Abstract
Previous investigations into the binding of substrates/cofactors to the PAH active site have only concentrated on Phe, thienylalanine and BH4. This is the first reported investigation to model aliphatic thioether amino acid substrates to PAH. The clearance of the thioether substrates (4.82-79.09% of Phe) in the rat and human (1.19-37.41% of Phe) showed species differences. The xenobiotic thioether substrates (SMC and SCMC) were predicted to be poor substrates for PAH by the molecular modelling investigation and this has now been confirmed by the in vitro enzyme kinetic data. However, reaction phenotyping investigations have found that PAH was the major enzyme involved in the metabolism of SCMC in vitro and in vivo.
Introduction
The role played by phenylalanine 4-monooxygenase (PAH) in the conversion of L-phenylalanine (Phe) to L-tyrosine (Tyr) has been under active investigation ever since it was first associated with the disease phenylketonuria (PKU) [Citation1]. The genetic mutations that result in PKU and hyperphenylalaninemia due to PAH deficiencies are now well known [Citation2]. The PAH enzyme is regulated by a variety of pre- and post-translational mechanisms in vivo and in vitro. At the pre-translational level PAH is activated by the glucocorticoid receptor [Citation1]. However, at the post-translational level PAH is activated by a number of processes [Citation3].
PAH is a homo tetrameric protein. The monomer subunits have no catalytic activity but the dimer and tetramers all possess enzyme activity and the full enzyme is in fact a dimer of dimers [Citation3]. Each monomer comprises, an N-terminal regulatory domain (amino acids 1-142), a catalytic domain (amino acids 143-410) and a C-terminal polymerization domain (amino acids 411-452). The major physiological control of PAH activity in vivo is by binding of Phe and/or phosphorylation of the allosteric enzyme [Citation3]. This results in changes in both Km and Vmax for Phe but does not alter the enzyme's substrate specificity. The binding of lysophosphatidycholine, modification of Cys237 or limited proteolysis all can activate PAH. These activation mechanisms not only alter the Km and Vmax for Phe, but also alter the substrate specificity of the enzyme as well [Citation3]. L-Methionine (Met), S-methyl-L-cysteine (SMC), S-carboxymethyl-L-cysteine (SCMC) and nor-leucine are all substrates for activated PAH [Citation3,4].
Both SMC and SCMC are mucolytic drugs used in the treatment of chronic obstructive pulmonary disease (COPD) and otitis media with effusions (OME). The clinical efficacy of these two drugs in patients has been the subject of debate for a number of years. However, it is clear that while some patients respond very well to the mucolytic drugs others show little or no benefit. This observed variation in response to these mucolytic agents could be linked to the human genetic polymorphism in the S-oxidation of SCMC in man [Citation5]. The major urinary metabolites of SCMC in man are the S-oxides (SCMC S-oxide, SMC S-oxide, N-acetyl SMC S-oxide, N-acetyl-SCMC S-oxide and thiodiglycolic acid S-oxide). Thus the polymorphism in the S-oxidation of SCMC will have a major effect on the clinical efficacy of SCMC since the mechanism of action of the drug is now attributed to its activity as a free radical scavenger. The thioether drug is converted to SCMC S-oxide by reactive oxygen intermediates [Citation6]. Since 65% of the population are extensive metabolisers, 32% are intermediate metabolisers and 3% are poor metabolisers with regard to the S-oxidation of SCMC[Citation7], the enzyme responsible for the S-oxidation of this drug will have a major effect on the level of the free radical scavenger in the body.
The role that PAH plays in the metabolism of the thioether mucolytic drugs is poorly understood, and the aims of this investigation were to model the binding of the thioether drugs to the PAH active site and to compare the predicted binding interactions with the Km and Vmax values determined experimentally.
Materials and methods
Molecular modelling of human PAH-substrate complexes
Model building and visualisation of the PAH-substrate complexes was performed using HyperChem™ version 7.1 and WebLab ViewerLite® version 5.0.
The template selected for the model building was provided by the 2.0 Å resolution crystal structure for the catalytic domain of human PAH ternary complex with norleucine and tetrahydrobiopterin (BH4)[Citation8]. Atomic co-ordinates for this complex were obtained from the Protein Data Bank [Citation9] (structure code: 1 mmt).
The docking of substrates into the PAH active site was performed manually but guided by the pose of the crystal structure ligand, norleucine. The resulting models were then refined by means of potential energy minimization using the AMBER force field [Citation10].
In vitro PAH enzyme assays
Enzyme assays
Direct PAH activity was determined by the method of Pey et al. [Citation11] with the following modifications. All incubations were carried out at 37°C for 10 min. Each enzyme assay contained (rat pooled cytosolic fractions, 2.0 mM phenylalanine/40.0 mM SCMC/50.0 mM SMC/20.0 mM Met, human pooled cytosolic fractions 2.0 mM phenylalanine/20.0 mM SCMC/40.0 mM SMC/15.0 mM Met), 50.0 mM potassium phosphate buffer (pH 6.8), catalase (7,800 units/mL), human/rat liver cytosol (200 μg/mL), 6.0 mM DTT, 100 μM BH4 in a total volume of 1.0 mL. Reactions were terminated by addition of 100 μl of 10% (w/v) TCA. Acid supernatants were prepared by centrifugation at 3,000 x g for 10 min. Blanks were assayed as described but using heat (100°C) in-activated rat or human cytosolic fractions.
Indirect PAH activity for the S-oxidation of Met, SCMC, SMC and S-methyl-ergothionine (SME) was determined using the method of Pey et al.[Citation11]. All incubations were carried out at 37°C. The Δ340 nm was measured for 1.0 min by using ε340 nm = 6220 M− 1· cm− 1. Each enzyme assay contained (rat pooled cytosolic fractions, 2.0 mM phenylalanine/40.0 mM SCMC/50.0 mM SMC/30.0 mM SME/20.0 mM Met, human pooled cytosolic fractions 2.0 mM phenylalanine/20.0 mM SCMC/40.0 mM SMC/ 2.0 mM SME/15.0 mM Met), 50.0 mM potassium phosphate buffer (pH 6.8), catalase (7,800 units/mL), human/rat liver cytosol (200 μg/mL), 200 μM NADH(H+), 1.0 units/mL dihydropteridine reductase, 100 μM BH4 in a total volume of 1.0 mL. Blanks were assayed as described but containing both 200 μg/mL PH8 (monoclonal antibody against PAH) and 1.0 mM 4-chlorophenylalanine (specific PAH inhibitor).
HPLC assays
The quantification of the S-oxides of Met, SCMC and SMC was carried out by HPLC as reported by Hirschberger et al. [Citation12], and the quantification of Tyr as reported by Doskeland et al. [Citation13].
Synthesis of S-carboxymethyl-L-cysteine and S-methyl-L-cysteine S-oxides
The synthesis of the mixture of SCMC (S/R) sulphoxide and SMC (S/R) diastereoisomers was based on the methods of Meese [Citation14] and Meese et al. [Citation15].
Synthesis of S-methyl-ergothionine
The synthesis of S-methyl-ergothionine (SME) was based on the method of Theodoropoulos [Citation16].
Data analysis
Enzyme kinetic data were analysed using WinNonLin version 4.1 and Leonora version 1.0. Statistical analysis was undertaken using SPSS version 10.0.
Results and discussion
Molecular modelling studies
Results obtained in the molecular modelling studies are summarised in and –1e. For each complex, attention was focused on the proximity of the substrate to the active site Fe2 + , BH4 cofactor, molecular oxygen and bound water. In addition to the modelling of the PAH-Phe, PAH-SMC, and PAH-SCMC complexes, studies were also made of the complex formed between the PAH enzyme and ergothionine (Erg). This latter compound was identified as a potential substrate for the enzyme following a manual search for commercially available compounds having the appropriate molecular properties and dimensions.
Table I. Molecular modelling of human PAH active site.
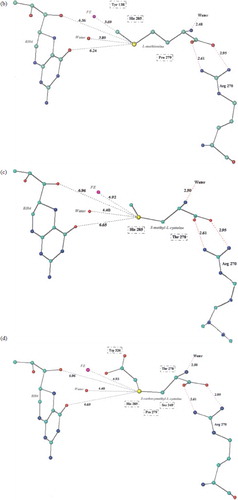
Figure 1. (a) 3-D schematic diagram of Phe-PAH interactions. (b) 3-D schematic diagram of Met-PAH interactions. (c) 3-D schematic diagram of SMC-PAH interactions. (d) 3-D schematic diagram of SCMC-PAH interactions. (e) 3-D schematic diagram of SME-protein interactions. Molecular models of the predicted complexes formed between PAH and the substrates Phe (a), Met (b), SMC (c), SCMC (d) and SME (e). Atoms are colour coded as C: green, N: blue, O: red, Fe: pink, S: yellow. Potential hydrogen bonds are indicated by dashed red lines, and PAH residues in van de Waals contact with the ligands are boxed. All inter atomic distances are given in Å.
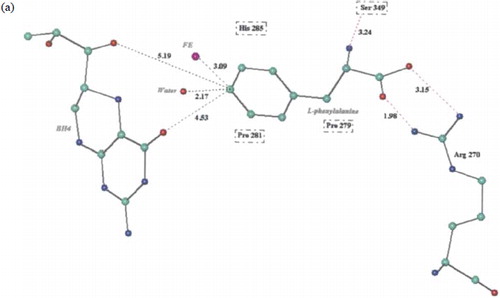

For the PAH-Phe complex ( and ) the Arg270 amino acid from one side of the active site channel is involved in an ionic interaction with the α-carboxyl group of the substrate. The distance between the carboxyl oxygen atoms and the amino nitrogen atoms of Arg270 are 1.98 Å and 3.15 Å. On the opposite side of the channel, the side chain OH of Ser349 hydrogen bonds with the α-amino group of Phe and the phenyl ring of Phe is in close proximity to Pro281. The site of oxidation of Phe is 2.17 Å from the bound water molecule and 3.09 Å from the Fe2 + ion. The latter distance is < 4.2 Å and thus deemed adequate for intercalation of molecular oxygen in the hydroxylation reaction[Citation17]. The site of oxidation on the phenyl ring is 4.33 Å and 5.19 Å from the oxygen atoms on the cofactor, BH4.
For the aliphatic amino acids, Met, SMC and SCMC, the distances of the site of oxidation from the Fe2 + were 3.69 Å, 4.92 Å and 4.93 Å respectively (). The predicted substrate Erg, has a sulphur atom at 3.14 Å from the Fe2 + . Thus from the modelling studies the preferred substrates for human PAH in terms of orientation and conformation within the catalytic site were Phe > Erg > Met > SMC ≈ SCMC.
Enzyme kinetic studies
The in vitro enzyme kinetics of both female rat and human PAH in pooled cytosolic fractions were investigated in order to assess the predicted substrate specificity. A summary of the results obtained in these is presented in .
Table II. Enzyme kinetic data for activated pooled rat cytosolic fraction PAH assays using Phe, Met, SME, SMC and SCMC as substrates.
The rate of clearance of the substrates via rat PAH was Phe > Met > SCMC > SMC (ClE 14.30 ± 0.72, 8.24 ± 0.41, 3.26 ± 0.16, 0.69 ± 0.03 μl min− 1 mg protein− 1). The predicted substrate, Erg was assayed via the coupled enzyme assay since no Erg sulphenic or sulphinic acid could be synthesed or found commercially. No activity was detected but when an inhibitor investigation of Erg and PAH was undertaken to determine the Ki and type of inhibition versus Phe, the predicted substrate was found to be an activator of rat PAH activity in vitro (results not shown). Since the sulphur containing substrates Met, SMC and SCMC are all sulphides and Erg was a sulphydryl compound, the sulphydryl group was converted to a sulphide group via methylation. The new potential substrate, S-methyl-ergothionine (SME) was found to be a substrate for rat PAH in the coupled assay with a clearance value of 11.31 ± 8.9 μl min− 1 mg protein− 1. Thus, from the clearance date in , the substrate specificity of rat PAH was Phe > SME > Met> SCMC > SMC. The inhibitor binding constant (Ki) and type of inhibition were also determined experimentally. All substrates (SME, Met, SCMC, SMC) were found (via Dixon plots) to be competitive inhibitors of the C-oxidation of Phe (results not shown). The experimentally determined Ki values for the inhibitors were not significantly different from their experimentally determined Km values (P>0.05, Student t-test) indicating that the binding constants for the compounds acting as substrates and inhibitors were the same. The assay of PAH using Phe, Met, SCMC and SMC was undertaken by two assays. The direct assay involved HPLC identification and quantification of the product of the enzyme reactions (Tyr, Met R/S S-oxides, SCMC R/S S-oxides and SMC R/S S-oxides). The same assays were repeated using the coupled assay (assaying the disappearance of the cofactor NADH(H+) at 340 nm). The correlation between the disappearance of cofactor and the appearance of product (coupling factor) was 0.94-0.95 for all substrates investigated. It was thus concluded that the coupled assay could be used to investigate substrate specificity when authentic standards of the products were not available.
When the experiments using pooled female Wistar rat cytosolic fractions were repeated in pooled human female cytosolic fractions, a similar pattern of results was obtained (). The rate of clearance of the substrates via human PAH was Phe > Met >SCMC > SMC (ClE 142.51 ± 0.71, 18.59 ± 1.01, 10.92 ± 0.16, 1.70 ± 0.09 μl min− 1 mg protein− 1). Again no activity was detected when Erg was investigated for substrate activity in the coupled assay but when an inhibitor investigation of Erg and PAH was undertaken to determine the Ki and type of inhibition versus Phe, the predicted substrate was also found to be an activator of human PAH activity in vitro (results not shown). The new potential substrate, S-methyl-ergothionine (SME) was found to be a substrate for human PAH in the coupled assay with a clearance value of 53.31 ± 2.67 μl min− 1 mg protein− 1. Thus, from the clearance date in , the substrate specificity of human PAH was Phe >SME > Met > SCMC > SMC. The inhibitor binding constant (Ki) and type of inhibition were also determined experimentally. All substrates (SME, Met, SCMC, SMC) were found (via Dixon plots) to be competitive inhibitors of the C-oxidation of Phe (results not shown). The experimentally determined Ki values for the inhibitors were again not significantly different from their experimentally determined Km values (P > 0.05, Student t-test), indicating that the binding constants for the compounds acting as substrates and inhibitors were the same. The assay of human PAH using Phe, Met, SCMC and SMC was again undertaken by both direct and coupled assays as with the rat PAH study. The correlation between the disappearance of cofactor and the appearance of product (coupling factor) was 0.94-0.96 for all substrates investigated. It was again concluded, therefore that the coupled assay could be used to investigate substrate specificity when authentic standards of products were not available in human and rat pooled cytosolic fractions for PAH activity.
Table III. Enzyme kinetic data for activated pooled human cytosolic fraction PAH assays using Phe, Met, SME, SMC and SCMC as substrates.
Acknowledgements
BB, GBS and BF would like to thank the Royal Thai Government for funding BB.
References
- S Kaufman. (1993). Adv Enzymol Rel Areas Mol Biol 67:77–264.
- PAHdb: www.mcgill.ca/pahdb.
- SE Hufton, IG Jennings, and RG Cotton. (1995). Biochem J 311:353–366.
- S Kaufman, and K Mason. (1982). J Biol Chem 257:14667–14678.
- SC Mitchell, RH Waring, CS Haley, JR Idle, and RL Smith. (1984). Br J Clin Pharmacol 18:507–521.
- L Brandolini, M Allegretti, V Berdini, MN Cervellera, P Mascagni, M Rinaldi, G Melillo, P Ghezzi, M Mengozzi, and R Bertini. (2003). Eur Cytokine Net 14:20–26.
- GB Steventon, S Sturman, RH Waring, and AC Williams. (2001). Drug Metab Drug Inter 18:79–98.
- OA Andersen, AJ Stokka, T Flatmark, and E Hough. (2003). J Mol Biol 333:747–757.
- H Merman, J Westbrook, Z Feng, G Gilliland, TN Bhat, H Weissig, IN Shindyalov, and PE Bourne. (2000). Nuc Acids Res 28:235–242.
- DA Case, TE Cheatham, T Darden, H Gohlke, R Luo, KM Merz, A Onufriev, C Simmerling, B Wang, and RJ Woods. (2005). J Comput Chem 26:1668–1688.
- AL Pey, A Martinez, R Charubala, DJ Maitland, K Teigen, A Calvo, W Pfeiderer, JM Wood, and KU Schallreuter. (2006). FASEB J 20:E1451–E1464.
- LL Hirschberger, J de la Rosa, and MH Stipanuk. (1985). J Chromatog 343:303–313.
- AP Doskeland, SO Doskeland, D Ogreid, and T Flatmark. (1984). J Biol Chem 259:11242–11248.
- CD Meese. (1987). Arch Pharm (Weinheim) 320:473–474.
- CD Meese, D Spechat, and U Hofmann. (1990). Arch Pharm (Weinheim) 323:957–965.
- D Theodoropoulos. (1959). Acta Chem Scand 13:383–384.
- K Teigen, NA Froystein, and A Martinez. (1999). J Mol Biol 294:807–823.
- OA Andersen, T Flatmark, and E Hough. (2002). J Mol Biol 320:1095–1108.