Abstract
Two new aconitine-type norditerpenoid alkaloids 6-dehydroacetylsepaconitine (1) and 13-hydroxylappaconitine (2), along with three known norditerpenoid alkaloids lycoctonine, delphatine and lappaconitine were isolated from the roots of the Aconitum heterophyllum Wall. These compounds exhibited significant antibacterial activity. The structure of compound 1 and 2 were deduced on the basis of their spectral data.
Introduction
Genus Aconitum is a rich source of diterpenoid alkaloids, many of which exhibited a broad spectrum of biological activities. Lappaconitine hydrobromide has been used as an antiarrhythmic drug [Citation1]. The methyllycaconitine perchlorate is used in curaremimetic preparation [Citation2]. Some aconitine and mesaconitine derivatives possess potent analgesic and anti-inflammatory activities [Citation3]. The methyllycaconitine and lycaconitine exhibited neuronal nicotinic acetylcholine receptor affinity [Citation4]. Lycaconitine, obtained from several Aconitum species, was found to be effective against multi-drug resistance cancers. Aconitum plants are widely used in Chinese and Indian traditional systems of medicine [Citation5–6]. Turkish Aconitum species are used externally in the treatment of rheumatic pain and sciatica and also against body lice [Citation7].
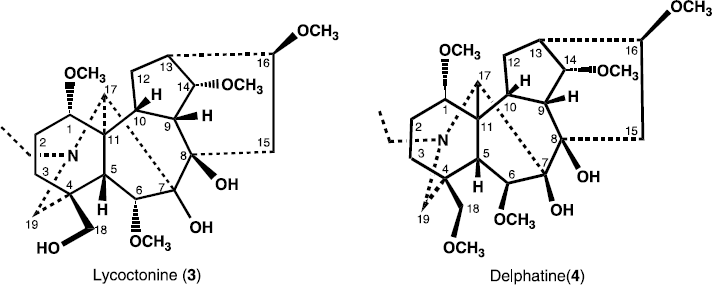
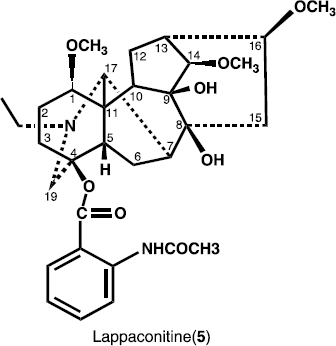
Previously, heterophyllisine, heterophylline, heterophyllidine, heteratisine, atisine, atidine, F-dihydroatisine, hetisine, benzoylheteratisine and atisenol were reported from A. heterophyllum [Citation8–11. In the present paper, we describe the isolation and structure elucidation of two new antibacterial norditerpenoid alkaloids 6-Dehydroacetylsepaconitine (1) and 13-hydroxylappaconitine (2), along with three known alkaloids lycoctonine (3), delphatine (4) and lappaconitine (5).
Experimental
General experimental
Optical rotations were measured on a JASCO DIP 360 polarimeter. IR spectra were recorded on a JASCO 302-A spectrophotometer. EI-MS and HREI-MS were recorded on JMS HX 110 with data system and on JMS-DA 500 mass spectrometers. The 1H- and 13C-NMR spectrums were recorded on Bruker NMR spectrometers operating at 400 MHz, (100 and 125 MHz for 13C). The chemical shifts values are reported in ppm (δ) units and the coupling constants ( J) are given in Hz.
Chromatographic conditions
For TLC, precoated aluminium sheets (silica gel 60F-254, E. Merck) were used. Visualization of the TLC plates was achieved under UV at 254 and 366 nm and by spraying with Dragendorff's reagent. Solvent system; “n-hexane–acetone–diethylamine 8:2:10 drops”, was used.
Plant material
The roots (5 kg dry wt) of Aconitum heterophyllum Wall, were collected from District Swat of Pakistan at an elevation of 2000 m in August 2005 and identified by Mr Mehboob ur Rahman (Assistant Professor) Department of Botany, Jahanzeb Post Graduate College, Saidu Sharif, Swat, NWFP, Pakistan. The voucher specimen (HA-014) is deposited in the herbarium of the botany department.
Extraction and isolation
Dried and powdered roots (5 Kg) of the plant were extracted exhaustively with n-hexane which solvent extract n-hexane (3 × 8 L) followed by 80% EtOH (3 × 10 L) extraction at room temperature for 7 days (3-times). The filtrate was evaporated in vacuo to yield 60 g of residue. The residue was acidified to pH 1.5 by 0.5 N H2SO4 and extracted with CH2Cl2 (3 × 2 L) collected alkaloidal mixture (18 g). The acidic aqueous solution was basified (pH 8–10) by using 10% KOH and extracted with CH2Cl2 (5 × 2 L) to yield 13.8 g of crude acidic compounds fraction. The crude acidic compounds fraction was fractionated on silica gel column (260 g), five combined fractions were obtained. On repeated flash column chromatography using solvent system n-hexane–acetone–diethylamine (9:1:10 drops per 100 ml) 6-dehydroacetylsepaconitine (1), 13-hydroxylappaconitine (2), along with lycoctonine, delphatine and lappaconitine known alkaloids were obtained.
6-dehydroacetylsepaconitine (1), amorphous powdered (13 mg). mp 122–125°C; ![]() +23.33° (c 0.8, CHCl3); UV (EtOH) λmax (log ε) 310 (3.75), 254 (4.22), and 225 (4.46) nm; IR νmaxCHCl3, 3492 (OH groups), 1700 (ester carbonyl, amide carbonyl), 1600, 1280, 1250, and 750 cm− 1 (1,2-substituted aromatic ring), 1083 (simple ether bonds); 1H-NMR (400 MHz, CDCl3): δ 1.17 (3H, t, J = 7.0 Hz, N-CH2CH3), δ 2.82 (1H, br s, C-17), δ 3.85 (1H, t, J = 7.9 Hz, C-1), δ 3.71 (1H, d, J = 4.8 Hz, C-14), δ 2.45 (1H, br s, C-5) δ 2.11 (1H, br s, C-7), δ 3.24 (1H, t, J = 8.4 Hz, C-16), δ 11.0 (NH). 13C-NMR (). HREI-MS (M+m/z, 614.6828)
+23.33° (c 0.8, CHCl3); UV (EtOH) λmax (log ε) 310 (3.75), 254 (4.22), and 225 (4.46) nm; IR νmaxCHCl3, 3492 (OH groups), 1700 (ester carbonyl, amide carbonyl), 1600, 1280, 1250, and 750 cm− 1 (1,2-substituted aromatic ring), 1083 (simple ether bonds); 1H-NMR (400 MHz, CDCl3): δ 1.17 (3H, t, J = 7.0 Hz, N-CH2CH3), δ 2.82 (1H, br s, C-17), δ 3.85 (1H, t, J = 7.9 Hz, C-1), δ 3.71 (1H, d, J = 4.8 Hz, C-14), δ 2.45 (1H, br s, C-5) δ 2.11 (1H, br s, C-7), δ 3.24 (1H, t, J = 8.4 Hz, C-16), δ 11.0 (NH). 13C-NMR (). HREI-MS (M+m/z, 614.6828)
Table I. 13C–NMR data of compounds 1 and 2 (CDCl3).
13-hydroxylappaconitine (2), amorphous powdered (10 mg). mp 145–147°C; ![]() +10.33° (c 1.0, CHCl3); IR νmaxCHCl3, 3492 (OH groups), 1720 (ester carbonyl, amide carbonyl), 1608, 1270, 1255, and 790 cm− 1 (1,2-substituted aromatic ring), 1089 (simple ether bonds); 1H-NMR (400 MHz, CDCl3): δ 1.03 (3H, t, J = 7.0 Hz, N-CH2CH3), δ 3.41 (1H, br s, H-17), δ 3.20 (2H, dd, J = 9.0 and 6.0 Hz, H-1 and H-16), δ 4.32 (1H, s, H-14), δ 2.45 (1H, br s, H-5), δ 2.35 (1H, br s, H-7), δ 8.68 and 7.92 (each 1H, dd, Ar-H) 7.50 and 7.04 (each 1H, t, Ar-H). 13C-NMR (). FAB ( + ve and − ve) (M+m/z, 601.0 and 599.0).
+10.33° (c 1.0, CHCl3); IR νmaxCHCl3, 3492 (OH groups), 1720 (ester carbonyl, amide carbonyl), 1608, 1270, 1255, and 790 cm− 1 (1,2-substituted aromatic ring), 1089 (simple ether bonds); 1H-NMR (400 MHz, CDCl3): δ 1.03 (3H, t, J = 7.0 Hz, N-CH2CH3), δ 3.41 (1H, br s, H-17), δ 3.20 (2H, dd, J = 9.0 and 6.0 Hz, H-1 and H-16), δ 4.32 (1H, s, H-14), δ 2.45 (1H, br s, H-5), δ 2.35 (1H, br s, H-7), δ 8.68 and 7.92 (each 1H, dd, Ar-H) 7.50 and 7.04 (each 1H, t, Ar-H). 13C-NMR (). FAB ( + ve and − ve) (M+m/z, 601.0 and 599.0).
Antibacterial activity
All of the isolated compounds were screened against strains of Escherichia coli, Bacillus subtilis, Shigella flexneri, Staphylococcus aureus, Pseudomonas aeruginosa, and Salmonella typhi. For antibacterial screening, 3 mg of sample was taken and dissolved in 3 ml of DMSO. Molten nutrient agar (45 mL) was poured on sterile petri plates, where it was allowed to solidify. Bacterial spread were made on these nutrient agar plates by dispensing 7 mL of sterile soft agar containing 100 μL of test-organism culture. Wells were dug with the help of a 6-mm sterile metallic borer at appropriate distance. Then, 100 μL of sample was poured into each well, and the plates were incubated at 37°C for 24 h. The results, in terms of inhibition zone, were noted. The drug Imipenem, a broad-spectrum β-lactam antibiotic, was used as a positive control. As a negative control, DMSO was used. The results are summarized in .
Table II. Table-2 Antibacterial activities of compounds 1-5. Relative to the standard drug Imipenem. For details, see Experimental.
Results and discussion
6-dehydroacetylsepaconitine (1) was obtained as a white amorphous powdered, and was assigned the molecular formula C32H42N2O10, on the basis of HREI-MS (m/z 614.6828, calcd. 614.6834). The mass spectrum of 1 was characteristic for diterpene bases of the C-18 series esterified through hydroxyl group at C-4 (lappaconitine, sepacnitine, N-deacetyllappacinitine, etc), where the maximum peak corresponds to the ejection of a molecule of acid from the molecular ion. In the mass spectrum of compound 1, the peak of the (M − 179)+ ion was the maximum peak and corresponded, as in the mass spectrum of N-acetylsepaconitine, to the ejection of acetylanthranilic acid [Citation12]. The IR spectrum showed of compound 1 showed absorption bands at 3492 (OH groups), 1700 broadened band, ester carbonyl, amide carbonyl), and bands at 1600, 1280, 1250, and 750 cm− 1 (1,2-substituted aromatic ring), 1083 (simple ether bonds). The 1H- and 13C-NMR spectra of 6-dehydroacetylsepaconitine (1) exhibited a close resemblance to that of the known compound N-acetylsepaconitine (3) [Citation12] except the presence of carbonyl group at C-6, instead of methylene group in compound 1.
The 1H-NMR spectrum of 6-dehydroacetylsepaconitine (1) exhibited signals for N-ethyl, three methoxy groups and several methine protons with geminal oxygen substituents. In the down field region of the spectrum a doublet of one proton at δ 3.71 (J = 4.8 Hz), characteristic for H-14. Triplet of three protons integration at δ 1.17 (J = 7.0 Hz), was due to the methyl of N-ethyl group. Similarly, in the down field region a triplet of one proton at δ 3.85 (J = 7.9 Hz), was assigned to the H-1, geminal to methoxy group. A broad singlet of one proton at δ 2.82 was assigned to the H-17, whereas, a broad singlet of one proton at δ 2.45, was assigned to the H-5, while, another singlet of one proton at δ 2.11 was assigned to H-7. The 13C-NMR spectrum (BB, DEPT) (), showed thirty two signals, including five methyls, six methylene, eleven methine, and ten quaternary carbons. Comparing the 13C-NMR data of compound 1 with those of the reported compound N-acetylsepaconitine (3), (), the appearance of a new quaternary carbon signal at δ 206.9, and the disappearance of CH2 signal at δ 24.5, indicated the presence of carbonyl group at C-6. The 1H- 13C correlation was determined by the HMQC spectrum, while the long range 1H- 13C connectivities were obtained through HMBC technique (). The H-5 (δ 2.45) showed 2J and 3J correlation with C-4 (δ 84.6), C-11 (δ 55.7), C-6 (δ 206.9) and C-17 (δ 61.5), whereas H-7 (δ 2.11), exhibited 1J and 2J correlation with C-6 (δ 206.9), C-7 (δ 46.8), C-8 (δ 74.5), and C-11 (δ 55.7), while, H-14 (δ 3.71), showed correlation with C-13 (δ 34.5), C-16 (δ 82.7).
Figure 1. Selective HMBC interactions in compound 1.
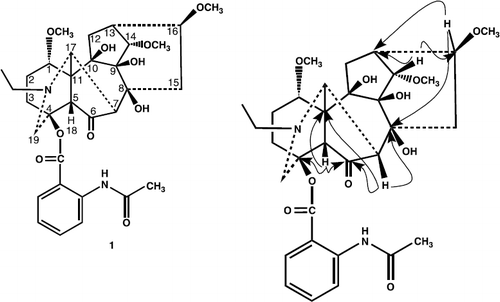
 Thus on the basis of above spectral data, the structure of compound 1 was deduced as 6-dehydroacetylsepaconitine.
Thus on the basis of above spectral data, the structure of compound 1 was deduced as 6-dehydroacetylsepaconitine.
13-hydroxylappaconitine (2) was obtained as a white amorphous powdered, and was assigned the molecular formula C32H44N2O9, on the basis of HREI-MS (m/z 600.7003, calcd. 600.7012). The IR spectrum showed of compound 2 showed absorption bands at 3492 (OH groups), 1700 broadened band, ester carbonyl, amide carbonyl), and bands at 1600, 1280, 1250, and 750 cm− 1 (1,2-substituted aromatic ring), 1083 (simple ether bonds). The mass fragmentation of 2 is characteristic of alkaloids with aconitine skeleton. The 1H- and 13C-NMR spectra of 13-hydroxylappaconitine (2) exhibited a close resemblance to that of the known compound lappaconitine (5) [Citation13], except the presence of hydroxyl group at C-13, instead of methine group in compound 2.
The 1H-NMR spectrum of 13-hydroxylappaconitine (2) exhibited signals for N-ethyl, three methoxy groups and several methine protons with geminal oxygen substituents. In the down field region of the spectrum a singlet of one proton at δ 4.32 was assigned to H-14. Similarly, a broad singlet of one proton in the down field region at δ 10.78 was assigned to the amide proton. Triplet of three protons at δ 1.03 (J = 7.0 Hz), was due to the methyl of N-ethyl group. Similarly, a double doublet of two protons at δ 3.21 (J = 9.0, 6.0 Hz), was assigned to the H-1 and H-16, geminal to methoxy group. A broad singlet of one proton at δ 2.45 was assigned to the H-5. The 13C-NMR spectrum (BB, DEPT) (), showed thirty two signals, including five methyls, seven methylene, eleven methine, and nine quaternary carbons. Comparing the 13C-NMR data of compound 2 with those of the reported compound lappaconitine (5), (), the appearance of a new quaternary carbon signal at δ 78.1, and the disappearance of CH signal at δ 49.0, indicated the presence of hydroxyl group at C-13. The 1H- 13C correlation was determined by the HMQC spectrum, while the long range 1H-13C connectivities were obtained through HMBC technique (). The H-5 (δ 2.45) showed correlation with C-4 (δ 84.6), C-11 (δ 53.1), and C-6 (δ 27.2), whereas H-7 (δ 2.11), exhibited correlation with C-6 (δ 27.2), C-7 (δ 47.8), C-8 (δ 68.4), and C- 17 (δ 58.0), while, H-14 (δ 3.71), showed correlation with C-13 (δ 78.1), C-14 (δ 87.4), C-9 (δ 79.2) and C-8 (δ 68.4), Similarly, H-16 (δ 3.33) exhibited interaction with C-16 (δ 82.2), C-15 (δ 32.4) and C-13 (δ 78.1).
Figure 2. Selective HMBC interactions in compound 2.
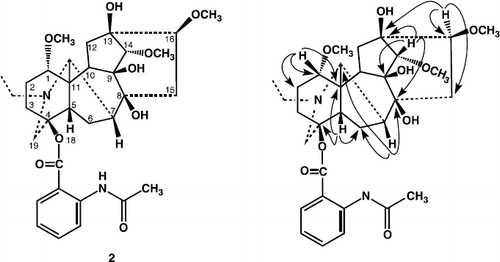
Thus on the basis of above spectral data, the structure of compound 2 was deduced as 13-hydroxylappaconitine.
Antibacterial activity: Compounds 1 and 2 showed significant antibacterial activity against Staphylococcus aureus. Compound 3 showed significant activities against Salmonella typhi and Pseudomonas aeruginosa, while compounds 4 and 5 showed significant activity against Salmonella typhi and Pseudomonas aeruginosa ().
Acknowledgements
We are thankful to Professor Mehboob-ur-Rahman for helping in the collection and identification of the plant.
References
- Atta-ur-Rahman, A Nasreen, F Akhtar, MS Shekhani, J Clardy, M Parvez, and MI Choudhary. (1997). J Nat Prod 56:872–874.
- MH Benn, and JM Jacuno In: SW Pelletier, editor. Alkaloids: Chemical and biological perspectives., Vol. 1 Chapter 4 New York: Wiley; (1984). 153.
- MG Hertog, EJ Feskens, PC Hollman, MB Katan, and D Kromhout. (1993). Lancet 342:1007–1011.
- JM Jacyno, JS Harwood, N-H Lin, JE Campbell, JP Sullivan, and MW Holladay. (1996). J Nat Prod 59:707.
- NG Bisset. (1981). J Ethnopharmacol 4:247.
- KS Mhaskar, E Blatter, and JF Caius. (2000). Ind Med Plants 1:28.
- T. BaytopTheraphy with Medicinal Plants in Turkey (Past and Present) Istanbul University, Publication No. 3255 1984. p 187.
- SW Pelletier, R Aneja, and KW Gopinath. (1968). Phytochemsitry 7:625–635.
- SW Pelletier, MMA Abdel, FM Janet, VM Naresh, and CS Lee. (1982). J Nat Prod 45 (6):779–781.
- WA Jacobs, and LC Craig. (1942). J Biol Chem 143:605.
- WA Jacobs, and LC Craig. (1943). J Biol Chem 147:571.
- VA Telnov, MS Yunusov, ND Abdullaev, and MG Zhamierashvili. (1989). Chem Nat Compounds 24 (4):472–475.
- A Ulubelen, AH Merely, F Mericli, U Kolak, M Arfan, M Ahmad, and H Ahmad. (2002). Pharmazie 57 (6):427.