulation is more indirect. Zhao and colleagues showed that in human hepatoma Huh7 and L-02 cells, HCV E2 mediated a specific activation of the MAPK/ERK-ATF-2 pathway, through interaction with CD81 and low density lipoprotein receptor (LDLR).Citation86 Phosphorylation levels of the upstream kinases Raf-1 and MEK1/2, as well as the downstream kinase p38/MAPK, were elevated following E2 treatment. Pretreatment of cells with the upstream kinase MEK1/2 inhibitor U0126, or blocking antibodies against CD81/LDLR, was enough to suppress the E2-induced MAPK/ERK activation.Citation86,Citation87
NS3–NS4A protein
The HCV NS3 protein is able to activate JNK kinase without affecting its expression, as seen in NS3-transfected HepG2 cells. This activation however, is not accompanied by p38/MAPK or ERK1/2 activation.Citation88 On the contrary, two different studies revealed that in hepatoma cells HCV NS3 expression activates the MAPK/ERK1/2 pathway. Feng and colleagues showed that ectopic expression of NS3 N-terminal part or the whole NS3 sequence augmented ERK1/2 phosphorylation without affecting their expression.Citation89 Nevertheless, the observed p38/MAPK and ERK1/2 activation was attributed to the NS3-induced amphiregulin expression.Citation90
The HCV NS3–4A complex, which contains NS3 and its cofactor NS4A, has recently been shown to interact with SMURF2, a negative regulator of TGF-β signaling. In cells expressing the HCV NS3–4A complex, TGF-β stimulation mediated an increased and prolonged TGF-β-induced phosphorylation of SMAD2/3 that was abrogated by SMURF2 overexpression. Neither NS3–4A protease activity nor SMURF2 ubiquitin–ligase activity were required to affect TGF-β signaling. Therefore, by targeting SMURF2, the HCV NS3–4A complex appeared to block the negative regulation of TGF-β signaling, thereby increasing the responsiveness of cells to TGF-β.Citation91
NS4B protein
A recent report demonstrated that the HCV NS4B protein also affects cell signaling pathways and in particular the STAT pathway. Huh7 cells transfected with the HCV NS4B protein exhibited increased STAT3 promoter activity, accompanied by increased protein expression, phosphorylation, and translocation of STAT3 to the nucleus. In addition, HCV NS4B overexpression resulted in suppression of STAT3 repressor, SOCS3. In order to elucidate the upstream signaling pathways implicated in the regulation of STAT3 expression, the authors discovered that the ERK1/2 and JNK kinases were involved, since NS4B transfection led to increased levels of phosphorylated ERK1/2 and JNK, while repression of these kinases abrogated STAT3 activation.Citation92
NS5A protein
HCV NS5A has been implicated in the activation of STAT pathway via increased STAT3 phosphorylation and translocation to the nucleus. In addition, HCV NS5A transactivates STAT3-regulated promoters.Citation9,Citation93 However, a direct association between those two molecules has not been observed. The effect has been attributed to the increased phosphorylation of the STAT3 upstream kinase JAK1, with which NS5A physically interacts.Citation93
The HCV NS5A protein can also modulate TGF-β signaling since the synthesis and secretion of TGF-β1 was found upregulated in HCV NS5A transfected cells. The effect was attributed to increased ROS production.Citation94 Conversely, Choi and Hwang showed that HCV NS5A inhibited the TGF-β-mediated signaling pathway in hepatoma cell lines via physical interaction with TβR-I. Furthermore, NS5A protein co-localized with TβR-I in the cytoplasm of Huh7 cells and inhibited TGF-β-mediated nuclear translocation of SMAD2, while it abrogated the phosphorylation of SMAD2 and the heterodimerization of SMAD3 and SMAD4.Citation95 Finally, HCV NS5A inhibits the activity of transcription factor activating protein-1 (AP1), which occurs through inhibition of ERK1/2 phosphorylation.Citation96,Citation97 This effect was attributed to the interaction of HCV NS5A protein with growth receptor-bound protein 2 (Grb2).Citation97
Iron and Lipid Metabolism in HCV Infection
Recently, numerous proteomic and microarray gene expression studies have revealed a link between lipid metabolism and iron homeostasis.Citation98 CHC presented with hepatitis C-associated metabolic syndrome (MetS), often accompanied by hypocholesterolemia,Citation99 low ApoB levels, hyperglycemia, enhanced risk for type 2 diabetes, and predisposition to obesity.Citation100 HCV particle assembly following replication happens on lipid droplets.Citation101 Indeed, HCV creates lipid-rich cellular surroundings to assist major viral functions, such as infection replication, virion assembly, and secretion, trafficking, and escape from host immune system. Viral particles associate and become complexed with VLDL during lipoprotein assembly. Then, following VLDL secretion, they circulate in patient serum in the form of Lipo-Viro-Particles (LVP). Consequently, the virion is not exposed to serum neutralizing antibodies and may escape immune surveillance. Later, it may be adsorbed by cellular LDL receptors, facilitating viral penetration into host cells.Citation102
Both host and viral factors seem to be involved in steatosis development, whose establishment and severity may be genotype-dependent.Citation103 Recent metabolomic studies have identified a great number of host genes implicated in lipid metabolic processes related to HCV life cycle, but hardly studied,Citation104 such as fatty acid metabolism and biogenesis, glycerosphingo- and phospholipid biosynthesis, and arachidonic acid metabolism, shedding new light in the relationship between the virus and host lipid metabolism. On the other hand, the main viral components responsible for the virus’s usurping of the host lipid metabolism seem to be HCV core protein along with NS5A. Specifically, HCV core has been found to accumulate on the surface of lipid droplets, the cell’s intracellular storage sites for triacylglycerols and cholesterol esters.Citation105 In addition, core transgenic mice suffer from aberrant liver inflammation, which progresses to severe hepatic steatosis and HCC development.Citation106 Finally, core together with NS5A act as powerful transcriptional modulators, either directly through physical interaction with the transcriptional machinery or indirectly by altering signaling pathways,Citation85,Citation107 epigenetic mechanisms,Citation108 and the activity of transcription factors that are responsible for maintaining physiological hepatic lipid metabolism. For example, core enhances lipogenesis by upregulating fatty acid synthase (FAS) through activation of nuclear retinoic X receptor α (RXRα).Citation109 Both molecules are crucial for de novo synthesis of fatty acids. In addition, core has been shown to activate all isoforms of sterol regulatory element binding proteins (SREBPs), the master regulators of fatty acid and cholesterol biosynthesis and cellular uptake of lipoproteins.Citation110 It also increases SREBP1c gene expression in an liver X receptor α (LXRα)/RXRα-dependent manner.Citation102 Increased LXR expression linked with LXR/RXR transactivation, have been associated with core- and NS5A-mediated hepatic lipogenesis through PI3K signaling.Citation111 Furthermore, core and NS5A mediate alterations in activity and expression of peroxisome proliferators-activated receptors α (PPARα) and γ (PPARγ), which are essential for β-oxidation, secretion of fatty acids, adipogenesis, and lipid oxidation.Citation112-Citation114 Finally, we have showed that HCV core downregulates the lipid regulatory factor ANGPTL-3 through HNF-1α loss of DNA binding, in an LXRα/β-dependent manner.Citation115
Slowly but steadily, a link between lipid metabolism and iron has emerged, mostly from large cohort studies, globally. High serum ferritin and/or transferrin have been positively correlated with elevated triglycerides and in some cases reduced HDL levels,Citation116,Citation117 both of which were related to increased insulin resistance and the MetS.Citation118 The link may be forged on the basis of disturbed hepatic iron stores and the iron-mediated hepatic steatosis that stems from it. Thus, the liver appears to be the key organ, where the interplay between iron and lipid metabolism takes place.Citation119 In fact, over one-third of the patients who suffer from non-alcoholic fatty liver disease (NAFLD), the major manifestation of MetS in the liver, have also been found to suffer from hyperferritinemia and possess increased hepatic iron stores.Citation120 Naturally, elevated hepcidin accompanies the biochemical alterations found in these patients.Citation116
Animal studies using mostly rats, have showed that iron deficiency is capable of altering several metabolic processes related to lipid metabolism in the liver.Citation57,Citation121-Citation123 Nevertheless, there is no general consensus over whether pathways of lipid metabolism are positively or negatively regulated by systemic iron restriction, mostly because different types of animal models, amount of delivered iron and varied experimental protocols have been used to generate the data.Citation119 In addition, some authors reported increased concentrations of serum and hepatic lipids and lipoprotein content, as well as qualitative and quantitative changes in saturated mono- and polyunsaturated fatty acid composition,Citation124 while others found that iron restriction caused lower plasma cholesterol, triglycerides, and phospholipid levels.Citation125 Despite these discrepancies, iron deficiency has been demonstrated to affect lipid metabolism largely through diminished activity of key, rate-limiting enzymes of various lipid metabolic pathways that use iron as a cofactor,Citation123 such as LCAT,Citation125 carnitine,Citation57 or stearyl coA desaturase.Citation125
As discussed earlier, ferrous iron is a catalyst for the formation of ROS thereby inducing cellular oxidative stress and lipid peroxidation. This burdened oxidative status is detrimental to individuals with increased iron body stores due to genetic conditions like hereditary hemochromatosis or even excess dietary iron intake. It manifests with impaired mitochondrial metabolism, changes in the ratio of saturated to unsaturated phospholipids and alterations in membrane fluidity, damage to cellular organelles and necroinflammation.Citation126,Citation127 A very important aspect of iron overload-induced oxidative stress is considered the activation of hepatic stellate cells (HSC), which in turn secrete fibrotic factors that promote hepatofibrinogenesis and later cirrhosis characteristic to NAFLD.Citation128 NAFLD often progresses to the less benign non-alcoholic steatohepatitis condition (NASH). In animal models of iron overload combined with a high fat diet, insulin resistance is also evident and is accompanied by low HDL levels and impaired glucose and lipid metabolism.Citation120
Finally, recent studies support the notion that iron may be capable of regulating broader lipid metabolism aspects of the MetS, such as atherosclerosis initiation and progression and type 2 diabetes development through manipulation of major lipid metabolism-specific transcription factors. For example, Bories and colleagues demonstrated that LXRα was activated by iron overload in atheroma-located macrophages, thereby initiating lipid and iron efflux from these cells.Citation129 Furthermore, administration of an iron-rich diet in hamsters resulted in elevated cholesterol serum levels through reduction of PPARα levels.Citation130
Several clinical studies have demonstrated the co-existence of elevated hepatic iron and serum ferritin with lipid peroxidation due to the expected increase in oxidative stress and hepatic steatosis.Citation131-Citation133 In fact, there is growing evidence that iron may modify lipid metabolism, quite likely through hepcidin.Citation134 Nevertheless, the interactions between these factors and their combined effect on the development of persistent or severe liver disease either under the instruction of HCV proteins or, when pre-existing, as facilitating agents for HCV infection and replication, have not been studied so far. It is the opinion of the authors that the iron–lipid interplay will constitute a new field of study in HCV pathobiology in the years to come.
Hepcidin in HCV Disease Progression and Response to Therapy
CHC is frequently associated with hepatic iron overload.Citation135 Hepatic steatosis, a histological hallmark of CHC, observed in 40–80% of the patients, is related to increased liver inflammation, progression to liver fibrosis, poor therapeutic responses to interferon α-based anti-viral treatment and liver carcinogenesis.Citation136-Citation139 Iron accumulation within the liver may contribute to liver disease. Thus, it has been considered to be a fundamental mechanism of HCC development.Citation33,Citation140 The role of hepcidin in human HCC deserves to be studied, since there have been only few reports on this matter.Citation141 However, it is known that CHC patients have low serum hepcidin levels, an occurrence that is linked with enhanced necroinflammation and fibrosis.Citation142,Citation143
The influence of intrahepatic iron deposition to disease severity and progression in chronic liver diseases not related to hemochromatosis, remains elusive. Several studies have addressed the association between CHC disease stage and hepatic iron overload; the majority but not all of these studies support the linkage between advanced fibrosis and increased iron in non-parenchymal reticuloendothelial system cells (for example sinusoidal, endothelial, and portal tracts).Citation40,Citation144,Citation145 In NAFLD patients, the extent of hepatic iron deposition may be associated with the dual regulatory mechanism (iron stores and inflammation) of hepcidin.Citation146 Moreover, parenchymal iron deposition is an important feature of the alcoholic liver disease (ALD), although in this case, iron accumulation in the macrophages is more noticeable toward the advanced stages of disease.Citation16 Finally, it has been shown that the HSC may be activated by oxidative stress;Citation147 nevertheless, its precise effect on these cells may vary, depending on the restricted conditions posed by the iron-filled cells.
More data are needed on liver hepcidin gene expression and serum hepcidin level as well as biochemical hepatic iron measurements combined with longitudinal follow-up studies, to define the involvement of hepatic iron to disease severity and advancement and the potential effect of iron exhaustion, for treating common disorders of the HCV-infected liver.
Recent data suggested a linkage between prohepcidin, concentrations and PEG-IFN/ribavirin therapy in CHC.Citation148 Comparable levels of serum prohepcidin between CHC and healthy individuals were demonstrated. Noteworthy, in HCV-3a patients, prohepcidin levels were found significantly upregulated, as compared with HCV-1 patients. This phenomenon could be attributed to the fact that genotype 3-infected patients display differences in iron metabolism in contrast to other genotypes. Sebastiani and colleaguesCitation149 observed that that genotype 3-infected patients exhibited elevated hepatic iron deposits more frequently, and this condition was related to viral-induced hepatic steatosis. Taking into account that liver steatosis is more prominent in HCV-3a-infected patients than in patients infected with other HCV genotypes, due to increased lipid droplet accumulation,Citation150 we could hypothesize that an interplay between iron homeostasis and lipids that alters the host response to interferon therapy may take place during the course of the disease. Ryan and colleaguesCitation151 demonstrated an IFN-α-mediated hepcidin upregulation in HCV patients and a subsequent plasma iron reduction caused following administration of a single PEG-IFN-α dose. The same effect was recorded in in vitro cell culture. This study showed that hypoferremia was associated with a better PEG-IFN-α response, pointing toward a possible antiviral mode of PEG-IFN action. The observed IFN-α-mediated hepcidin upregulation was due to the JAK/STAT3 signaling pathway, with elevated serum hepcidin recorded in HCV patients. This could be attributed to the fact that hepcidin is an acute phase protein secreted in response to inflammatory stimuli. Individuals with acute infection or inflammation showed increased hepcidin levels and thus lowering of systemic iron levels, which may constitute a key innate immune response mechanism. More studies are needed to investigate whether antiviral drugs or other events, occurring simultaneously such as the increase in serum iron levels, are responsible for the noticed early hepcidin rise.
In conclusion, a better delineation of hepcidin regulation in HCV infection could help in developing novel therapeutic schemes. It is hoped that delineation of the mechanisms that govern the interplay between iron homeostasis, lipid metabolism and the cellular signaling alterations will provide novel targets for virus control and treatment of HCV-related disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Steinbicker AU, Muckenthaler MU. Out of balance--systemic iron homeostasis in iron-related disorders. Nutrients 2013; 5:3034 - 61; http://dx.doi.org/10.3390/nu5083034; PMID: 23917168
- Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, Ho LP, Townsend AR, Drakesmith H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011; 118:4129 - 39; http://dx.doi.org/10.1182/blood-2011-04-351957; PMID: 21873546
- MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal 2008; 10:997 - 1030; http://dx.doi.org/10.1089/ars.2007.1893; PMID: 18327971
- Tomosugi N, Kawabata H, Wakatabe R, Higuchi M, Yamaya H, Umehara H, Ishikawa I. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood 2006; 108:1381 - 7; http://dx.doi.org/10.1182/blood-2005-10-4043; PMID: 16621968
- Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis 2008; 40:132 - 8; http://dx.doi.org/10.1016/j.bcmd.2007.07.009; PMID: 17905609
- Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276:7806 - 10; http://dx.doi.org/10.1074/jbc.M008922200; PMID: 11113131
- Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101:2461 - 3; http://dx.doi.org/10.1182/blood-2002-10-3235; PMID: 12433676
- Ganz T. Hepcidin--a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol 2005; 18:171 - 82; http://dx.doi.org/10.1016/j.beha.2004.08.020; PMID: 15737883
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306:2090 - 3; http://dx.doi.org/10.1126/science.1104742; PMID: 15514116
- Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037 - 44; http://dx.doi.org/10.1172/JCI0215686; PMID: 12370282
- Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med 2013; 19:850 - 8; http://dx.doi.org/10.1038/nm.3184; PMID: 23836235
- Hoffman B, Liu Q. Hepatitis C viral protein translation: mechanisms and implications in developing antivirals. Liver Int 2011; 31:1449 - 67; http://dx.doi.org/10.1111/j.1478-3231.2011.02543.x; PMID: 21745290
- Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 2013; 19:837 - 49; http://dx.doi.org/10.1038/nm.3248; PMID: 23836234
- Valenti L, Pulixi EA, Arosio P, Cremonesi L, Biasiotto G, Dongiovanni P, Maggioni M, Fargion S, Fracanzani AL. Relative contribution of iron genes, dysmetabolism and hepatitis C virus (HCV) in the pathogenesis of altered iron regulation in HCV chronic hepatitis. Haematologica 2007; 92:1037 - 42; http://dx.doi.org/10.3324/haematol.11281; PMID: 17640859
- Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol 2007; 69:69 - 85; http://dx.doi.org/10.1146/annurev.physiol.69.031905.164337; PMID: 17014365
- Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol 2008; 88:7 - 15; http://dx.doi.org/10.1007/s12185-008-0120-5; PMID: 18594779
- Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000; 275:19906 - 12; http://dx.doi.org/10.1074/jbc.M000713200; PMID: 10747949
- Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000; 403:776 - 81; http://dx.doi.org/10.1038/35001596; PMID: 10693807
- McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000; 5:299 - 309; http://dx.doi.org/10.1016/S1097-2765(00)80425-6; PMID: 10882071
- Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, Muckenthaler MU. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010; 95:1261 - 8; http://dx.doi.org/10.3324/haematol.2009.020123; PMID: 20179090
- Brasse-Lagnel C, Karim Z, Letteron P, Bekri S, Bado A, Beaumont C. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 2011; 140:1261 - 71, e1; http://dx.doi.org/10.1053/j.gastro.2010.12.037; PMID: 21199652
- Franchini M, Montagnana M, Lippi G. Hepcidin and iron metabolism: from laboratory to clinical implications. Clin Chim Acta 2010; 411:1565 - 9; http://dx.doi.org/10.1016/j.cca.2010.07.003; PMID: 20620132
- Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009; 9:217 - 27; http://dx.doi.org/10.1016/j.cmet.2009.01.010; PMID: 19254567
- Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005; 2:399 - 409; http://dx.doi.org/10.1016/j.cmet.2005.10.010; PMID: 16330325
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood 2011; 117:4425 - 33; http://dx.doi.org/10.1182/blood-2011-01-258467; PMID: 21346250
- Castoldi M, Muckenthaler MU. Regulation of iron homeostasis by microRNAs. Cell Mol Life Sci 2012; 2012:9; PMID: 22678662
- Zumbrennen-Bullough KWQ, Chen W, Babitt J. MicroRNA-130a Downregulates Hepcidin Expression during Iron Deficiency by Targeting ALK2. Proceedings of Fifth Congress of the International BioIron Society (IBIS), Biennial World Meeting (BioIron 2013); London, UK 14–18 April 2013 2013.
- Zhang X, Rovin BH. Beyond anemia: hepcidin, monocytes and inflammation. Biol Chem 2013; 394:231 - 8; http://dx.doi.org/10.1515/bchm-2012-0217; PMID: 23314535
- Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood 2002; 99:3505 - 16; http://dx.doi.org/10.1182/blood.V99.10.3505; PMID: 11986201
- Cairo G, Recalcati S. Iron-regulatory proteins: molecular biology and pathophysiological implications. Expert Rev Mol Med 2007; 9:1 - 13; http://dx.doi.org/10.1017/S1462399407000531; PMID: 18053288
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J 2011; 434:365 - 81; http://dx.doi.org/10.1042/BJ20101825; PMID: 21348856
- Muckenthaler MU. Fine tuning of hepcidin expression by positive and negative regulators. Cell Metab 2008; 8:1 - 3; http://dx.doi.org/10.1016/j.cmet.2008.06.009; PMID: 18590684
- Pietrangelo A. Metals, oxidative stress, and hepatic fibrogenesis. Semin Liver Dis 1996; 16:13 - 30; http://dx.doi.org/10.1055/s-2007-1007215; PMID: 8723320
- Videla LA, Fernández V, Tapia G, Varela P. Oxidative stress-mediated hepatotoxicity of iron and copper: role of Kupffer cells. Biometals 2003; 16:103 - 11; http://dx.doi.org/10.1023/A:1020707811707; PMID: 12572670
- Braliou GG, Verga Falzacappa MV, Chachami G, Casanovas G, Muckenthaler MU, Simos G. 2-Oxoglutarate-dependent oxygenases control hepcidin gene expression. J Hepatol 2008; 48:801 - 10; http://dx.doi.org/10.1016/j.jhep.2007.12.021; PMID: 18313788
- Chaston TB, Matak P, Pourvali K, Srai SK, McKie AT, Sharp PA. Hypoxia inhibits hepcidin expression in HuH7 hepatoma cells via decreased SMAD4 signaling. Am J Physiol Cell Physiol 2011; 300:C888 - 95; http://dx.doi.org/10.1152/ajpcell.00121.2010; PMID: 21289291
- Deugnier Y, Turlin B. Pathology of hepatic iron overload. World J Gastroenterol 2007; 13:4755 - 60; PMID: 17729397
- Ko C, Siddaiah N, Berger J, Gish R, Brandhagen D, Sterling RK, Cotler SJ, Fontana RJ, McCashland TM, Han SH, et al. Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: results from the National Hemochromatosis Transplant Registry. Liver Int 2007; 27:1394 - 401; http://dx.doi.org/10.1111/j.1478-3231.2007.01596.x; PMID: 17927713
- Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H, Kobayashi Y, Iwasa M, Watanabe S, Adachi Y, et al. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med 2007; 13:97 - 104; http://dx.doi.org/10.2119/2006-00057.Fujita; PMID: 17515961
- Sikorska K, Stalke P, Izycka-Swieszewska E, Romanowski T, Bielawski KP. The role of iron overload and HFE gene mutations in the era of pegylated interferon and ribavirin treatment of chronic hepatitis C. Med Sci Monit 2010; 16:CR137 - 43; PMID: 20190684
- Drakesmith H, Prentice A. Viral infection and iron metabolism. Nat Rev Microbiol 2008; 6:541 - 52; http://dx.doi.org/10.1038/nrmicro1930; PMID: 18552864
- Maliken BD, Nelson JE, Kowdley KV. The hepcidin circuits act: balancing iron and inflammation. Hepatology 2011; 53:1764 - 6; http://dx.doi.org/10.1002/hep.24267; PMID: 21520181
- De Domenico I, Ward DM, Kaplan J. Hepcidin regulation: ironing out the details. J Clin Invest 2007; 117:1755 - 8; http://dx.doi.org/10.1172/JCI32701; PMID: 17607352
- Poli M, Luscieti S, Gandini V, Maccarinelli F, Finazzi D, Silvestri L, Roetto A, Arosio P. Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation. Haematologica 2010; 95:1832 - 40; http://dx.doi.org/10.3324/haematol.2010.027003; PMID: 20634490
- Fontana RJ, Israel J, LeClair P, Banner BF, Tortorelli K, Grace N, Levine RA, Fiarman G, Thiim M, Tavill AS, et al. Iron reduction before and during interferon therapy of chronic hepatitis C: results of a multicenter, randomized, controlled trial. Hepatology 2000; 31:730 - 6; http://dx.doi.org/10.1002/hep.510310325; PMID: 10706565
- Bonkovsky HL, Naishadham D, Lambrecht RW, Chung RT, Hoefs JC, Nash SR, Rogers TE, Banner BF, Sterling RK, Donovan JA, et al, HALT-C Trial Group. Roles of iron and HFE mutations on severity and response to therapy during retreatment of advanced chronic hepatitis C. Gastroenterology 2006; 131:1440 - 51; http://dx.doi.org/10.1053/j.gastro.2006.08.036; PMID: 17101320
- Meynard D, Babitt JL, Lin HY. The liver: conductor of systemic iron balance. Blood 2014; 123:168 - 76; http://dx.doi.org/10.1182/blood-2013-06-427757; PMID: 24200681
- Di Bisceglie AM, Simpson LH, Lotze MT, Hoofnagle JH. Development of hepatocellular carcinoma among patients with chronic liver disease due to hepatitis C viral infection. J Clin Gastroenterol 1994; 19:222 - 6; http://dx.doi.org/10.1097/00004836-199410000-00011; PMID: 7528758
- Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver. Viruses 2013; 5:439 - 69; http://dx.doi.org/10.3390/v5020439; PMID: 23358390
- Buratti E, Tisminetzky S, Zotti M, Baralle FE. Functional analysis of the interaction between HCV 5’UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res 1998; 26:3179 - 87; http://dx.doi.org/10.1093/nar/26.13.3179; PMID: 9628916
- Cho H, Lee HC, Jang SK, Kim YK. Iron increases translation initiation directed by internal ribosome entry site of hepatitis C virus. Virus Genes 2008; 37:154 - 60; http://dx.doi.org/10.1007/s11262-008-0250-0; PMID: 18566883
- Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 2001; 7:194 - 206; http://dx.doi.org/10.1017/S1355838201001790; PMID: 11233977
- Theurl I, Zoller H, Obrist P, Datz C, Bachmann F, Elliott RM, Weiss G. Iron regulates hepatitis C virus translation via stimulation of expression of translation initiation factor 3. J Infect Dis 2004; 190:819 - 25; http://dx.doi.org/10.1086/422261; PMID: 15272411
- Johnson KR, Merrick WC, Zoll WL, Zhu Y. Identification of cDNA clones for the large subunit of eukaryotic translation initiation factor 3. Comparison of homologues from human, Nicotiana tabacum, Caenorhabditis elegans, and Saccharomyces cerevisiae. J Biol Chem 1997; 272:7106 - 13; http://dx.doi.org/10.1074/jbc.272.11.7106; PMID: 9054404
- Izumi N, Enomoto N, Uchihara M, Murakami T, Ono K, Noguchi O, Miyake S, Nouchi T, Fujisawa K, Marumo F, et al. Hepatic iron contents and response to interferon-alpha in patients with chronic hepatitis C. Relationship to genotypes of hepatitis C virus. Dig Dis Sci 1996; 41:989 - 94; http://dx.doi.org/10.1007/BF02091542; PMID: 8625774
- Liu Y, An D, Sun R, Jin L, Wang Q. Inhibition of translation initiation factors might be the potential therapeutic targets for HCV patients with hepatic iron overload. Med Hypotheses 2012; 78:142 - 3; http://dx.doi.org/10.1016/j.mehy.2011.10.011; PMID: 22047986
- Bartolomei G, Cevik RE, Marcello A. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J Gen Virol 2011; 92:2072 - 81; http://dx.doi.org/10.1099/vir.0.032706-0; PMID: 21593278
- Fillebeen C, Pantopoulos K. Iron inhibits replication of infectious hepatitis C virus in permissive Huh7.5.1 cells. J Hepatol 2010; 53:995 - 9; http://dx.doi.org/10.1016/j.jhep.2010.04.044; PMID: 20813419
- Fillebeen C, Rivas-Estilla AM, Bisaillon M, Ponka P, Muckenthaler M, Hentze MW, Koromilas AE, Pantopoulos K. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C Virus. J Biol Chem 2005; 280:9049 - 57; http://dx.doi.org/10.1074/jbc.M412687200; PMID: 15637067
- Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology 2003; 124:1509 - 23; http://dx.doi.org/10.1016/S0016-5085(03)00275-0; PMID: 12730889
- Tung BY, Emond MJ, Bronner MP, Raaka SD, Cotler SJ, Kowdley KV. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology 2003; 124:318 - 26; http://dx.doi.org/10.1053/gast.2003.50046; PMID: 12557137
- Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sakai A, Okuda M, Hidaka I, et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 2008; 134:226 - 38; http://dx.doi.org/10.1053/j.gastro.2007.10.011; PMID: 18166355
- Moriya K, Miyoshi H, Shinzawa S, Tsutsumi T, Fujie H, Goto K, Shintani Y, Yotsuyanagi H, Koike K. Hepatitis C virus core protein compromises iron-induced activation of antioxidants in mice and HepG2 cells. J Med Virol 2010; 82:776 - 92; http://dx.doi.org/10.1002/jmv.21661; PMID: 20336713
- Choi SO, Cho YS, Kim HL, Park JW. ROS mediate the hypoxic repression of the hepcidin gene by inhibiting C/EBPalpha and STAT-3. Biochem Biophys Res Commun 2007; 356:312 - 7; http://dx.doi.org/10.1016/j.bbrc.2007.02.137; PMID: 17349976
- Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008; 48:1420 - 9; http://dx.doi.org/10.1002/hep.22486; PMID: 18671304
- Nagashima M, Kudo M, Chung H, Ishikawa E, Hagiwara S, Nakatani T, Dote K. Regulatory failure of serum prohepcidin levels in patients with hepatitis C. Hepatol Res 2006; 36:288 - 93; http://dx.doi.org/10.1016/j.hepres.2006.08.006; PMID: 16979376
- Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007; 109:353 - 8; http://dx.doi.org/10.1182/blood-2006-07-033969; PMID: 16946298
- Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006; 108:3204 - 9; http://dx.doi.org/10.1182/blood-2006-06-027631; PMID: 16835372
- Miyachi H, Kobayashi Y, Relja B, Fujita N, Iwasa M, Gabazza EC, Takei Y. Effect of suppressor of cytokine signaling on hepcidin production in hepatitis C virus replicon cells. Hepatol Res 2011; 41:364 - 74; http://dx.doi.org/10.1111/j.1872-034X.2011.00777.x; PMID: 21348906
- Fillebeen C, Muckenthaler M, Andriopoulos B, Bisaillon M, Mounir Z, Hentze MW, Koromilas AE, Pantopoulos K. Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J Hepatol 2007; 47:12 - 22; http://dx.doi.org/10.1016/j.jhep.2007.01.035; PMID: 17399844
- Kanda T, Yokosuka O, Omata M, Hepatitis C. Virus and Hepatocellular Carcinoma. Biology 2013; 2:304 - 16; http://dx.doi.org/10.3390/biology2010304
- Fukutomi T, Zhou Y, Kawai S, Eguchi H, Wands JR, Li J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology 2005; 41:1096 - 105; http://dx.doi.org/10.1002/hep.20668; PMID: 15841445
- Kochlios E, Foka P, Tsitoura E, Doumba P, Koskinas J, Mavromara P. Effect of HCV core and core+1/S on pro- and anti-inflammatory cytokine and chemokine gene expression. Bologna, Italy: Medimond SRL Publ., 2010.
- Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem 2011; 286:10847 - 55; http://dx.doi.org/10.1074/jbc.M110.217653; PMID: 21282107
- Yoshida T, Hanada T, Tokuhisa T, Kosai K, Sata M, Kohara M, Yoshimura A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J Exp Med 2002; 196:641 - 53; http://dx.doi.org/10.1084/jem.20012127; PMID: 12208879
- Cozza G, Mazzorana M, Papinutto E, Bain J, Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene M, et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem J 2009; 421:387 - 95; http://dx.doi.org/10.1042/BJ20090069; PMID: 19432557
- Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, Chung RT. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 2005; 128:1034 - 41; http://dx.doi.org/10.1053/j.gastro.2005.02.006; PMID: 15825084
- Taniguchi H, Kato N, Otsuka M, Goto T, Yoshida H, Shiratori Y, Omata M. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J Med Virol 2004; 72:52 - 9; http://dx.doi.org/10.1002/jmv.10545; PMID: 14635011
- Pavio N, Battaglia S, Boucreux D, Arnulf B, Sobesky R, Hermine O, Brechot C. Hepatitis C virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with Smad3 and inhibit the TGF-beta pathway. Oncogene 2005; 24:6119 - 32; http://dx.doi.org/10.1038/sj.onc.1208749; PMID: 16007207
- Battaglia S, Benzoubir N, Nobilet S, Charneau P, Samuel D, Zignego AL, Atfi A, Bréchot C, Bourgeade MF. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS One 2009; 4:e4355; http://dx.doi.org/10.1371/journal.pone.0004355; PMID: 19190755
- Tsutsumi T, Suzuki T, Moriya K, Shintani Y, Fujie H, Miyoshi H, Matsuura Y, Koike K, Miyamura T. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology 2003; 38:820 - 8; http://dx.doi.org/10.1002/hep.1840380408; PMID: 14512869
- Serti E, Doumba PP, Thyphronitis G, Tsitoura P, Katsarou K, Foka P, Konstandoulakis MM, Koskinas J, Mavromara P, Georgopoulou U. Modulation of IL-2 expression after uptake of hepatitis C virus non-enveloped capsid-like particles: the role of p38 kinase. Cell Mol Life Sci 2011; 68:505 - 22; http://dx.doi.org/10.1007/s00018-010-0466-8; PMID: 20680391
- Hayashi J, Aoki H, Kajino K, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 2000; 32:958 - 61; http://dx.doi.org/10.1053/jhep.2000.19343; PMID: 11050045
- Aoyagi K, Ohue C, Iida K, Kimura T, Tanaka E, Kiyosawa K, Yagi S. Development of a simple and highly sensitive enzyme immunoassay for hepatitis C virus core antigen. J Clin Microbiol 1999; 37:1802 - 8; PMID: 10325327
- Katsarou K, Lavdas AA, Tsitoura P, Serti E, Markoulatos P, Mavromara P, Georgopoulou U. Endocytosis of hepatitis C virus non-enveloped capsid-like particles induces MAPK-ERK1/2 signaling events. Cell Mol Life Sci 2010; 67:2491 - 506; http://dx.doi.org/10.1007/s00018-010-0351-5; PMID: 20358251
- Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS, Qi ZT. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp Cell Res 2005; 305:23 - 32; http://dx.doi.org/10.1016/j.yexcr.2004.12.024; PMID: 15777784
- Zhao LJ, Zhao P, Chen QL, Ren H, Pan W, Qi ZT. Mitogen-activated protein kinase signalling pathways triggered by the hepatitis C virus envelope protein E2: implications for the prevention of infection. Cell Prolif 2007; 40:508 - 21; http://dx.doi.org/10.1111/j.1365-2184.2007.00453.x; PMID: 17635518
- Hassan M, Ghozlan H, Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology 2005; 333:324 - 36; http://dx.doi.org/10.1016/j.virol.2005.01.008; PMID: 15721365
- Feng DY, Sun Y, Cheng RX, Ouyang XM, Zheng H. Effect of hepatitis C virus nonstructural protein NS3 on proliferation and MAPK phosphorylation of normal hepatocyte line. World J Gastroenterol 2005; 11:2157 - 61; PMID: 15810084
- Pei R, Chen H, Lu L, Zhu W, Beckebaum S, Cicinnati V, Lu M, Chen X. Hepatitis C virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. J Gen Virol 2011; 92:2237 - 48; http://dx.doi.org/10.1099/vir.0.032581-0; PMID: 21653755
- Verga-Gérard A, Porcherot M, Meyniel-Schicklin L, André P, Lotteau V, Perrin-Cocon L. Hepatitis C virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF-β signaling. FASEB J 2013; 27:4027 - 40; http://dx.doi.org/10.1096/fj.13-229187; PMID: 23781096
- Weizer-Stern O, Adamsky K, Margalit O, Ashur-Fabian O, Givol D, Amariglio N, Rechavi G. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol 2007; 138:253 - 62; http://dx.doi.org/10.1111/j.1365-2141.2007.06638.x; PMID: 17593032
- Sarcar B, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A mediated STAT3 activation requires co-operation of Jak1 kinase. Virology 2004; 322:51 - 60; http://dx.doi.org/10.1016/j.virol.2004.01.008; PMID: 15063116
- Presser LD, Haskett A, Waris G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-β1: role of TGF-β1 in HCV replication. Virology 2011; 412:284 - 96; http://dx.doi.org/10.1016/j.virol.2010.12.051; PMID: 21296375
- Choi SH, Hwang SB. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J Biol Chem 2006; 281:7468 - 78; http://dx.doi.org/10.1074/jbc.M512438200; PMID: 16407286
- Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J Biol Chem 2003; 278:17775 - 84; http://dx.doi.org/10.1074/jbc.M210900200; PMID: 12621033
- Georgopoulou U, Caravokiri K, Mavromara P. Suppression of the ERK1/2 signaling pathway from HCV NS5A protein expressed by herpes simplex recombinant viruses. Arch Virol 2003; 148:237 - 51; http://dx.doi.org/10.1007/s00705-002-0925-0; PMID: 12556990
- Nelson JE, Klintworth H, Kowdley KV. Iron metabolism in Nonalcoholic Fatty Liver Disease. Curr Gastroenterol Rep 2012; 14:8 - 16; http://dx.doi.org/10.1007/s11894-011-0234-4; PMID: 22124850
- Aso Y, Wakabayashi S, Yamamoto R, Matsutomo R, Takebayashi K, Inukai T. Metabolic syndrome accompanied by hypercholesterolemia is strongly associated with proinflammatory state and impairment of fibrinolysis in patients with type 2 diabetes: synergistic effects of plasminogen activator inhibitor-1 and thrombin-activatable fibrinolysis inhibitor. Diabetes Care 2005; 28:2211 - 6; http://dx.doi.org/10.2337/diacare.28.9.2211; PMID: 16123492
- Younossi ZM, McCullough AJ. Metabolic syndrome, non-alcoholic fatty liver disease and hepatitis C virus: impact on disease progression and treatment response. Liver Int 2009; 29:Suppl 2 3 - 12; http://dx.doi.org/10.1111/j.1478-3231.2008.01949.x; PMID: 19187068
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 2007; 9:1089 - 97; http://dx.doi.org/10.1038/ncb1631; PMID: 17721513
- Moriishi K, Mochizuki R, Moriya K, Miyamoto H, Mori Y, Abe T, Murata S, Tanaka K, Miyamura T, Suzuki T, et al. Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc Natl Acad Sci U S A 2007; 104:1661 - 6; http://dx.doi.org/10.1073/pnas.0607312104; PMID: 17234812
- Björnsson E, Angulo P. Hepatitis C and steatosis. Arch Med Res 2007; 38:621 - 7; http://dx.doi.org/10.1016/j.arcmed.2006.09.001; PMID: 17613353
- Roe B, Kensicki E, Mohney R, Hall WW. Metabolomic profile of hepatitis C virus-infected hepatocytes. PLoS One 2011; 6:e23641; http://dx.doi.org/10.1371/journal.pone.0023641; PMID: 21853158
- Roingeard P, Hourioux C. Hepatitis C virus core protein, lipid droplets and steatosis. J Viral Hepat 2008; 15:157 - 64; http://dx.doi.org/10.1111/j.1365-2893.2007.00953.x; PMID: 18086178
- Koike K, Moriya K, Matsuura Y. Animal models for hepatitis C and related liver disease. Hepatol Res 2010; 40:69 - 82; http://dx.doi.org/10.1111/j.1872-034X.2009.00593.x; PMID: 20156300
- Sato Y, Kato J, Takimoto R, Takada K, Kawano Y, Miyanishi K, Kobune M, Sato Y, Takayama T, Matunaga T, et al. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 2006; 55:1801 - 8; http://dx.doi.org/10.1136/gut.2005.070417; PMID: 16581947
- Gómez-Gonzalo M, Benedicto I, Carretero M, Lara-Pezzi E, Maldonado-Rodríguez A, Moreno-Otero R, Lai MM, López-Cabrera M. Hepatitis C virus core protein regulates p300/CBP co-activation function. Possible role in the regulation of NF-AT1 transcriptional activity. Virology 2004; 328:120 - 30; http://dx.doi.org/10.1016/j.virol.2004.06.044; PMID: 15380363
- Tsutsumi T, Suzuki T, Shimoike T, Suzuki R, Moriya K, Shintani Y, Fujie H, Matsuura Y, Koike K, Miyamura T. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 2002; 35:937 - 46; http://dx.doi.org/10.1053/jhep.2002.32470; PMID: 11915042
- Waris G, Felmlee DJ, Negro F, Siddiqui A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol 2007; 81:8122 - 30; http://dx.doi.org/10.1128/JVI.00125-07; PMID: 17507484
- García-Mediavilla MV, Pisonero-Vaquero S, Lima-Cabello E, Benedicto I, Majano PL, Jorquera F, González-Gallego J, Sánchez-Campos S. Liver X receptor α-mediated regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liver steatosis and HCV replication. Lab Invest 2012; 92:1191 - 202; http://dx.doi.org/10.1038/labinvest.2012.88; PMID: 22641099
- Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L, Podevin P, Conti F, Canva V, Philippe D, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 2005; 128:334 - 42; http://dx.doi.org/10.1053/j.gastro.2004.11.016; PMID: 15685545
- Kim K, Kim KH, Ha E, Park JY, Sakamoto N, Cheong J. Hepatitis C virus NS5A protein increases hepatic lipid accumulation via induction of activation and expression of PPARgamma. FEBS Lett 2009; 583:2720 - 6; http://dx.doi.org/10.1016/j.febslet.2009.07.034; PMID: 19631645
- Kim KH, Hong SP, Kim K, Park MJ, Kim KJ, Cheong J. HCV core protein induces hepatic lipid accumulation by activating SREBP1 and PPARgamma. Biochem Biophys Res Commun 2007; 355:883 - 8; http://dx.doi.org/10.1016/j.bbrc.2007.02.044; PMID: 17331464
- Foka P, Karamichali E, Dalagiorgou G, Serti E, Doumba PP, Pissas G, Kakkanas A, Kazazi D, Kochlios E, Gaitanou M, et al. Hepatitis C virus modulates lipid regulatory factor Angiopoietin-like 3 gene expression by repressing HNF-1alpha activity. J Hepatol 2014; 60:30 - 8; http://dx.doi.org/10.1016/j.jhep.2013.08.016; PMID: 23978712
- Dongiovanni P, Fracanzani AL, Fargion S, Valenti L. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol 2011; 55:920 - 32; http://dx.doi.org/10.1016/j.jhep.2011.05.008; PMID: 21718726
- Wlazlo N, Greevenbroek MM. Lipid metabolism: a role for iron?. Curr Opin Lipidol 2012; 23:258 - 9; http://dx.doi.org/10.1097/MOL.0b013e328354752d; PMID: 22576585
- Haap M, Machann J, von Friedeburg C, Schick F, Stefan N, Schwenzer NF, Fritsche A, Häring HU, Thamer C. Insulin sensitivity and liver fat: role of iron load. J Clin Endocrinol Metab 2011; 96:E958 - 61; http://dx.doi.org/10.1210/jc.2010-2682; PMID: 21430023
- Ahmed U, Latham PS, Oates PS. Interactions between hepatic iron and lipid metabolism with possible relevance to steatohepatitis. World J Gastroenterol 2012; 18:4651 - 8; http://dx.doi.org/10.3748/wjg.v18.i34.4651; PMID: 23002334
- Choi JS, Koh IU, Lee HJ, Kim WH, Song J. Effects of excess dietary iron and fat on glucose and lipid metabolism. J Nutr Biochem 2013; 24:1634 - 44; http://dx.doi.org/10.1016/j.jnutbio.2013.02.004; PMID: 23643521
- Lewis M, Iammarino RM. Lipemia in rodent iron-deficiency anemia. J Lab Clin Med 1971; 78:546 - 54; PMID: 5114051
- Sherman AR. Lipogenesis in iron-deficient adult rats. Lipids 1978; 13:473 - 8; http://dx.doi.org/10.1007/BF02533616; PMID: 692295
- Tosco A, Fontanella B, Danise R, Cicatiello L, Grober OM, Ravo M, Weisz A, Marzullo L. Molecular bases of copper and iron deficiency-associated dyslipidemia: a microarray analysis of the rat intestinal transcriptome. Genes Nutr 2010; 5:1 - 8; http://dx.doi.org/10.1007/s12263-009-0153-2; PMID: 19821111
- Stangl GI, Kirchgessner M. Different degrees of moderate iron deficiency modulate lipid metabolism of rats. Lipids 1998; 33:889 - 95; http://dx.doi.org/10.1007/s11745-998-0285-8; PMID: 9778136
- Rao GA, Crane RT, Larkin EC. Reduced plasma lecithin cholesterol acyl transferase activity in rats fed iron-deficient diets. Lipids 1983; 18:673 - 6; http://dx.doi.org/10.1007/BF02534575; PMID: 6656532
- Bacon BR, Britton RS, O’Neill R. Effects of vitamin E deficiency on hepatic mitochondrial lipid peroxidation and oxidative metabolism in rats with chronic dietary iron overload. Hepatology 1989; 9:398 - 404; http://dx.doi.org/10.1002/hep.1840090309; PMID: 2920996
- Feldman ES, Bacon BR. Hepatic mitochondrial oxidative metabolism and lipid peroxidation in experimental hexachlorobenzene-induced porphyria with dietary carbonyl iron overload. Hepatology 1989; 9:686 - 92; http://dx.doi.org/10.1002/hep.1840090505; PMID: 2707735
- Ramm GA, Crawford DH, Powell LW, Walker NI, Fletcher LM, Halliday JW. Hepatic stellate cell activation in genetic haemochromatosis. Lobular distribution, effect of increasing hepatic iron and response to phlebotomy. J Hepatol 1997; 26:584 - 92; http://dx.doi.org/10.1016/S0168-8278(97)80424-2; PMID: 9075666
- Bories G, Colin S, Vanhoutte J, Derudas B, Copin C, Fanchon M, Daoudi M, Belloy L, Haulon S, Zawadzki C, et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ Res 2013; 113:1196 - 205; http://dx.doi.org/10.1161/CIRCRESAHA.113.301656; PMID: 24036496
- Bonomo LdeF, Silva M, Oliveira RdeP, Silva ME, Pedrosa ML. Iron overload potentiates diet-induced hypercholesterolemia and reduces liver PPAR-α expression in hamsters. J Biochem Mol Toxicol 2012; 26:224 - 9; http://dx.doi.org/10.1002/jbt.21410; PMID: 22570273
- Farinati F, Cardin R, De Maria N, Della Libera G, Marafin C, Lecis E, Burra P, Floreani A, Cecchetto A, Naccarato R. Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J Hepatol 1995; 22:449 - 56; http://dx.doi.org/10.1016/0168-8278(95)80108-1; PMID: 7545199
- Kageyama F, Kobayashi Y, Kawasaki T, Toyokuni S, Uchida K, Nakamura H. Successful interferon therapy reverses enhanced hepatic iron accumulation and lipid peroxidation in chronic hepatitis C. Am J Gastroenterol 2000; 95:1041 - 50; http://dx.doi.org/10.1111/j.1572-0241.2000.01979.x; PMID: 10763957
- Konishi M, Iwasa M, Araki J, Kobayashi Y, Katsuki A, Sumida Y, Nakagawa N, Kojima Y, Watanabe S, Adachi Y, et al. Increased lipid peroxidation in patients with non-alcoholic fatty liver disease and chronic hepatitis C as measured by the plasma level of 8-isoprostane. J Gastroenterol Hepatol 2006; 21:1821 - 5; http://dx.doi.org/10.1111/j.1440-1746.2006.04420.x; PMID: 17074020
- Barisani D, Pelucchi S, Mariani R, Galimberti S, Trombini P, Fumagalli D, Meneveri R, Nemeth E, Ganz T, Piperno A. Hepcidin and iron-related gene expression in subjects with Dysmetabolic Hepatic Iron Overload. J Hepatol 2008; 49:123 - 33; http://dx.doi.org/10.1016/j.jhep.2008.03.011; PMID: 18462824
- Hézode C, Cazeneuve C, Coué O, Roudot-Thoraval F, Lonjon I, Bastie A, Duvoux C, Pawlotsky JM, Zafrani ES, Amselem S, et al. Liver iron accumulation in patients with chronic active hepatitis C: prevalence and role of hemochromatosis gene mutations and relationship with hepatic histological lesions. J Hepatol 1999; 31:979 - 84; http://dx.doi.org/10.1016/S0168-8278(99)80308-0; PMID: 10604569
- Lok AS, Everhart JE, Chung RT, Kim HY, Everson GT, Hoefs JC, Greenson JK, Sterling RK, Lindsay KL, Lee WM, et al, HALT-C Trial Group. Evolution of hepatic steatosis in patients with advanced hepatitis C: results from the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial. Hepatology 2009; 49:1828 - 37; http://dx.doi.org/10.1002/hep.22865; PMID: 19291787
- Macaluso FS, Maida M, Minissale MG, Li Vigni T, Attardo S, Orlando E, Petta S. Metabolic factors and chronic hepatitis C: a complex interplay. Biomed Res Int 2013; 2013:564645; http://dx.doi.org/10.1155/2013/564645; PMID: 23956991
- Hino K, Nishina S, Hara Y. Iron metabolic disorder in chronic hepatitis C: mechanisms and relevance to hepatocarcinogenesis. J Gastroenterol Hepatol 2013; 28:Suppl 4 93 - 8; http://dx.doi.org/10.1111/jgh.12243; PMID: 24251712
- Adinolfi LE, Durante-Mangoni E, Zampino R, Ruggiero G. Review article: hepatitis C virus-associated steatosis--pathogenic mechanisms and clinical implications. Aliment Pharmacol Ther 2005; 22:Suppl 2 52 - 5; http://dx.doi.org/10.1111/j.1365-2036.2005.02597.x; PMID: 16225474
- Mueller S, Afdhal NH, Schuppan D. Iron, HCV, and liver cancer: hard metal setting the pace?. Gastroenterology 2006; 130:2229 - 34; http://dx.doi.org/10.1053/j.gastro.2006.05.006; PMID: 16762645
- Ward DG, Roberts K, Brookes MJ, Joy H, Martin A, Ismail T, Spychal R, Iqbal T, Tselepis C. Increased hepcidin expression in colorectal carcinogenesis. World J Gastroenterol 2008; 14:1339 - 45; http://dx.doi.org/10.3748/wjg.14.1339; PMID: 18322945
- Piperno A, Mariani R, Trombini P, Girelli D. Hepcidin modulation in human diseases: from research to clinic. World J Gastroenterol 2009; 15:538 - 51; http://dx.doi.org/10.3748/wjg.15.538; PMID: 19195055
- Tsochatzis E, Papatheodoridis GV, Koliaraki V, Hadziyannis E, Kafiri G, Manesis EK, Mamalaki A, Archimandritis AJ. Serum hepcidin levels are related to the severity of liver histological lesions in chronic hepatitis C. J Viral Hepat 2010; 17:800 - 6; http://dx.doi.org/10.1111/j.1365-2893.2009.01244.x; PMID: 20002304
- Mitsuyoshi H, Yasui K, Yamaguchi K, Minami M, Okanoue T, Itoh Y. Pathogenic Role of Iron Deposition in Reticuloendothelial Cells during the Development of Chronic Hepatitis C. Int J Hepatol 2013; 2013:686420; http://dx.doi.org/10.1155/2013/686420; PMID: 23653861
- Pietrangelo A, Montosi G, Garuti C, Contri M, Giovannini F, Ceccarelli D, Masini A. Iron-induced oxidant stress in nonparenchymal liver cells: mitochondrial derangement and fibrosis in acutely iron-dosed gerbils and its prevention by silybin. J Bioenerg Biomembr 2002; 34:67 - 79; http://dx.doi.org/10.1023/A:1013874804911; PMID: 11860182
- Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, Kowdley KV, Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011; 53:448 - 57; http://dx.doi.org/10.1002/hep.24038; PMID: 21274866
- Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology 1999; 30:987 - 96; http://dx.doi.org/10.1002/hep.510300433; PMID: 10498651
- Jaroszewicz J, Rogalska M, Flisiak I, Flisiak R. Successful antiviral therapy is associated with a decrease of serum prohepcidin in chronic hepatitis C. World J Gastroenterol 2010; 16:1747 - 52; http://dx.doi.org/10.3748/wjg.v16.i14.1747; PMID: 20380007
- Sebastiani G, Vario A, Ferrari A, Pistis R, Noventa F, Alberti A. Hepatic iron, liver steatosis and viral genotypes in patients with chronic hepatitis C. J Viral Hepat 2006; 13:199 - 205; http://dx.doi.org/10.1111/j.1365-2893.2005.00662.x; PMID: 16475996
- Hourioux C, Patient R, Morin A, Blanchard E, Moreau A, Trassard S, Giraudeau B, Roingeard P. The genotype 3-specific hepatitis C virus core protein residue phenylalanine 164 increases steatosis in an in vitro cellular model. Gut 2007; 56:1302 - 8; http://dx.doi.org/10.1136/gut.2006.108647; PMID: 17213339
- Ryan JD, Altamura S, Devitt E, Mullins S, Lawless MW, Muckenthaler MU, Crowe J. Pegylated interferon-α induced hypoferremia is associated with the immediate response to treatment in hepatitis C. Hepatology 2012; 56:492 - 500; http://dx.doi.org/10.1002/hep.25666; PMID: 22334511