
ABSTRACT
Introduction
Prader-Willi syndrome (PWS) is a complicated neurodevelopmental genetic disorder stemming from the loss of expression of imprinted genes within the 15q11-q13 region. It is characterized by impaired hypothalamic development and function. Infants with PWS typically present hypotonia and feeding difficulties, which in later stages of childhood progress to hyperphagia, obesity, and endocrine dysfunctions. However, early diagnosis and treatment have proven effective in mitigating obesity and related co-morbidities in patients with PWS. Moreover, the precise molecular classification of PWS is crucial to tailor the appropriate treatment strategies and provide valuable genetic counseling.
Areas covered
This review contains various conventional and novel PWS diagnostic methods, assessing each method’s underlying mechanisms, advantages and disadvantages. Furthermore, our review presents a genetic testing workflow for PWS diagnosis and explores promising techniques for newborn and prenatal screening, which facilitate early diagnosis and intervention. This review synthesizes pertinent studies from 1990 to 2022, gathered from databases including PubMed, Web of Science, EBSCO, and Cochrane.
Expert opinion
Starting with MS-MLPA is the most efficient way to detect underlying genetic mechanisms. However, it is essential to note that certain rare instances, such as balanced chromosomal rearrangements, may require complementary diagnostic techniques to identify accurately.
1. Introduction
Prader-Willi syndrome (PWS) is a multisystem disorder caused by the absence of expression of the paternally active genes on chromosome 15q11-q13 region [Citation1]. Conversely, Angelman syndrome (AS) is caused by the absence of the active maternal gene in chromosome 15 [Citation2].
So far, Multiple genes have been identified and mapped within the PWS region, including MKRN3, MAGEL2, NDN, PWRN1, NPAP1, SNURF-SNRPN and small nucleolar RNA cluster (). Due to genomic imprinting, SNRPN, MAGEL2 and NDN genes are differentially methylated in CpG islands of their promoter regions [Citation3]. This CpG island at the 5’ end is extensively methylated on the maternal chromosome and unmethylated on the paternal allele, leading to the transcriptional silencing of the maternal allele [Citation4,Citation5].
Figure 1. Overview of the critical region for PWS. Blue spots represent biallelically expressed genes, orange boxes/vertical lines represent paternally expressed genes, and green vertical lines represent maternally expressed genes. It is adapted from [Citation6].
![Figure 1. Overview of the critical region for PWS. Blue spots represent biallelically expressed genes, orange boxes/vertical lines represent paternally expressed genes, and green vertical lines represent maternally expressed genes. It is adapted from [Citation6].](/cms/asset/0f04f834-a986-4a3a-84ec-ada6a74dae92/ieod_a_2262104_f0001_oc.jpg)
PWS affects males and females equally, with prevalence rates varying between 1 in 10,000 births to 1 in 25,000 births [Citation7]. Hypothalamus dysfunction and abnormal development are primarily responsible for most PWS phenotypes [Citation8]. Miller et al. identified seven nutritional phases in PWS. Phase 0 is associated with intrauterine growth restriction. Phase 1 is characterized by hypotonia. In phase 1a (0–9 months), the infant presents feeding difficulty with or without failure to thrive. While in phase 1b (9–25 months), the infant has a normal appetite and weight increase rate. Phase 2 is characterized by weight gain, with no significant change in appetite in subphase 2a (about 2–4.5 years) but an increased food interest in subphase 2b (about 4.5–8 years). In phase 3 (from 8 years to adult), the patients with PWS show marked hyperphagia and insatiable appetite. However, some adults may progress to phase 4, characterized by a return to normal appetite [Citation9]. Additionally, characteristic craniofacial features, behavior problems, cognitive disabilities, spinal deformity, and hip dysplasia are also presented in PWS [Citation2,Citation10,Citation11].
Due to hypothalamic dysfunction, most cases of PWS present endocrine dysfunctions, including growth hormone (GH) deficiency (GHD), central hypothyroidism, glucose metabolism disorders, corticotropin deficiency and hypogonadism. GHD is common in children with PWS (about 40–100%) and may be associated with short stature, excessive body fat, decreased muscle mass and energy expenditure [Citation12,Citation13]. Additionally, GHD and hypogonadism may cause a marked bone phenotype in PWS, including low bone mineral density, reduced bone mineral content, and a high frequency of osteoporosis and fractures [Citation14].
GH treatment could improve growth body composition, muscle strength, motor function and cognitive level [Citation15]. Early GH therapy has more favorable outcomes in body composition without increasing adverse effects. In addition, early treatment has been proven to improve learning and speech problems [Citation16–18]. Thus, early diagnosis and treatment are necessary for patients with PWS. The diagnosis of age has recently dropped significantly, with most cases diagnosed in the first year of life [Citation19]. However, misdiagnosis and missed diagnoses are still noted in some countries [Citation20]. This review aims to evaluate each method’s underlying mechanisms, advantages and disadvantages, and explore an effective diagnostic strategy for PWS.
2. Literature search
This review comprehensively assessed relevant studies from 1990 to 2022, sourced from PubMed, Web of Science, EBSCO, and Cochrane databases, using the keywords: ‘Prader-Willi syndrome’ OR ‘PWS’ AND [‘diagnosis’ OR ‘diagnose’ OR ‘genetic analysis’ OR ‘molecular analysis’].
3. Genetic typing
There are three main molecular classes in PWS. Approximately 70–75% of cases of PWS result from the deletion of paternal 15q11.2–13. The typical deletion is subdivided into two main subgroups, which involve a common distal breakpoint (BP3) and two proximal breakpoints (BP1, BP2) (). Longer type I, approximately 6Mb in size, extends from BP1 to BP3. While shorter type II, about 5.3MB in size, spans from BP2 to BP3 [Citation21,Citation22]. Patients with deletion more frequently present feeding problems, hypopigmentation, sleep disturbance and speech articulation defects () [Citation23]. Furthermore, patients with type I deletion present more behavior and cognitive problems than type II due to the loss of four genes from BP1 to BP2 [Citation24]. In addition, about 8% of the deletion subjects with PWS are caused by unique or atypical deletions (neither type I nor type II). A smaller or larger deletion could lead to a milder or more severe phenotype than the typical deletion [Citation25].
Table 1. Genotypes and phenotypes in PWS.
About 25–30% of cases of PWS arise from maternal uniparental disomy (mUPD) of chromosome 15 (mUPD15) [Citation26]. According to the pathogenetic mechanism, mUPD15 has three types: maternal heterodisomy, maternal isodisomy and segmental isodisomy [Citation27]. Compared with typical deletions, PWS with mUPD have higher verbal IQ scores and better social skills but are more prone to autism [Citation28]. However, hundreds of recessive genes are located on chromosome 15. If the mother carries some recessive disease genes on chromosome 15, individuals with mUPD15 have a higher risk of having other genetic diseases, including cardiac abnormalities, seizures, hearing loss, or metabolic defects [Citation27]. Furthermore, the remaining cases of PWS are mainly due to imprinting defects (IDs), accounting for about 1–3% [Citation29]. The most common cause of ID is epimutation, with a small recurrence risk [Citation30]. However, about 15% of IDs result from microdeletion in the imprinting center (IC), which is regulated by epigenetic modification and located at gene SNRPN and promoter. The recurrence risk in this situation is 50% [Citation31,Citation32]. The clinical features in ID are similar to mUPD15 [Citation28].
Chromosome 15 rearrangements (translocation or inversions) are also reported in some sporadic cases [Citation33]. In addition, about 1% of PWS deletion patients are caused by unbalanced chromosome 15 rearrangements, which could lead to a 50% recurrence risk when the rearrangement is paternally inherited [Citation21].
4. Clinical diagnosis
Consensus clinical diagnostic criteria for PWS were established in 1993 before the availability of genetic diagnosis, as shown in [Citation34]. However, with the availability of genetic tests, the purpose of clinical diagnosis has gradually changed into raising diagnostic suspicion. Thus, Meral et al. proposed a revised criterion () with a lower threshold to prompt DNA testing for PWS [Citation11]. However, genetic methods are still necessary to diagnose PWS.
Table 2. Consensus clinical diagnostic criteria for PWS.
Table 3. Revised criteria to prompt DNA testing for PWS.
5. Cytogenetic diagnosis
5.1. High resolution cytogenetics
Before the availability of genetic tests, high resolution cytogenetics provided the first laboratory diagnostic test for PWS. However, it is less used nowadays. Deletions of paternal 15q11.2–13 were reported in about 60% of patients with PWS with high resolution cytogenetic techniques. In addition, about 3–5% of patients showed other chromosomal abnormalities involving chromosome 15. However, one-third of patients with PWS presented a normal karyotype in high resolution cytogenetics due to submicroscopic deletions and mUPD [Citation35,Citation36].
5.2. Fluorescence in-situ hybridization (FISH)
The fluorescence in-situ hybridization (FISH) technique is used to detect the absence of the PWS region on chromosome 15. It is based on the hybridization of single-stranded DNA to a complementary sequence of the PWS region. It can be performed on cells after routine cytogenetic analyses or fresh preparations. The probe labeled with fluorochrome hybridizes to the complementary site. Thus, a colored signal at the hybridization site can be visualized by fluorescence microscopy [Citation37]. For unaffected individuals, signals show on both chromosomes 15, while in PWS patients with deletions, the signal shows only on one chromosome 15 [Citation38]. In addition, FISH can distinguish type I from type II deletion with appropriate probes [Citation39,Citation40]. However, it could not be performed in a high throughput manner and fails to detect UPD or differentiate between PWS and AS deletions.
5.3. Chromosomal microarray analysis (CMA)
Chromosomal microarray analysis (CMA) detects microdeletions and microduplications effectively, which can distinguish changes as small as 100 to 200 kb [Citation41]. It has two main techniques: comparative genomic hybridization (CGH) and single nucleotide polymorphisms (SNP). The array CGH (aCGH) compares the patient’s DNA sample to the reference sample to identify copy number changes. The fragment of the patient’s and control DNA samples, labeled with distinct fluorescent colors such as green and red, are mixed in equal proportions and hybridized to the probe on the array. By digital imaging software, the fluorescence intensity of every probe would be measured. Thus, deletion or duplication will be differentiated by the ratio of the fluorescence intensities. In addition, the resolution of aCGH depends on the number, type, and distribution of probes [Citation42].
In the SNP array, the patient’s sample is labeled and hybridized into the probe, which is selected from the known DNA locations distributed across the human genome. The copy number changes and single nucleotide polymorphism will be detected by comparing the absolute fluorescence probe intensities to the normal control. In addition, the SNP array can detect long contiguous stretches of homozygosity (LCSH) to distinguish UPD [Citation43].
CMA is proficient in detecting deletion size and additional chromosomal anomalies. However, balanced chromosomal rearrangements (such as balanced translocations or inversions) cannot be identified [Citation44].
6. Molecular genetic diagnosis
6.1. Southern blotting
In Southern blot methylation analysis, it is necessary to choose probes to assess the differential methylation status of the gene SNRPN rather than any other locus. The DNA sample was digested with restriction endonucleases, followed by gel electrophoresis for separation. Subsequently, the mixture would be transferred to the porous membrane. Specific sequences would be detected by hybridizing with labeled probes [Citation45]. Thus, Southern blot analysis can identify large deletions and UPD for PWS and AS. However, the site of restriction endonucleases could be affected by a rare restriction fragment length polymorphism, which leads to false positive results [Citation4]. In addition, it is time-consuming and gradually replaced by polymerase chain reaction (PCR).
6.2. Polymerase chain reaction
6.2.1. Reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR is a sensitive method to detect low-abundance RNA and analyze gene expression. Rachel and Francke used RT-PCR to test the expression of the SNRPN gene to diagnose PWS. The SNRPN expression could be detected in unaffected people while negative in whatever forms of patients with PWS [Citation46]. However, Muralidhar et al. reported weak SNRPN expression in two PWS subjects, and no noticeable clinical features were identified between the weak and negative gene expression [Citation47]. Except for the SNRPN gene, many RNA transcripts exist in the PWS region. Nevertheless, unreliable RNA extraction and unstable RNA limit its application.
6.2.2. PCR after restriction endonuclease digestion
Based on differential digestion of expressed SNRPN sequences, Chotai and Payne developed a novel PCR test to diagnose PWS and AS. The methylation requiring nuclease McrBC digests methylated sequences. While the methylation-sensitive endonuclease NotI digests unmethylated DNA. Following treatment with either NotI or McrBC, the DNA samples are amplified by PCR. After McrBC digestion, the SNRPN sequence is absent in patients with PWS. Conversely, the SNRPN sequence is absent in AS patients after NotI digestion. The potential advantages of this method are rapid, simple and cost-effective. However, a false negative is the most critical problem in this method because of partial digests. In addition, this method misses PWS or AS patients with chromosomal abnormalities and fails to distinguish between deletions, UPD or ID [Citation48].
6.2.3. Methylation-sensitive polymerase chain reaction (MS-PCR)
MS-PCR is considered a rapid and analytically sensitive technique. By treating DNA with sodium bisulfite, unmethylated cytosine converts to uracil, while methylated cytosine remains non-reactive. Thus, the differential methylation status of gene SNRPN is converted into sequence differences, followed by amplification with primers specific for methylated and unmethylated DNA in PCR reactions [Citation49]. Zeschnigk et al. developed a method with one common primer binding to parental alleles and two different primers specifically binding to paternal or maternal alleles [Citation50]. Compared with Southern blotting, this approach is economical and avoids restriction enzymes. It detects deletions, UPD and ID and correctly detects more than 99% of suspected cases, although it does not provide more information on molecular mechanisms, and further molecular genetic tests are necessary to distinguish the underlying molecular cause. However, Morandi et al. reported that a mosaic mUPD 15 girl with incomplete PWS presented a negative result of MS-PCR [Citation51]. Thus, Baumer et al. used MS-PCR followed by denaturing high performance liquid chromatography (DHPLC) to detect low cell mosaicisms [Citation52]. Due to single base variants or small deletions, DNA sequence analysis is needed for negative MS-PCR results. In addition, allelic dropout due to the rare presence of polymorphisms could cause a false-positive result, while the competition for reagents contributes to false-negative results [Citation53,Citation54].
6.3. Pyrosequencing
Pyrosequencing technology is a robust, high-throughput sequencing method that analyzes short- to medium-length DNA sequences. The sequence of methylated and unmethylated alleles is different after treating with bisulfite. Pyrosequencing distinguishes sequence differences by detecting the signal of pyrophosphate release, which was produced by incorporating nucleotides into the template strand [Citation55,Citation56]. Thus, White et al. used pyrosequencing assays to quantify CpG islands within the SNRPN gene to identify PWS and AS. This method could be used individually and in combination to diagnose PWS and AS. In addition, it could also detect cases of mosaicism, and the cost and time are similar to the MS-PCR [Citation57]. However, absolute quantification is affected by DNA concentration, PCR amplification bias, and bisulfite treatment.
6.4. Melting curve analysis
The melting curve analysis is a powerful method based on PCR. The difference in nucleic acid melting temperature (Tm) highly depends on the sequence variants. Thus, Worm et al. first described the methylation-specific melting curve analysis (MS-MA) that integrates bisulfite-treated DNA amplification and melting analysis to detect PWS and AS [Citation58]. Bisulfite-treated unmethylated cytosine is converted to uracil, decreasing the stability of the heteroduplex structure and Tm. Methylated alleles had high Tm than the unmethylated alleles. Therefore, characteristic melting curves of PWS and AS are acquired. Compared with normal individuals with two marked peaks, PWS individuals only show a single peak at the higher melting temperature (corresponding to the maternal allele) [Citation59,Citation60].
To detect the deletion and non-deletion genotype, Wang et al. used the LIS1 gene as a reference gene for PWS melting curve analysis. The LIS1 gene is located on chromosome 17p13.3. The copy number changes of LIS1 are rare, and the clinical phenotypes are distinct from PWS. Thus, deletion or non-deletion PWS could be identified by comparing relative peak height ratios of maternal SNRPN: LIS1. The non-deletion patients with PWS had a higher ratio than the deletion patients with PWS. However, peak height ratio variants were observed within each genotype in the replicate assay run, and positive control samples are recommended to be set for each assay run [Citation61]. In addition, Hung et al. used real-time PCR with melting curve analysis to distinguish between deletion and non-deletion PWS individuals [Citation59].
In MS-MA, DNA binding dye SYBR Green is unsaturated and inhibits PCR amplification. While EvaGreen is a saturated concentration dye and can be used at much higher dye concentrations without inhibiting PCR amplification. Thus, White et al. established a methylation-sensitive high-resolution melting-curve analysis (MS-HRM) with EvaGreen to distinguish PWS, which detected a lower abundance of methylated DNA. In MS-HRM, mutation or mosaicism may cause unusual melting curve shapes for PWS and AS samples. However, one PWS sample with a variant melting curve was not detected as mosaicism or mutation [Citation62,Citation63]. Thus, samples that cannot be unambiguously assigned to three diagnostic categories should be further investigated.
HRM is a nondestructive test that other techniques can subsequently analyze. In addition, as a post-PCR technique, HRM could decrease the risk of PCR contamination and dispense electrophoresis gel analysis [Citation64].
6.5. Methylation sensitive multiplex ligation-dependent probe amplification (MS-MLPA)
MS-MLPA could simultaneously detect methylation status and copy number changes of the 15q11-q13 region. The reactions were divided into two parts. One is treated with probe ligation and HhaI endonuclease for methylation detection, and the other is only treated with probe ligation for copy number changes [Citation65]. The HhaI enzyme only digests paternal unmethylated genomic DNA, and no PCR product will be generated. In contrast, the methylated sequences will prevent the digestion and amplify in the subsequent PCR. In addition, additional methylation-sensitive control probes outside chromosome 15 are needed to ensure complete digestion [Citation60]. Therefore, the methylation status and copy number changes could be detected by comparing probe-height ratios. In unaffected individuals, the probe-height ratio in Hhal digested samples is half that in undigested samples (0.5/1). However, PWS samples with typical deletions show a half reduction for probes within the 15q11-q13 region, whether digested or not (0.5/0.5). For type I deletions, the number of probes decreases in the region from BP1 to BP3. While for type II deletions, the copy number is normal from BP1 to BP2 [Citation66]. Patients with PWS with UPD have two methylated allies. Thus, the number of probes in undigested samples is similar to that in digested samples (1/1). Furthermore, IC microdeletion can also be detected with appropriate probes [Citation67]. If probes could not detect the microdeletion in IC, however, MS-MLPA will not distinguish UPD from ID, and further microsatellite analysis is necessary to perform. In addition, single-base variations in the probe-binding regions or the restriction enzyme recognition site would lead to false positive or negative results [Citation60].
6.6. Microsatellite analysis (MSA)
Microsatellites, also named short tandem repeats (STR), are short, highly polymorphous and tandemly repeated simple sequences ranging in size from 1 to 6 base pairs. With the occurrence of PCR, the high sensitive microsatellite analysis has been widely used in discovering genes and diagnosing diseases, which is fast and requires less DNA [Citation68]. STR markers are located in the typical PWS and AS deletion region and the distal region near the telomere. Microsatellite loci are identified by primers labeled with fluorescence and amplified with PCR. However, the proband and parental DNA samples are all required, which is impractical in some cases [Citation69,Citation70]. In addition, an interpretation of the result should build on more than one informative marker. Microsatellite analysis is a time-costuming method that could distinguish between UPD heterodisomy and deletion. Nevertheless, it failed to identify deletion and UPD isodisomy.
6.7. RNA
Due to the unreliable and unstable extracted RNA, it is uncommonly used for PWS diagnosis. However, Yin et al. discovered long non-coding RNAs (lncRNAs) with small nuclear RNAs (snoRNAs) ends and named snoRNA-related lncRNAs (sno-lncRNAs), which were processed from intron. Moreover, the PWS critical region encodes five classes of sno-lncRNAs, which are highly expressed in unaffected individuals while not expressed in patients with PWS [Citation71]. Depletion of PWS region sno-lncRNAs not changes gene expression but leads to altered alternative splicing of several hundred mRNAs [Citation72]. In addition, sno-lncRNAs are reliable and have slow degradation compared to other RNAs. These findings suggest that measuring the expression of sno-lncRNAs in the blood is a promising tool for diagnosing PWS.
7. Prenatal screening
Prenatal screening for PWS is a promising prospect, which may lead to early treatment with improved quality of life. In addition, it is essential for those PWS families with high recurrence risks.
Prenatal characteristics associated with PWS include abnormal fetal growth, polyhydramnios, and notably decreased fetal movements [Citation73,Citation74]. While these features are nonspecific, the simultaneous presence of abnormal fetal growth alongside decreased fetal movements or polyhydramnios should raise high suspicions for PWS.
Chorionic villus sampling and amniocentesis are the most common sources of genetic testing in prenatal diagnosis. However, these samples are more hypomethylated than other tissue, which makes methylation analysis difficult in PWS prenatal screening. Glenn et al. found that the SNRPN gene maintains the imprint throughout a wide range of tissue (including chorionic villus and amniocentesis) and is suitable for methylation analysis [Citation75].
Noninvasive prenatal testing (NIPT) could detect chromosome aneuploidies with high sensitivity and specificity, which utilizes cell-fetal DNA from maternal plasma or serum and is safe for the fetus. Wapner et al. utilized the SNP-based NIPT to detect five fetal microdeletion syndromes (including PWS and AS) [Citation76]. However, a positive predictive value is expected to be low because of these rare diseases, and the application of NIPT is required to be further studied before it is widely adopted [Citation77]. In addition, preimplantation genetic testing (PGT) could be used in some families with IC microdeletions.
If trisomy 15 or trisomy 15 mosaicism is detected, PWS with maternal UPD or AS with paternal UPD should be alerted due to the trisomic rescue event in early pregnancy. In this instance, DNA methylation or CMA should be highly considered [Citation78]. If 15q12 missing was detected, FISH, CMA, or MS-MLPA should be considered due to PWS/AS critical deletion [Citation79].
8. Newborn screening
Whole blood is the most typical DNA source in many methods for detecting PWS and AS. However, it is difficult for whole blood to massively screen newborns. Dried blood spot (DBS), a part of newborn screening programs in many countries, is simple to collect and transport [Citation80]. Thus, Mahmoud et al. used MS-PCR and MS-MLPA to diagnose PWS on DBS. This pilot study showed that PWS could be correctly identified once high-quality DNA was extracted successfully. However, some samples extracted low DNA concentration and did not meet the requirements for methylation analysis. Although, these samples have a long storage time of 8–10 years, which may be affected by inappropriate storage conditions and bacteria containment [Citation81]. In addition, Ferreira et al. used MS-HRM to screen PWS on DBS and assessed three different DNA isolation methods from DBS (Qiagen-DBS, Mem-DBS, and Chellex-DBS). In MS-HIRM, none of the isolation methods significantly changed the melting temperature curve. Mem-DBS and Chellex-DBS methods provided high DNA concentration, while the Qiagen-DBS method showed high DNA purity and quality, which benefits high amplification in MS-HRM [Citation82]. In addition, Godlier et al. used methylation-specific quantitative melt analysis (MS-QMA) to screen chromosome 15 imprinting disorders in DBS [Citation83]. The MS-QMA combines HRM and real-time PCR (RT-PCR) to provide quantification of DNA methylation [Citation84]. Thus, it is an emerging field for PWS newborn screening.
9. Conclusion
Early diagnosis and treatment of PWS could effectively mitigate obesity and related co-morbidities, significantly improving life quality for patients with PWS and their families. In this review, we concluded the advantages and disadvantages of each method, as shown in . Furthermore, prenatal and newborn screening could potentially improve diagnostic age [Citation85,Citation86]. In addition, the discovery of sno-lncRNAs offers a novel approach for screening or diagnosing PWS due to their stability and slow degradation. Combining sno-lncRNA with neonatal screening is a promising prospect to be further studied.
Table 4. Advantages and limitations of PWS diagnosis.
10. Expert opinion
Recently, the age of diagnosis for PWS has significantly decreased, with most cases diagnosed within the first year of life, leading to early human growth hormone treatment and management for PWS. However, there are four keys to be considered in PWS clinical and laboratory diagnosis. Firstly, it is essential to distinguish the relatively few patients with PWS from the vast majority of laboratory referrals that present with similar symptoms, both at birth and later in life. Secondly, after diagnosing PWS, it is necessary to identify those few patients with a high recurrence risk (IC deletion) from most patients with a low recurrence risk (deletions and UPD). Thirdly, the testing process should be as ‘user-friendly’ as possible. It is notoriously difficult to obtain blood or tissues from probands and their families, and family members are not always available for additional genetic testing. Fourthly, the testing cost and time-consuming should be recognized. The fewer tests performed, the more cost-effective and time-efficient the diagnosis becomes [Citation40].
Therefore, we present a genetic testing workflow for PWS diagnosis (). The initial requirement is to identify or recognize suspected patients. Classic clinical features of abnormal growth, polyhydramnios, decreased fetal movements, hypotonia with poor suck, excessive eating with obesity, and global developmental delay could help to identify suspected patients. When patients are suspected to be PWS, starting with MS-MLPA is the most efficient way. This method could detect more than 99% of patients with PWS and exclude AS immediately. MS-MLPA could detect deletion (including large deletion and IC microdeletion), providing a definitive diagnosis for about 70%-75% of patients with PWS. If IC microdeletion is confirmed, deletion analysis should be performed in families (especially for the father) to rule out familiar IC deletion.
Figure 2. Genetic testing strategies for the Prader-Willi syndrome.
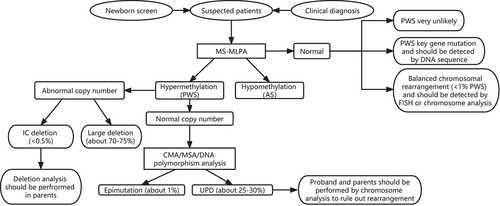
If the result shows a normal copy number with abnormal methylation status, it may indicate UPD or ID. Then CMA, MSA, or DNA polymorphisms should be performed to distinguish UPD and ID. If PWS with mUPD is confirmed, the chromosome analysis should be performed both in probands and parents to rule out cytogenetic rearrangement. However, this instance is rare. If the ID is performed, the recurrence risk is small due to ID epimutation.
If the result of methylation analysis is normal, PWS is very unlikely. However, methylation analysis could not detect balanced chromosomal rearrangement and key gene mutation in PWS critical region (<1% PWS), which should be detected by chromosome analysis and DNA sequence [Citation20].
In addition, PWS screening is increasingly being recognized as a promising prospect for further study. In prenatal screening, prenatal features are critical for clinicians to conduct appropriate diagnostic analysis as soon as possible. In addition, there is a growing interest in expanding NIPT due to its safety in prenatal screening. However, NIPT is more demanding and requires further research before it is widely adopted. In newborn screening, the combination of methylation analysis and DBS gradually becomes an emerging field due to its convenience for sample collection and transportation. Pilot studies on neonatal screening could be carried out in countries with large populations first.
Due to the reliance and slow degradation of sno-lncRNAs, measuring the expression of sno-lncRNA in blood presents a novel tool for screening or diagnosis of PWS. However, the method of extracting sno-lncRNAs is technically demanding, requiring further research before it can be widely implemented.
Article highlights
The importance of early diagnosis and treatment for Prader-Willi Syndrome.
Advantages and disadvantages of each method.
Genetic testing strategies for Prader-Willi syndrome.
Prenatal and newborn screening is a promising prospect to be further studied.
Measuring the expression of sno-lncRNAs in the blood is a novel tool to screen or diagnose PWS
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or material discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or mending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Authors’ contributions
CCZ conceptualized and designed the study and reviewed and revised the manuscript. YG drafted the initial manuscript. MLZ, YLD, and YHJ collected and reviewed the information about PWS diagnosis. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Additional information
Funding
References
- Butler MG, Miller JL, Forster JL. Prader-Willi syndrome - clinical genetics, diagnosis and treatment approaches: an update. Curr Pediatr Rev. 2019;15(4):207–244. doi: 10.2174/1573396315666190716120925
- Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015 Dec;38(12):1249–1263. doi: 10.1007/s40618-015-0312-9
- Beygo J, Buiting K, Ramsden SC, et al. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. 2019 Sep;27(9):1326–1340. doi: 10.1038/s41431-019-0435-0
- Glenn CC, Saitoh S, Jong MT, et al. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996 Feb;58(2):335–346.
- Cheon CK. Genetics of Prader-Willi syndrome and Prader-Will-like syndrome. J Pediatr Endocrinol Metab. 2016 Sep;21(3):126–135. doi: 10.6065/apem.2016.21.3.126
- Yang L, Shu X, Mao S, et al. Genotype–Phenotype Correlations in Angelman Syndrome. Genes. 2021;12(7):987. doi: 10.3390/genes12070987
- Buiting K, Cassidy SB, Driscoll DJ, et al. Clinical utility gene card for: prader-Willi syndrome. Eur J Hum Genet. 2014 Sep;22(9):1153–1153. doi: 10.1038/ejhg.2014.66
- Tauber M, Hoybye C. Endocrine disorders in Prader-Willi syndrome: a model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021 Apr;9(4):235–246. doi: 10.1016/S2213-8587(21)00002-4
- Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011 May;155a(5):1040–1049. doi: 10.1002/ajmg.a.33951
- Yang L, Zhou Q, Ma B, et al. Perinatal features of Prader-Willi syndrome: a Chinese cohort of 134 patients. Orphanet J Rare Diseases. 2020 Jan 21;15(1):24. doi: 10.1186/s13023-020-1306-z
- Gunay-Aygun M, Schwartz S, Heeger S, et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001 Nov;108(5):E92. doi: 10.1542/peds.108.5.e92
- Grugni G, Sartorio A, Crinò A. Growth hormone therapy for Prader-Willi syndrome: challenges and solutions. Ther Clin Risk Manag. 2016;12:873–881. doi: 10.2147/TCRM.S70068
- Bridges N. What is the value of growth hormone therapy in Prader Willi syndrome? Archives of disease in childhood. Arch Dischildhood. 2014 Feb;99(2):166–170. doi: 10.1136/archdischild-2013-303760
- Brunetti G, Grugni G, Piacente L, et al. Analysis of circulating mediators of bone remodeling in Prader–Willi syndrome. Calcif Tissue Int. 2018 Jun;102(6):635–643. doi: 10.1007/s00223-017-0376-y
- Cheng R. Early recombinant human growth hormone treatment improves mental development and alleviates deterioration of motor function in infants and young children with Prader–Willi syndrome. World J Pediatr. 2023;19(5):438–449. doi: 10.1007/s12519-022-00653-y
- Kimonis VE, Tamura R, Gold JA, et al. Early diagnosis in Prader–Willi syndrome reduces obesity and associated co-morbidities. Genes. 2019 Nov 6;10(11):898. doi: 10.3390/genes10110898
- Bakker NE, Siemensma EP, van Rijn M, et al. Beneficial effect of growth hormone treatment on health-related quality of life in children with Prader-Willi syndrome: a randomized controlled trial and longitudinal study. Hormone Res Paediatrics. 2015;84(4):231–239. doi: 10.1159/000437141
- Lecka-Ambroziak A, Wysocka-Mincewicz M, Doleżal-Ołtarzewska K, et al. Effects of recombinant human growth hormone treatment, depending on the therapy start in different nutritional phases in paediatric patients with Prader–Willi Syndrome: a Polish multicentre study. JCM. 2021 Jul 19;10(14):3176. doi: 10.3390/jcm10143176
- Bacheré N, Diene G, Delagnes V, et al. Early diagnosis and multidisciplinary care reduce the hospitalization time and duration of tube feeding and prevent early obesity in PWS infants. Hormone Res. 2008;69(1):45–52. doi: 10.1159/000111795
- Yang-Li D, Fei-Hong L, Hui-Wen Z, et al. Recommendations for the diagnosis and management of childhood Prader-Willi syndrome in China. Orphanet J Rare Diseases. 2022 Jun 13;17(1):221. doi: 10.1186/s13023-022-02302-z
- Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med. 2012 Jan;14(1):10–26. doi: 10.1038/gim.0b013e31822bead0
- Bittel DC, Butler MG. Prader–Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005 Jul 25;7(14):1–20.
- Torrado M, Araoz V, Baialardo E, et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A. 2007 Mar 1;143(5):460–468. doi: 10.1002/ajmg.a.31520
- Kim Y, Wang SE, Jiang YH. Epigenetic therapy of Prader-Willi syndrome. Transl Res. 2019 Jun;208:105–118. doi: 10.1016/j.trsl.2019.02.012
- Kim SJ, Miller JL, Kuipers PJ, et al. Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012 Mar;20(3):283–290. doi: 10.1038/ejhg.2011.187
- Butler MG, Hartin SN, Hossain WA, et al. Molecular genetic classification in Prader-Willi syndrome: a multisite cohort study. J Med Genet. 2019 Mar;56(3):149–153. doi: 10.1136/jmedgenet-2018-105301
- Alves C, Franco RR. Prader-Willi syndrome: endocrine manifestations and management. Arch Endocrinol Metab. 2020 May;64(3):223–234. doi: 10.20945/2359-3997000000248
- Roof E, Stone W, MacLean W, et al. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. 2000 Feb;44(1):25–30. doi: 10.1046/j.1365-2788.2000.00250.x
- Whittington JE, Butler JV, Holland AJ. Changing rates of genetic subtypes of Prader–Willi syndrome in the UK. Eur J Hum Genet. 2007 Jan;15(1):127–130. doi: 10.1038/sj.ejhg.5201716
- Glenn CC, Driscoll DJ, Yang TP, et al. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod. 1997 Apr;3(4):321–332. doi: 10.1093/molehr/3.4.321
- Ohta T, Gray TA, Rogan PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet. 1999 Feb;64(2):397–413. doi: 10.1086/302233
- Ming JE, Blagowidow N, Knoll JH, et al. Submicroscopic deletion in cousins with Prader-Willi syndrome causes a grandmatrilineal inheritance pattern: effects of imprinting. Am J Med Genet A. 2000 May 1;92(1):19–24.
- Casamassima AC, Shapiro LR, Wilmot PL, et al. Prader-Willi syndrome and Robertsonian translocations involving chromosome 15. Clin Genet. 1991 Apr;39(4):294–297. doi: 10.1111/j.1399-0004.1991.tb03028.x
- Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993 Feb;91(2):398–402. doi: 10.1542/peds.91.2.398
- Smith A, Prasad M, Deng ZM, et al. Comparison of high resolution cytogenetics, fluorescence in situ hybridisation, and DNA studies to validate the diagnosis of Prader-Willi and Angelman’s syndromes. Arch Dischildhood. 1995 May;72(5):397–402. doi: 10.1136/adc.72.5.397
- Borelina D, Engel N, Esperante S, et al. Combined cytogenetic and molecular analyses for the diagnosis of Prader-Willi/Angelman syndromes. J Biochem Mol Biol. 2004 Sep 30;37(5):522–526. doi: 10.5483/BMBRep.2004.37.5.522
- Gozzetti A, Le Beau MM. Fluorescence in situ hybridization: uses and limitations. Semin Hematol. 2000 Oct;37(4):320–333. doi: 10.1016/S0037-1963(00)90013-1
- Schad CR, Jalal SM, Thibodeau SN. Genetic testing for Prader-Willi and Angelman syndromes. Mayo Clin Proc. 1995 Dec;70(12):1195–1196. doi: 10.4065/70.12.1195
- Sukarova-Angelovska E, Piperkova K, Sredovska A, et al. Implementation of fluorescent in situ hybridization (FISH) as a method for detecting microdeletion syndromes - our first experiences. Prilozi. 2007 Dec;28(2):87–98.
- Smith A, Hung D. The dilemma of diagnostic testing for Prader-Willi syndrome. Transl Pediatr. 2017 Jan;5(1):46–56. doi: 10.21037/tp.2016.07.04
- Van den Veyver IB, Beaudet AL. Comparative genomic hybridization and prenatal diagnosis. Current Opinion In Obstetrics & Gynecology. 2006 Apr;18(2):185–191. doi: 10.1097/01.gco.0000192986.22718.cc
- Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Sterility. 2018 Feb;109(2):201–212. doi: 10.1016/j.fertnstert.2018.01.005
- Stosic M, Levy B, Wapner R. The use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol Clin North Am. 2018 Mar;45(1):55–68. doi: 10.1016/j.ogc.2017.10.002
- Lo JO, Shaffer BL, Feist CD, et al. Chromosomal microarray analysis and prenatal diagnosis. Obstet Gynecol Surv. 2014 Oct;69(10):613–621. doi: 10.1097/OGX.0000000000000119
- Southern E. Southern blotting. Nat Protoc. 2006;1(2):518–525. doi: 10.1038/nprot.2006.73
- Wevrick R, Francke U. Diagnostic test for the Prader-Willi syndrome by SNRPN expression in blood. Lancet (London, England). 1996 Oct 19;348(9034):1068–1069.
- Muralidhar B, Marney A, Butler MG. Analysis of imprinted genes in subjects with Prader-Willi syndrome and chromosome 15 abnormalities. Genet Med. 1999 May;1(4):141–145. doi: 10.1097/00125817-199905000-00005
- Chotai KA, Payne SJ. A rapid, PCR based test for differential molecular diagnosis of Prader-Willi and Angelman syndromes. J Med Genet. 1998 Jun;35(6):472–475. doi: 10.1136/jmg.35.6.472
- Herman JG, Graff JR, Myöhänen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996 Sep 3;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821
- Zeschnigk M, Lich C, Buiting K, et al. A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet. 1997 Mar;5(2):94–98. doi: 10.1159/000484740
- Morandi A, Bonnefond A, Lobbens S, et al. A girl with incomplete Prader-Willi syndrome and negative MS-PCR, found to have mosaic maternal UPD-15 at SNP array. Am J Med Genet A. 2015 Nov;167a(11):2720–2726. doi: 10.1002/ajmg.a.37222
- Baumer A, Wiedemann U, Hergersberg M, et al. A novel MSP/DHPLC method for the investigation of the methylation status of imprinted genes enables the molecular detection of low cell mosaicisms. Human Mutation. 2001 May;17(5):423–430. doi: 10.1002/humu.1118
- Hussain Askree S, Hjelm LN, Ali Pervaiz M, et al. Allelic dropout can cause false-positive results for Prader-Willi and Angelman syndrome testing. J Mol Diagn. 2011 Jan;13(1):108–112. doi: 10.1016/j.jmoldx.2010.11.006
- Hamajima N, Saito T, Matsuo K, et al. Competitive amplification and unspecific amplification in polymerase chain reaction with confronting two-pair primers. J Mol Diagn. 2002 May;4(2):103–107. doi: 10.1016/S1525-1578(10)60688-5
- Ronaghi M, Uhlén M, Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998 Jul 17;281(5375):363, 365.
- Dupont JM, Tost J, Jammes H, et al. De Novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004 Oct 1;333(1):119–127. doi: 10.1016/j.ab.2004.05.007
- White HE, Durston VJ, Harvey JF, et al. Quantitative analysis of SNRPN(correction of SRNPN) gene methylation by pyrosequencing as a diagnostic test for Prader-Willi syndrome and Angelman syndrome. Clin Chem. 2006 Jun;52(6):1005–1013. doi: 10.1373/clinchem.2005.065086
- Worm J, Aggerholm A, Guldberg P. In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem. 2001;47(7):1183–1189. doi: 10.1093/clinchem/47.7.1183
- Hung CC, Lin SY, Lin SP, et al. Quantitative and qualitative analyses of the SNRPN gene using real-time PCR with melting curve analysis. J Mol Diagn. 2011 Nov;13(6):609–613. doi: 10.1016/j.jmoldx.2011.06.005
- Procter M, Chou LS, Tang W, et al. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006 Jul;52(7):1276–1283. doi: 10.1373/clinchem.2006.067603
- Wang W, Law HY, Chong SS. Detection and discrimination between deletional and non-deletional Prader-Willi and Angelman syndromes by methylation-specific PCR and quantitative melting curve analysis. J Mol Diagn. 2009 Sep;11(5):446–449. doi: 10.2353/jmoldx.2009.090015
- White HE, Hall VJ, Cross NC. Methylation-sensitive high-resolution melting-curve analysis of the SNRPN gene as a diagnostic screen for Prader-Willi and Angelman syndromes. Clin Chem. 2007 Nov;53(11):1960–1962. doi: 10.1373/clinchem.2007.093351
- Dahl C, Guldberg P. High-resolution melting for accurate assessment of DNA methylation. Clin Chem. 2007 Nov;53(11):1877–1878. doi: 10.1373/clinchem.2007.094854
- Ribeiro Ferreira I, Darleans Dos Santos Cunha W, Henrique Ferreira Gomes L, et al. A rapid and accurate methylation-sensitive high-resolution melting analysis assay for the diagnosis of Prader Willi and Angelman patients. Mol Gene Genomic Med. 2019 Jun;7(6):e637. doi: 10.1002/mgg3.637
- Moelans CB, Atanesyan L, Savola SP, et al. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA). Methods in molecular biology. Vol. 1708, Clifton (NJ); 2018p. 537–549.
- Bittel DC, Kibiryeva N, Butler MG. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet Test. 2007;11(4):467–475. Winter. doi: 10.1089/gte.2007.0061
- Henkhaus RS, Kim SJ, Kimonis VE, et al. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers. 2012 Mar;16(3):178–186. doi: 10.1089/gtmb.2011.0115
- Schlötterer C. Evolutionary dynamics of microsatellite DNA. Chromosoma. 2000 Sep;109(6):365–371. doi: 10.1007/s004120000089
- Devriendt K, Matthijs G, Claes S, et al. Prader-Willi syndrome in a child with mosaic trisomy 15 and mosaic triplo-X: a molecular analysis. J Med Genet. 1997 Apr;34(4):318–322. doi: 10.1136/jmg.34.4.318
- Mao L. Microsatellite analysis. Applications and pitfalls. Ann N Y Acad Sci. 2000 Apr;906(1):55–62. doi: 10.1111/j.1749-6632.2000.tb06591.x
- Yin QF, Yang L, Zhang Y, et al. Long noncoding RNAs with snoRNA ends. Molecular Cell. 2012 Oct 26;48(2):219–230. doi: 10.1016/j.molcel.2012.07.033
- Xing YH, Chen LL. Processing and roles of snoRNA-ended long noncoding RNAs. Critical reviews in biochemistry and molecular biology. Crit Rev Biochem Mol Biol. 2018 Dec;53(6):596–606. doi: 10.1080/10409238.2018.1508411
- Bigi N, Faure JM, Coubes C, et al. Prader-Willi syndrome: is there a recognizable fetal phenotype? Prenat Diagn. 2008 Sep;28(9):796–799. doi: 10.1002/pd.1973
- Srebnik N, Gross Even-Zohar N, Salama A, et al. Recognizing the unique prenatal phenotype of Prader-Willi syndrome (PWS) indicates the need for a diagnostic methylation test. Prenat Diagn. 2020 Jun;40(7):878–884. doi: 10.1002/pd.5712
- Glenn CC, Deng G, Michaelis RC, et al. DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn. 2000 Apr;20(4):300–306. doi: 10.1002/(SICI)1097-0223(200004)20:4<300:AID-PD803>3.0.CO;2-A
- Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015 Mar;212(3):332.e1–9. doi: 10.1016/j.ajog.2014.11.041
- Vora NL, O’Brien BM. Noninvasive prenatal testing for microdeletion syndromes and expanded trisomies: proceed with caution. Obstet & Gynecol. 2014 May;123(5):1097–1099. doi: 10.1097/AOG.0000000000000237
- Butler MG. Benefits and limitations of prenatal screening for Prader-Willi syndrome. Prenat Diagn. 2017 Jan;37(1):81–94. doi: 10.1002/pd.4914
- Chang CW, Hsu HK, Kao CC, et al. Prenatal diagnosis of Prader-Willi syndrome and Angelman syndrome for fetuses with suspicious deletion of chromosomal region 15q11-q13. Int J Gynaecol Obstet Off Organ Int Fed Gynaecol Obstet. 2014 Apr;125(1):18–21. doi: 10.1016/j.ijgo.2013.09.028
- Hollegaard MV, Grauholm J, Nielsen R, et al. Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. Mol Gene Metabol. 2013 Sep;110(1–2):65–72. doi: 10.1016/j.ymgme.2013.06.004
- Mahmoud R, Singh P, Weiss L, et al. Newborn screening for Prader-Willi syndrome is feasible: early diagnosis for better outcomes. Am J Med Genet A. 2019 Jan;179(1):29–36. doi: 10.1002/ajmg.a.60681
- Ferreira IR, Costa RA, Gomes LHF, et al. A newborn screening pilot study using methylation-sensitive high resolution melting on dried blood spots to detect Prader-Willi and Angelman syndromes. Sci Rep. 2020 Aug 3;10(1):13026. doi: 10.1038/s41598-020-69750-0
- Godler DE, Ling L, Gamage D, et al. Feasibility of screening for chromosome 15 imprinting disorders in 16 579 newborns by using a novel genomic workflow. JAMA Netw Open. 2022 Jan 4;5(1):e2141911. doi: 10.1001/jamanetworkopen.2021.41911
- Inaba Y, Schwartz CE, Bui QM, et al. Early detection of fragile X syndrome: applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin Chem. 2014 Jul;60(7):963–973. doi: 10.1373/clinchem.2013.217331
- Xiao H, Zhang J, Dong X, et al. Secondary genomic findings in the 2020 China Neonatal Genomes Project participants. World J Pediatr. 2022;18(10):687–694. doi: 10.1007/s12519-022-00558-w
- Tong F. Application of next generation sequencing in the screening of monogenic diseases in China, 2021: a consensus among Chinese newborn screening experts. World J Pediatr. 2022;18(4):235–242. doi: 10.1007/s12519-022-00522-8