
Abstract
As a result of an increasing aging population, the number of individuals taking multiple medications simultaneously has grown considerably. For these individuals, taking multiple medications has increased the risk of undesirable drug–drug interactions (DDIs), which can cause serious and debilitating adverse drug reactions (ADRs). A comprehensive understanding of DDIs is needed to combat these deleterious outcomes. This review provides a synopsis of the pharmacokinetic (PK) and pharmacodynamic (PD) mechanisms that underlie DDIs. PK-mediated DDIs affect all aspects of drug disposition: absorption, distribution, metabolism and excretion (ADME). In this review, the cells that play a major role in ADME and have been investigated for DDIs are discussed. Key examples of drug metabolizing enzymes and drug transporters that are involved in DDIs and found in these cells are described. The effect of inhibiting or inducing these proteins through DDIs on the PK parameters is also reviewed. Despite most DDI studies being focused on the PK effects, DDIs through PD can also lead to significant and harmful effects. Therefore, this review outlines specific examples and describes the additive, synergistic and antagonistic mechanisms of PD-mediated DDIs. The effects DDIs on the maximum PD response (Emax) and the drug dose or concentration (EDEC50) that lead to 50% of Emax are also examined. Significant gaps in our understanding of DDIs remain, so innovative and emerging approaches are critical for overcoming them.
Introduction
The aging population in the USA is expected to rise in the foreseeable future and this group often deals with multiple health conditions.Citation1,Citation2 To treat these multiple health conditions, individuals within this group have been required to take two or more prescription drugs simultaneously. This has led to a significant increase in the number of Americans that need to take multiple medications.Citation3 For those over 65 years old, nearly half take more than five drugs simultaneously.Citation4 In many cases, these individuals take drugs that they do not need.Citation4 Ultimately, taking multiple medications simultaneously increases an individual’s risk for undesirable drug–drug interactions (DDIs) that lead to serious and debilitating adverse drug reactions (ADRs).Citation5–Citation7
A comprehensive understanding of DDIs is critical for safer coadministration of drugs and reduced risk of ADRs. In this review, a synopsis of the pharmacokinetic (PK) and pharmacodynamic (PD) mechanisms that underlie DDIs is provided. In this work, the drug that causes the DDI will be called the “perpetrator” drug, while the drug of interest will be called the “victim” drug.Citation8–Citation10 In PK, DDIs touch all aspects of drug disposition: absorption, distribution, metabolism and excretion (ADME). This review will discuss the cells that play a role in each aspect of ADME and have been the focus of DDI investigations. DDIs through PK occur through drug metabolizing enzymes and drug transporters that are found within these cells. This work provides representative DDI examples with a few key proteins and examines their effect on PK parameters. Although most current investigations have focused on how PK is affected by DDIs, significant DDIs have also been noted with PD. DDIs through PD can occur through additive, synergistic and antagonistic mechanisms. Several illustrative examples of DDIs from each of these mechanisms is provided in this work. Their effect on the maximum PD response (Emax) and the drug dose or concentration at 50% of Emax (EDEC50) is discussed.
Pharmacokinetics (PK)
PK is what the body does to a drug and it includes ADME.Citation11
Depending on the process of ADME that is affected, PK-mediated DDIs can lead to elevated free plasma concentrations of the “victim” drug that can cause undesirable ADRs and toxicity. They can also lead to depressed free plasma concentrations of the “victim” drug and reduced therapeutic efficacy. The major cells, drug-metabolizing enzymes and drug transporters that have been implicated in DDIs and are discussed in this work are shown in . The effect of DDIs on the PK is described below and summarized with additional detail in .
Figure 1 Drug-metabolizing enzymes and drug transporters in (A) an enterocyte, (B) a brain endothelial cell, (C) a placental trophoblast, (D) hepatocyte and biliary endothelial cells, and (E) a renal proximal tubule cell.
Abbreviations: BCRP, breast cancer resistance protein; CYP1A2, CYP/CYP450 1A2; CYP3A4, CYP/CYP450 3A4; MCT1, monocarboxylate transporter 1; OAT1, organic anionic transporter 1; OAT3, organic anionic transporter 3; OATP1A2, organic anionic transporting polypeptide 1A2; OCTs, organic cationic transporters; MRP4, multidrug resistance protein 4; Pgp, P-glycoprotein.
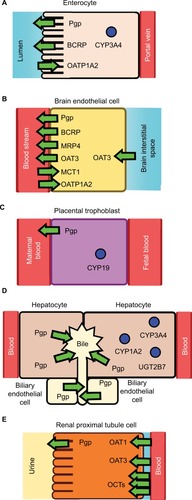
Table 1 Pharmacokinetically mediated DDIs
Absorption
Drug absorption can occur through both oral and extraoral routes such as through the skin.Citation11 Because oral drug administration is the preferred route for administration,Citation11 this review is focused on drug absorption through the gastrointestinal (GI) tract. The GI tract is composed of the mouth, esophagus, stomach, small intestines and the colon.Citation11 Of these anatomical structures, most of the drug absorption occurs in the small intestines as a result of its relatively large surface area.Citation11,Citation12 The large surface area is due in large part to cells that contain microvilli called enterocytes that line the small intestine.Citation11,Citation12 Drug absorption in these cells is controlled by passive diffusion across the plasma membrane and the presence of drug metabolizing enzymes and drug transporters ().Citation11
Drug transporters
There are many drug transporters on the apical (lumen-side) and basal (blood-side) membrane surfaces of the enterocytes ().Citation13 Two highly expressed drug efflux transporters on the apical side of the enterocytes are Pgp and the BCRP.Citation14 Pgp and BCRP are part of the ATP-Binding Cassette (ABC) transporter superfamily and function by effluxing drugs out from the cytosol to the intestinal lumen.Citation15,Citation16 Each of these transporters has distinct specificities with Pgp and BCRP being functional monomers and dimers, respectively.Citation15,Citation16 Also found on the apical side of enterocytes is the organic anionic transporting polypeptide 1A2 (OATP1A2), which is a drug influx transporter.Citation17–Citation19 In contrast to Pgp and BCRP, this transporter influxes a diverse range of ligands including fexofenadine and imatinib from the intestinal lumen to the cytosol of the enterocyte.Citation17–Citation19
Inhibition and induction of the transporters from DDIs within the enterocyte can lead to significant effects on drug absorption. In a randomized crossover clinical study, the Pgp inhibitor and antibiotic erythromycin was coadministered with the cardiovascular drug and Pgp substrate talinolol.Citation20 Erythromycin inhibition of Pgp reduced the secretion of talinolol into the intestinal lumen and increased its oral bioavailability.Citation20 In contrast, induction of Pgp by the antibiotic rifampicin in another clinic study with talinolol found that it decreased the oral bioavailability.Citation21 Because BCRP is an efflux transporter like Pgp, one might expect the oral bioavailability of drugs to increase for substrates, when BCRP is inhibited. Indeed, inhibition of the BCRP by the tyrosine kinase inhibitor fostamatinib did increase the oral bioavailability of rosuvastatin in addition to doubling the peak serum concentration (Cmax) and area under the curve (AUC).Citation22,Citation23 In contrast to Pgp and BCRP, the influx transporter OATP1A2 moves drugs in the opposite direction in the enterocyte.Citation17–Citation19 Selective inhibition of the OATP1A2 by naringin from grapefruit juice leads to significant decreases in the AUC, Cmax and the oral bioavailability of the antihistamine fexofenadine.Citation24
Intestinal cytochromes P450 (CYPs)
The smooth endoplasmic reticulum of enterocytes contains drug metabolizing monooxygenases, which are also found in high concentrations in the liver, called cytochromes P450 (CYPs).Citation25 These enzymes are highly promiscuous and have several distinct isoforms.Citation26 The enterocytes contain mostly the CYP3A (82%) subfamily enzymes with some the CYP2C9 (14%) isoform and minor concentrations of other CYPs.Citation27,Citation28
The expression level of CYP3A enzymes was found to vary across the small intestine with the highest levels in the duodenum.Citation29,Citation30 These enzymes are responsible for metabolism of a significant portion of commercially available drugs and have exhibited a significant amount of DDIs.Citation31 However, determining specific DDIs with CYP3A enzymes is complicated by the fact that they are also present in the liver.Citation27,Citation28,Citation32 However, a clinical study found that inhibition of intestinal CYP3A4 by the antifungal drug itraconazole and its metabolites increased the gut oral bioavailability (Fg) of the sedative midazolam.Citation33 In another clinical study, inhibition of CYP3A4 by the macrolide antibiotic troleandomycin led to large increases in the AUC and increased gut oral bioavailability of the opioid drug alfentanil.Citation34 The opposite effect occurred with the inducer rifampicin, which reduced both the AUC and the gut oral bioavailability.Citation34
Distribution
Drug distribution involves the transfer of drugs from the systemic circulation to the rest of the body. All tissues in the body facilitate the distribution of drugs to varying degrees.Citation35 Distribution can occur by passive diffusion and membrane protein-mediated transport.Citation35 The membrane barriers that provide the largest roles in drug distribution are the blood– brain barrier (BBB), blood–testis barrier (BTB), blood–cerebrospinal fluid barrier (BCSFB) and the placental barrier.Citation35 DDIs from drug distribution within these membrane barriers have been known to occur, when protein-mediated transport is inhibited or induced.Citation11 These DDIs will result in changes in the apparent volume distribution (Vd).Citation11 The most studied membrane barriers in terms of DDIs and distribution is the BBB followed by the placental barrier, so they both will be the focus of this review.
Blood–brain barrier
The BBB functions as a barrier for drugs into the brain and central nervous system (CNS) from the blood stream ().Citation36,Citation37 The BBB is a complex structure that is composed of endothelial cells and pericytes that are surrounded by the end-feet of astrocytes.Citation36,Citation37 In addition, the endothelial cells that form the inner half of the BBB are held together by proteins that form tight junctions between the cells.Citation36,Citation37 These tight junctions prevent molecules from going between the cells. However, drugs can cross into the brain through the BBB by passive diffusion or through protein transporters located on the plasma membrane. The rate at which a drug passively diffuses across the BBB depends on its lipophilicity,Citation38–Citation41 its hydrogen bond donors and acceptor characteristicsCitation40,Citation42 and its size.Citation43 On the apical surface (lumenal or blood-side) are a variety of drug transporters that control the flow of drugs into the brain.Citation44 On the apical plasma membrane, transport from the cytosol to the blood stream is mediated by BCRP, Pgp, the multidrug resistance protein 4 (MRP4) and the organic anionic transporter 3 (OAT3).Citation44 Transport from the blood stream to the cytosol is controlled by the monocarboxylate transporter 1 (MCT1) and the organic anionic transporting peptide 1A2 (OATP1A2).Citation44 On the opposite (basal) side of the endothelial side is the OAT3 transporter.Citation44
The most studied BBB transporter is Pgp. The strongest evidence that Pgp is involved in drug transport across the BBB is from studies with Pgp knockout mice.Citation45,Citation46 Studies with the mouse mdr1a variant of Pgp showed large differences in drug distribution were observed between the brain and other tissues.Citation45,Citation46 On the other hand, there have been some question of the clinical relevance of Pgp-mediated DDIs at the BBB.Citation47,Citation48 In two positron emission tomography (PET) studies, there were large increases in verapamil concentration in the brain of rats and only modest changes in the human brain in the presence of the Pgp inhibitor cyclosporine A.Citation49,Citation50 These differences were attributed to differences in the transporter makeup between human and rodent BBB.Citation47 In humans, the most convincing evidence of Pgp-mediated drug transport comes from PET studies with potent Pgp inhibitor tariquidar.Citation48,Citation51,Citation52 This inhibitor was shown to increase brain penetration of verapamil, loperamide and a loperamide metabolite.Citation48,Citation51,Citation52 In a group of healthy individuals, tariquidar increased the apparent brain volume distribution of the cardiovascular drug verapamil.Citation53
Placental barrier
The placenta is a layer of tissue that separates the mother from the unborn child.Citation54 Distribution of drugs across the placenta are regulated by phase I and phase II drug metabolizing enzymes and transporters within the syncytiotrophoblast.Citation55 Unfortunately, our current understanding of DDIs with the placenta remains limited.Citation140
The phase I drug metabolizing enzyme CYP/CYP450 19 (CYP19) (a.k.a. aromatase) has the highest level of mRNA expression in the placenta.Citation55 The enzyme has substrates, inhibitors and CYP19 expression modulators that could potentially lead to DDIs.Citation56 For example, the opioid drug methadone was found to be a substrate and mechanism-based inhibitor of CYP19.Citation57 As a result, methadone has significant risk for DDIs with CYP19 substrates including the anti-diabetic drug glyburide.Citation58 In terms of CYP19 expression modulators, the drug betamethasone, which is commonly administered to pregnant women, was shown to down regulate mRNA expression of CYP19.Citation59 The corticosteroid dexamethasone, which is often administered to pregnant women, was found to have the opposite effect and induce CYP19 expression.Citation60
The placenta is also known to have a variety of drug transporters.Citation140 Most DDI investigations with the placenta have been focused on Pgp.Citation140 Pgp has been shown to play a protective role against drugs in wild type and Pgp knockout mice.Citation61,Citation62 A study done with pregnant nonhuman primates showed that Pgp activity increased with gestational age for the mother and within the placental barrier suggesting that it might be an important factor to consider during pregnancy.Citation63
Most Pgp DDI investigations have focused on Pgp inhibition.Citation140 In a perfused human placenta model, Pgp inhibition has increased drug penetration in the placenta including the anti-HIV drug lopinavir.Citation140 In a PET study of a nonhuman primate, the Pgp inhibitor cyclosporine A and the cardiovascular drug [11C]-verapamil showed increased tissue distribution in the maternal brain and the fetal liver.Citation64 There is some in vitro evidence that Pgp may behave differently in the placenta than other tissues. The Pgp inhibitors, quinidine and verapamil, had no effect on Pgp-mediated digoxin transport in an isolated perfused human placenta.Citation65 However, verapamil did inhibit Pgp-mediated talinolol transport in the same in vitro system.Citation66
Metabolism
Drug metabolizing enzymes can be found throughout the body including the brain,Citation67 heart,Citation68 lungs,Citation69 intestinesCitation70 and even the skin.Citation71 Metabolism in ADME typically refers to enzymatic biotransformations that occur in the liver.Citation11 In the liver, drug metabolizing enzymes are expressed in the hepatocytes and the biliary epithelium ().Citation25,Citation72 There are both phase I and phase II drug metabolizing enzymes in these cells.Citation73 CYPs dominate the phase I drug metabolizing enzymes, while UDP glucuronosyl transferases (UGTs) and sulfotransferases (SULTs) dominate the phase II drug metabolizing enzymes.Citation73 Unlike the intestines, the major CYP isoforms and subfamilies are evenly distributed throughout the liver and include CYP1A2, CYP2B6, CYP2C, CYP2D6, CYP2E1, CYP4F and CYP3A.Citation29,Citation32,Citation74 The major UGT subfamilies found in the liver are UGT1A and UGT2B.Citation75 The SULT1A1 is the major sulfotransferase isoform in the liver.Citation76
Hepatic CYPs
While intestinal CYPs affect drug absorption, the hepatic CYPs affect drug elimination.Citation11 DDIs from CYPs in the liver occur as a result of inhibiting or inducing expression of the CYPs.Citation11 Inhibiting CYP will increase the Cmax, AUC and the elimination half-time (t1/2), while inducing it will have the opposite effect on those PK parameters.Citation11 Many CYP DDI studies have focused on the CYP3A4 isoform.Citation77,Citation78 Deciphering specific DDIs from liver CYP3A4 is complicated by the fact that there is significant expression of CYP3A4 in the liver and intestines.Citation27,Citation28,Citation32 There have been attempts to separate their individual effects.Citation79 For example, in a PK study, the anti-fungal drug and CYP3A4 inhibitor itraconazole was coadministered with the cardiovascular drug felodipine.Citation80 Itraconazole was found to increase the Cmax and the AUC of felodipine several fold.Citation80 A later analysis suggested that intestinal and hepatic CYP3A4 caused similar increases in felodipine bioavailability.Citation79,Citation80 In another clinical study, the effects from intestinal and liver nifedipine metabolism by CYP3A4 after induction by rifampicin was examined.Citation81 Rifampicin induction of CYP3A4 caused the AUC and the elimination t1/2 to decrease about 30%.Citation81 Rifampicin primarily affected the intestinal extraction ratio revealing that most of the DDIs are due to intestinal CYP3A4 and not hepatic CYP3A4.Citation81
Of the CYPs, the CYP1A2 isoform makes significant contributions to the liver, but only plays a minor role in the intestine.Citation28,Citation32 Therefore, clinically observed DDIs with this CYP will reflect liver metabolism and not biotransformations within the intestines. The serotonin reuptake inhibitor (SSRI) fluvoxamine is a specific inhibitor of CYP1A2.Citation82 When fluvoxamine was coadministered with caffeine, there were significant increases to the Cmax and AUC of caffeine, and there was more than an 11-fold increase in the elimination t1/2 of the stimulant.Citation83
UDP-glucuronosyltransferases (UGTs)
UGTs are a family of conjugating phase II enzymes that catalyzes the transfer of glucuronic acid to hydroxyl, carboxyl, or amine functional groups of drugs.Citation84 UGTs in the liver are membrane anchored proteins that reside within the smooth endoplasmic reticulum in hepatocytes and biliary endothelial cells.Citation85 Glucuronidation through the UGT2B7 isoform is the primary pathway for metabolism of antiretroviral drug azidothymidine (AZT) making it an ideal probe substrate for UGT DDIs.Citation86–Citation89 Both in vitro and in vivo investigations with AZT have noted significant DDIs with the drug.Citation90 In an in vitro study, the anti-seizure drug valproate inhibited glucuronidation of AZT by Chinese hamster lung fibroblasts overexpressing UGT2B7.Citation88 This in vitro result correlates well with a clinical study performed on HIV-infected individuals.Citation91 They found a significant reduction in the AUC and Cmax of the glucuronidated product and an increase in the AUC and Cmax of the parent drug.Citation91 UGTs in the liver can also be induced by rifampicin.Citation92 In a couple of clinical studies with HIV-infected patients, rifampicin induction of the UGT1A1 isoform caused significant reduction in the AUC and Cmax of the AZT.Citation93,Citation94
Excretion
In this review, drug excretion is defined as the removal of drugs from the body, so that it is not confused with elimination, which includes biotransformation in the liver. More than two thirds of drug excretion occurs through the kidneys with most of the remaining excretion occurring through the liver via the bile.Citation11 Only a minor amount of drugs are excreted through the sweat and the lungs, so it will not be discussed in this review.Citation11
Renal excretion
Renal drug excretion is primarily mediated by drug transporters within renal tubular cells (RTCs) of the proximal convoluted tubule found in the nephron ().Citation95,Citation96 The RTCs have a wide range of cationic and anionic drug transporters.Citation95,Citation96 In this section, a brief discussion of DDIs is provided of Pgp, organic anionic transporters (OATs) and organic cationic transporters (OCTs).
One of the most investigated drug transporters in the kidney is the Pgp transporter. It is found in the brush-border apical membrane of RTCs and effluxes drugs from the cytosol into the urine.Citation95 Many DDI investigations have been performed with the cardiovascular drug digoxin due to its low therapeutic index, its propensity for DDIs with Pgp, and the fact that it is primarily excreted through the kidneys by Pgp.Citation97 Drug transport is inhibited by a wide range of “perpetrator” drugs including the anti-cancer drug paclitaxel and cholesterol lowering statin drugs.Citation97 Inhibition of digoxin transport by Pgp leads to significant increases in the AUC and the elimination t1/2 with corresponding decreases in the renal clearance (CLR).Citation97 Unlike other Pgp substrates, administration of the Pgp inducer rifampicin did not affect the CLR or t1/2 of digoxin.Citation98
The OAT transporter family of exchange transporters are responsible for transporting anionic drugs including diuretics, antivirals and antibiotics.Citation99,Citation100 The OAT 1–3 isoforms are located on the S2 segment of the proximal tubule on the basolateral membrane, while OAT4 and the urate/anion exchanger (URAT1) isoforms are located on the apical membrane.Citation95,Citation99 This transporter family is an exchange transporter that exchanges drugs for carboxylates.Citation99 The OAT1 and OAT3 transporters are responsible for the transport of the diuretic furosemide.Citation101 When furosemide transport by OAT1 and OAT3 are inhibited by probenecid, there is significant elevations in the AUC of furosemide and reduction in its CLR.Citation96,Citation101
Members of the OCT transporter family can transport cationic drugs such as the antihypertensive drug atenolol and the antiviral drug lamivudine.Citation96 These transporters are located on the basolateral membranes of the RTCs.Citation95 One of the most studied drugs within this transporter family is the antidiabetic drug metformin, which is a substrate for several isoforms of OCT transporters.Citation102 The drug is advantageous for studying renal DDIs because it does not undergo significant hepatic metabolism.Citation103 The proton pump and OCT inhibitor lansoprazole increased the AUC, increased the elimination t1/2 and reduced the CLR.Citation102 Metformin PK parameters were similarly affected by DDIs with the histamine H2 receptor antagonist cimetidine and the drug pyrimethamine.Citation96
Biliary excretion
Less than a third of drugs are excreted through the bile.Citation11 Excretion in the liver occurs through transporters that are located within hepatocytes and biliary endothelial cells of the bile ducts.Citation104 There is a diverse range of transporters in these cells including OATs and OCTs.Citation104 The most studied transporter is Pgp, which is located on apical membrane of hepatocytes and endothelial cells of the bile duct.Citation104 The opioid agonist thienorphine undergoes significant biliary excretion in rats.Citation105 When Pgp inhibitor tarquidar was coadministered to rats, the Cmax and AUC of thienorphine increased, while the mean residence time (MRT) and the elimination t1/2 decreased.Citation106 The “paradoxically” reduced MRT and elimination t1/2 of thienorphine was attributed to interrupted enterohepatic circulation through the bile ducts.Citation106
Pharmacodynamics
PD defines what a drug does to the body that leads to a physiological response.Citation11 Drugs can induce a PD response by interacting with protein receptors such as the case of agonists with the GABAA receptor.Citation107 They may also interact with proteins in the second messenger system such as the case of tyrosine kinase inhibitors that target the protein kinase.Citation108 PD can also occur with enzymes such as the nonsteroidal anti-inflammatory drugs (NSAIDs) and the platelet cyclooxygenase to inhibit platelet activation.Citation109
PD DDIs occur when a coadministered drug alters the PD effect of another drug outside of their PK effects.Citation110 Like PK DDIs, these DDIs occur when two or more drugs are coadministered to a patient.Citation110 The drugs can antagonize or synergistically interact.Citation110 DDIs can also occur with drugs that have similar modes of action such as lowering blood pressure.Citation111 PD DDIs can have additive, synergistic or antagonistic effects on PD responses.Citation110
The Emax and the EDEC50 can change in response to PD-mediated DDIs. A decrease in the EDEC50 leads to a “leftward” shift of the dose response curve and indicates synergism.Citation110,Citation112 No change in the EDEC50 of the dose response curve and an Emax that reflects the sum of individual PD responses indicates additivity.Citation110,Citation112 An increase in the EDEC50, which will lead to a “rightward” shift in the dose response curve, reveals competitive antagonism.Citation110,Citation112 Depending on the PD DDI mechanism, a decrease in the Emaxcan be due to noncompetitive or uncompetitive antagonism.Citation110,Citation112,Citation113
Beyond analyses of dose response curves, there are analytical methods that can be used to quantitatively determine PD DDIs.Citation110 DDIs can be graphically assessed using an isobologram with the drug doses at 50% of Emax plotted along the axes.Citation110 Additive PD DDIs will typically appear as a diagonal straight line on these graphs, but can be nonlinear in cases where one of the drugs is a partial agonist.Citation110,Citation114 Synergistic and antagonistic DDIs from PD will have leftward and rightward curvature, respectively, with respect to a diagonal in these graphs.Citation110 Competitive antagonists can also be assessed using Schild plots, which exploit the observed “rightward” shift of the dose response curves.Citation110 They will lead to positive linear slopes in these graphs.Citation110 summarizes the PD-mediated DDIs for representative drugs and their effect on PD Parameters. Clinical examples of additive, synergistic and antagonistic PD DDIs are described below.
Table 2 Pharmacodynamically mediated DDIs
Additive
An additive PD DDI is when the overall PD response is the sum of the individual PD responses from the individual drugs.Citation115 This DDI is in contrast with a synergistic DDI where the overall PD response is greater than the sum of the individual PD responses.Citation110 For example, liraglutide is a derivative metabolic hormone that acts as a long-acting glucagon-like peptide-1 receptor agonist that lowers glucose,Citation116 while insulin detemir is a long acting insulin derivative that also functions to reduce blood glucose.Citation117 When the drugs are coadministered, the glucose lowering effect of the two drugs is additive and equal to the sum of the individual PD responses.Citation118 The drug interactions of phenprocoumon and nonsteroidal anti-inflammatory drugs (NSAIDs) is another example of an additive DDI.Citation115 Phenprocoumon is a vitamin K antagonist that inhibits vitamin K oxide reductase and indirectly prevents the activation of several clotting factors.Citation119 NSAIDs inhibit platelet cyclooxygenase, which prevents platelet activation.Citation109 The net effect of taking these drugs together is that their anti-coagulant effects are additive leading to an increased risk of bleeding.Citation120
Synergistic
When the two or more drugs are taken together and the resulting DDIs are synergistic, the resulting PD response can be greater than the sum of the individual PD responses.Citation121 For example, the combination of diphenhydramine and ethanol leads to synergism in the PD response.Citation122 Ethanol acts as a GABAA receptor agonist increasing chloride conductance on the post synaptic neuron.Citation123 Diphenhydramine is a muscuranic acetylcholine receptor antagonist causing a reduction in positive charge of the neuron.Citation124 This leads to a net increase in the negative charge across the neuron and increased mental impairment.Citation122 Another example of PD synergism is with the drugs tramadol and acetaminophen.Citation125 These drugs exhibited a greater reduction in pain and an enhanced antihyperalgesic effect than the drugs taken alone.Citation125
Antagonistic
Drug antagonism occurs when a “perpetrator” drug dampens or inhibits the PD response of a “victim” drug. Antagonism can be competitive, noncompetitive and uncompetitive. As mentioned above, competitive antagonism occurs when a “perpetrator” drug increases the EDEC50, but has no effect on the Emax.Citation126 In contrast, noncompetitive antagonism causes a reduction in the Emax, but has no significant effect on the EDEC50 (eg,Citation127). Uncompetitive antagonism can have similar effects on Emax and EDEC50 as noncompetitive antagonism, but requires interaction by the “victim” drug first to the receptor.Citation113
A good use of competitive antagonism is with the drug naloxone (Narcan®). Naloxone counteracts the effect of opioids, which are µ-opioid receptor agonists, by competing for the µ-opioid receptor.Citation128 In rats, naloxone was more effective at blocking the effects of the µ-opioid receptor agonists methadone and fentanyl than the µ-opioid receptor agonists heroin and morphine.Citation129 In a clinical study with opioid-dependent individuals, naloxone was found to counteract the PD response of the µ-opioid agonist buprenorphine.Citation130 Noncompetitive PD antagonism can be allosteric in that it occurs at an alternate site than the “victim” drug or it can be irreversible. The interaction of ruthenium red with capsaicin is an example of noncompetitive allosteric antagonism.Citation127 Ruthenium red was found to reduce contractile response induced by capsaicin in rat tissues.Citation127 The antagonistic effect is likely due to competition between these molecules at different sites on the transient receptor potential (TRP) channels.Citation131 The drug omeprazole (Prilosec®) is a noncompetitive irreversible antagonist by covalently modifying the H+,K+-ATPase in the stomach.Citation132,Citation133 The Alzheimer’s drug memantine is an example of an uncompetitive antagonist, which interacts with the N-methyl-d-aspartate (NMDA) receptor and is used in the treatment of Alzheimer’s disease.Citation134,Citation135 The uncompetitive antagonism of memantine differs from noncompetitive antagonists in that it requires activation of the NMDA receptor before memantine binding can occur.Citation113
Conclusion and outlook
Understanding DDIs remains an ongoing challenge and significant gaps in our understanding remain. This review was focused on representative DDIs between two drugs. However, it is quite common for individuals, especially the elderly, to be taking considerably more drugs at a time.Citation5,Citation7 In this case, the DDIs may be very complex and exceedingly difficult to deconvolute. Several novel analytical approaches are emerging that will allow deconvolution of complex drug interactions from multiple drug targets simultaneously. An ensemble approach for multiple drug target deconvolution was recently used to decipher the interactions of inhibitors to multiple kinases.Citation136 A random walk algorithm was developed to unravel the protein–protein interaction network that underlies PD-mediated DDIs.Citation137 In silico methods have been developed to predict both PK and PD DDIs of arbitrary molecules.Citation138,Citation139 Ultimately, novel future approaches to investigate and deconvolute DDIs will lead to safer and more efficacious coadministration of drugs.
Acknowledgments
We would like to thank the National Cancer Institute for a NIH R01 grant (1R01CA204846-01A1) and National Institute of General Medical Sciences for an NIH R15 grant (1R15GM107913-01A1) for funding this work.
Disclosure
The authors report no conflicts of interest in this work.
References
- OrtmanJMVelkoffVAHoganHAn Aging Nation: The Older Population in the United StatesSuitland, MDUS Census Bureau2014
- SaliveMEMultimorbidity in older adultsEpidemiol Rev201335758323372025
- GuQDillonCFBurtVLPrescription drug use continues to increase: U.S. prescription drug data for 2007-2008NCHS Data Brief20104218
- RambhadeSChakarbortyAShrivastavaAPatilUKRambhadeAA survey on polypharmacy and use of inappropriate medicationsToxicol Int2012191687322736907
- MaherRLHanlonJTHajjarERClinical consequences of polypharmacy in elderlyExpert Opin Drug Saf2014131
- RodriguesMCSde OliveiraCDrug-drug interactions and adverse drug reactions in polypharmacy among older adults: an integrative review 1Rev Lat Am Enfermagem201624
- ShahBMHajjarERPolypharmacy, adverse drug reactions, and geriatric syndromesClin Geriatr Med201228217318622500537
- OgilvieBWParkinsonADrugs as victims and perpetrators and the pharmacokinetic concept of maximum exposureHandb Metab Pathways Xenobiot2014103123
- PolasekTMFpyLMinersJODoogueMPPerpetrators of pharmacokinetic drug–drug interactions arising from altered cytochrome P450 activity: a criteria-based assessmentBr J Clin Pharmacol201171572773621223357
- PrueksaritanontTChuXGibsonCDrug–drug interaction studies: regulatory guidance and an industry perspectiveAAPS J201315362964523543602
- RowlandMTozerTNClinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications4thPhiladelphia, PALippincott Williams & Wilkins2010
- HelanderHFFändriksLSurface area of the digestive tract - revisitedScand J Gastroenterol201449668168924694282
- MüllerJKeiserMDrozdzikMOswaldSExpression, regulation and function of intestinal drug transporters: an updateBiol Chem20163982175192
- VaessenSFvan LipzigMMPietersRHKrulCAWortelboerHMvan de SteegERegional expression levels of drug transporters and metabolizing enzymes along the pig and human intestinal tract and comparison with Caco-2 cellsDrug Metab Dispos201745435336028153842
- WilkensSStructure and mechanism of ABC transportersF1000Prime Rep201571425750732
- NiZBikadiZRosenbergMFMaoQStructure and function of the human breast cancer resistance protein (BCRP/ABCG2)Curr Drug Metab201011760361720812902
- KalliokoskiANiemiMImpact of OATP transporters on pharmacokineticsBr J Pharmacol2009158369370519785645
- ObaidatARothMHagenbuchBThe expression and function of organic anion transporting polypeptides in normal tissues and in cancerAnnu Rev Pharmacol Toxicol20125213515121854228
- RothMObaidatAHagenbuchBOATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamiliesBr J Pharmacol201216551260128722013971
- SchwarzUIGramattéTKrappweisJOertelRKirchWP-glycoprotein inhibitor erythromycin increases oral bioavailability of talinolol in humansInt J Clin Pharmacol Ther200038416116710783825
- WestphalKWeinbrennerAZschiescheMInduction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: A new type of drug/drug interactionClin Pharmacol Ther200068434535511061574
- ElsbyRMartinPSurryDSharmaPFennerKSolitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug-drug interaction with rosuvastatin by causing up to a 2-fold increase in statin exposureDrug Metab Dispos201644339840826700956
- MartinPGillenMRitterJEffects of fostamatinib on the pharmacokinetics of oral contraceptive, warfarin, and the statins rosuvastatin and simvastatin: results from phase I clinical studiesDrugs in R&D20161619310726748647
- BaileyDGDresserGKLeakeBFKimRBNaringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juiceClin Pharmacol Ther200781449550217301733
- MurrayGIBarnesTSSewellHFEwenSWMelvinWTBurkeMDThe immunocytochemical localisation and distribution of cytochrome P-450 in normal human hepatic and extrahepatic tissues with a monoclonal antibody to human cytochrome P-450Br J Clin Pharmacol19882544654753289600
- Ortiz de MontellanoPRCytochrome P450: Structure, Mechanism, and Biochemistry4thSwitzerlandSpringer International Publishing2015
- GaletinAGertzMHoustonJBContribution of intestinal cytochrome P450-mediated metabolism to drug-drug inhibition and induction interactionsDrug Metab Pharmacokinet2010251284720208387
- PaineMFHartHLLudingtonSSHainingRLRettieAEZeldinDCThe human intestinal cytochrome P450 “pie.”Drug Metab Dispos200634588088616467132
- NakanishiYMatsushitaAMatsunoKRegional distribution of cytochrome P450 mRNA expression in the liver and small intestine of cynomolgus monkeysDrug Metab Pharmacokinet201025329029720610888
- ThummelKEKunzeKLShenDDEnzyme-catalyzed processes of first-pass hepatic and intestinal drug extractionAdv Drug Deliv Rev1997272-39912710837554
- HaddadADavisMLagmanRThe pharmacological importance of cytochrome CYP3A4 in the palliation of symptoms: review and recommendations for avoiding adverse drug interactionsSupport Care Cancer200715325125717139496
- MichaelsSWangMZThe revised human liver cytochrome P450 “Pie”: absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomicsDrug Metab Dispos20144281241125124816681
- TempletonIPengC-CThummelKDavisCKunzeKIsoherranenNAccurate prediction of dose-dependent CYP3A4 inhibition by itraconazole and Its metabolites from in vitro inhibition dataClin Pharmacol Ther201088449950520739919
- Kharasch EvanDAlysaWChristineHPamelaSIntravenous and oral alfentanil as in vivo probes for hepatic and first-pass cytochrome P450 3A activity: noninvasive assessment by use of pupillary miosisClin Pharmacol Ther200476545246615536460
- GroverABenetLZEffects of drug transporters on volume of distributionAAPS J200911225026119399628
- BallabhPBraunANedergaardMThe blood–brain barrier: an overview: structure, regulation, and clinical implicationsNeurobiol Dis200416111315207256
- ZhaoZNelsonARBetsholtzCZlokovicBVEstablishment and dysfunction of the blood-brain barrierCell201516351064107826590417
- AbbottNJAnatomy and physiology of the blood–brain barriersHammarlund-UdenaesMde LangeEThorneRGDrug Delivery to the Brain: Physiological Concepts, Methodologies and ApproachesNew YorkSpringer2014321
- BradburyMWBPhysiology and Pharmacology of the Blood-Brain BarrierBerlin, HeidelbergSpringer Science & Business Media2012
- PardridgeWMTransport of protein-bound hormones into tissues in vivoEndocr Rev1981211031237028469
- SerlinYShelefIKnyazerBFriedmanAAnatomy and physiology of the blood-brain barrierSemin Cell Dev Biol2015382625681530
- ChikhaleEGK-YNBurtonPSBorchardtRTHydrogen bonding potential as a determinant of the in vitro and in situ blood–brain barrier permeability of peptidesPharm Res19941134124198008709
- LevinVARelationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeabilityJ Med Chem19802366826847392035
- UrquhartBLKimRBBlood−brain barrier transporters and response to CNS-active drugsEur J Clin Pharmacol20096511106319727692
- SchinkelAHMayerUWagenaarENormal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteinsProc Natl Acad Sci U S A1997948402840339108099
- SchinkelAHSmitJJvan TellingenODisruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugsCell19947744915027910522
- EyalSHsiaoPUnadkatJDDrug interactions at the blood-brain barrier: fact or fantasy?Pharmacol Ther200912318010419393264
- SugimotoHHirabayashiHAmanoNMoriwakiTRetrospective analysis of P-glycoprotein-mediated drug-drug interactions at the blood-brain barrier in humansDrug Metab Dispos201341468368823340958
- HsiaoPSasongkoLLinkJMVerapamil P-glycoprotein transport across the rat blood-brain barrier: cyclosporine, a concentration inhibition analysis, and comparison with human dataJ Pharmacol Exp Ther2006317270471016415090
- SasongkoLLinkJMMuziMImaging P-glycoprotein transport activity at the human blood-brain barrier with positron emission tomographyClin Pharmacol Ther200577650351415961982
- BauerMKarchRZeitlingerMApproaching complete inhibition of P-glycoprotein at the human blood–brain barrier: an (R)-[11C] verapamil PET studyJ Cereb Blood Flow Metab201535574374625669913
- CantoreMBenadibaMElsingaPH11C- and 18F-labeled radioligands for P-Glycoprotein imaging by positron emission tomographyChemMedChem201611110811826563728
- BauerMZeitlingerMKarchRP-glycoprotein mediated interaction between (R)-[11C]verapamil and tariquidar at the human blood-brain barrier studied with positron emission tomography, a comparison with rat dataClin Pharmacol Ther201291222723322166851
- GriffithsSKCampbellJPPlacental structure, function and drug transferContin Educ Anaesth Crit Care Pain20151528489
- NishimuraMYagutiHYoshitsuguHNaitoSSatohTTissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCRYakugaku Zasshi2003123536937512772594
- HongYChoMYuanY-CChenSMolecular basis for the interaction of four different classes of substrates and inhibitors with human aromataseBiochem Pharmacol20087551161116918184606
- WjLBiesRKamdenLKDestaZFlockhartDAMethadone: a substrate and mechanism-based inhibitor of CYP19 (aromatase)Drug Metab Dispos20103881308131320410453
- ZharikovaOLFokinaVMNanovskayaTNIdentification of the major human hepatic and placental enzymes responsible for the biotransformation of glyburideBiochem Pharmacol200978121483149019679108
- PaakkiPKirkinenPHelinH-LPelkonenORaunioHPasanenMAntepartum glucocorticoid therapy suppresses human placental xenobiotic and steroid metabolizing enzymesPlacenta200021224124610736248
- BerkovitzGDCarterKMLevineMAMigeonCJAbnormal induction of aromatase activity by dexamethasone in fibroblasts from a patient with cortisol resistanceJ Clin Endocrinol Metab1990706160816112347895
- LankasGRWiseLDCartwrightMEPippertTUmbenhauerDRPlacental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in miceReprod Toxicol19981244574639717696
- SmitJWHuismanMTvan TellingenOWiltshireHRSchinkelAHAbsence or pharmacological blocking of placental P-glycoprotein profoundly increases fetal drug exposureJ Clin Invest1999104101441144710562306
- ChungFEyalSMuziMPositron emission tomography imaging of tissue P-glycoprotein activity during pregnancy in the non-human primateBr J Pharmacol2010159239440420002098
- EyalSChungFSMuziMSimultaneous PET imaging of P-glycoprotein inhibition in multiple tissues in the pregnant non-human primateJ Nucl Med200950579880619403878
- HolcbergGSapirOTsadkinMLack of interaction of digoxin and P-glycoprotein inhibitors, quinidine and verapamil in human placenta in vitroEur J Obstet Gynecol Reprod Biol200310913313712860328
- MayKMinarikovaVLinnemannKRole of the multidrug transporter proteins ABCB1 and ABCC2 in the diaplacental transport of talinolol in the term human placentaDrug Metab Dispos200836474074418195111
- MiksysSLTyndaleRFDrug-metabolizing cytochrome P450s in the brainJ Psychiatry Neurosci200227640641512491573
- Chaudhary KetulRNagarjunBSSeubert JohnMCytochrome P450 enzymes and the heartIUBMB Life2009611095496019787709
- OlssonBBondessonEBorgströmLPulmonary drug metabolism, clearance, and absorptionSmythHDCHickeyHJControlled Pulmonary Drug Delivery Advances in Delivery Science and TechnologyNew YorkSpringer20112150
- ThummelKEGut instincts: CYP3A4 and intestinal drug metabolismJ Clin Invest2007117113173317617975661
- SvenssonCKBiotransformation of drugs in human skinDrug Metab Dispos200937224725319005027
- FatimaLDominiqueWVéroniqueBPhase I and Phase II drug-metabolizing enzymes are expressed and heterogeneously distributed in the biliary epitheliumHepatology200330614981506
- EvansWERellingMVPharmacogenomics: translating functional genomics into rational therapeuticsScience1999286543948749110521338
- WatkinsPBMurraySAThomasPEWrightonSADistribution of cytochromes P-450, cytochrome b5, and NADPH-cytochrome P-450 reductase in an entire human liverBiochem Pharmacol19903934714762106322
- AchourBDantonioANiosiMQuantitative characterization of major hepatic UDP-glucuronosyltransferase enzymes in human liver microsomes: comparison of two proteomic methods and correlation with catalytic activityDrug Metab Dispos201745101102111228768682
- GamageNBarnettAHempelNHuman sulfotransferases and their role in chemical metabolismToxicol Sci200690152216322073
- OguCCMaxaJLDrug interactions due to cytochrome P450Proc2000134421423
- DresserGKSpenceJDBaileyDGPharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibitionClin Pharmacokinet2000381415710668858
- KatoMChibaKHisakaAThe intestinal first-pass metabolism of substrates of CYP3A4 and P-glycoprotein—quantitative analysis based on information from the literatureDrug Metab Pharmacokinet200318636537215618757
- Kirsi-maijaJOlkkola KlausTNeuvonen PerttiJItraconazole greatly increases plasma concentrations and effects of felodipineClin Pharmacol Ther19976144104159129558
- HoltbeckerNFrommMFKroemerHKOhnhausEEHeidemannHThe nifedipine-rifampin interaction. Evidence for induction of gut wall metabolismDrug Metab Dispos19962410112111238894514
- RasmussenBBNielsenTLBrøsenKFluvoxamine is a potent inhibitor of the metabolism of caffeine in vitroPharmacol Toxicol19988362402459868741
- Culm-MerdekKEvon MoltkeLLHarmatzJSGreenblattDJFluvoxamine impairs single-dose caffeine clearance without altering caffeine pharmacodynamicsBr J Clin Pharmacol200560548649316236038
- OdaSFukamiTYokoiTNakajimaMA comprehensive review of UDP-glucuronosyltransferase and esterases for drug developmentDrug Metab Pharmacokinet2015301305125760529
- LiuYCoughtrieMWHRevisiting the latency of uridine diphosphate-glucuronosyltransferases (UGTs)—How does the endoplasmic reticulum membrane influence their function?Pharmaceutics20179332
- BarbierOTurgeonDGirardC3′-azido-3′-deoxythimidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7Drug Metab Dispos200028549750210772627
- CourtMHKrishnaswamySHaoQEvaluation of 3′-azido-3′-deoxythymidine, morphine, and codeine as probe substrates for UDP-glucuronosyltransferase 2B7 (UGT2B7) in human liver micro-somes: specificity and influence of the UGT2B7*2 polymorphismDrug Metab Dispos20033191125113312920168
- EthellBTAndersonGDBurchellBThe effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferasesBiochem Pharmacol20036591441144912732356
- VealGJBackDJMetabolism of zidovudineGen Pharmacol-Vasc S199526714691475
- KiangTKEnsomMHChangTKUDP-glucuronosyltransferases and clinical drug-drug interactionsPharmacol Ther200510619713215781124
- Lertora JuanJLRege ArvindBGreenspan DebraLPharma-cokinetic interaction between zidovudine and valproic acid in patients infected with human immunodeficiency virusClin Pharmacol Ther19945632722787924122
- ChenJRaymondKRoles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptorAnn Clin Microbiol Antimicrob20065316480505
- BurgerDMMeenhorstPLKoksCHBeijnenJHPharmacokinetic interaction between rifampin and zidovudineAntimicrob Agents Chemother1993377142614318363370
- GallicanoKDSahaiJShuklaVKInduction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patientsBr J Clin Pharmacol199948216817910417493
- Launay-VacherVIzzedineHKarieSHulotJSBaumelouADerayGRenal tubular drug transportersNephron Physiol20061033p97p10616554667
- YinJWangJRenal drug transporters and their significance in drug– drug interactionsActa Pharm Sin B20166536337327709005
- LedwitchKVRobertsAGCardiovascular ion channel inhibitor drug-drug interactions with P-glycoproteinAAPS J201719240942028028729
- GreinerBEichelbaumMFritzPThe role of intestinal P-glycoprotein in the interaction of digoxin and rifampinJ Clin Invest1999104214715310411543
- AnzaiNKanaiYEndouHOrganic anion transporter family: current knowledgeJ Pharmacol Sci2006100541142616799257
- BurckhardtGDrug transport by organic anion transporters (OATs)Pharmacol Ther2012136110613022841915
- BurckhardtGBurckhardtBCIn vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapyHdb Exp Pharmacol201120129104
- DingYJiaYSongYThe effect of lansoprazole, an OCT inhibitor, on metformin pharmacokinetics in healthy subjectsEur J Clin Pharmacol201470214114624170325
- SirtoriCRFranceschiniGGalli-KienleMDisposition of metformin (N,N-dimethylbiguanide) in manClin Pharmacol Ther1978246683693710026
- FaberKNMüllerMJansenPLMDrug transport proteins in the liverAdv Drug Deliv Rev200355110712412535576
- DengJZhuangXShenGLiHGongZBiliary excretion and enterohepatic circulation of thienorphine and its glucuronide conjugate in ratsActa Pharm Sin B201222174180
- KongL-LShenG-LWangZ-YInhibition of P-glycoprotein and multidrug resistance-associated protein 2 regulates the hepatobiliary excretion and plasma exposure of thienorphine and its glucuronide conjugateFront Pharmacol2016724227555820
- SigelESteinmannMEStructure, function, and modulation of GABA(A) receptorsJ Biol Chem201228748402244023123038269
- HartmannJTHaapMKoppHGLippHPTyrosine kinase inhibitors – a review on pharmacology, metabolism and side effectsCurr Drug Metab200910547048119689244
- SchaferAIAndrewS IEffects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasisJ Clin Pharmacol19953532092197608308
- HinderMPharmacodynamic drug-drug interactionsVogelHGMaasJGebauerADrug Discovery and Evaluation: Methods in Clinical PharmacologyBerlin, HeidelbergSpringer2011367376
- MignatCUngerTACE inhibitorsDrug interactions of clinical significanceDrug Saf19951253343477669262
- ZhaoLAuJLWientjesMGComparison of methods for evaluating drug-drug interactionFront Biosci20102241249
- LiptonSAFailures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insultsNeuroRx20041110111015717010
- TallaridaRJAn overview of drug combination analysis with isobologramsJ Pharmacol Exp Ther200631911716670349
- CascorbiIDrug interactions--principles, examples and clinical consequencesDtsch Arztebl Int201210933-3454655623152742
- DharmalingamMSriramUBaruahMPLiraglutide: a review of its therapeutic use as a once daily GLP-1 analog for the management of type 2 diabetes mellitusIndian J Endocrinol Metab201115191721584160
- KeatingGMInsulin detemir: a review of its use in the management of diabetes mellitusDrugs201272172255228723110609
- MorrowLHompeschMGuthrieHChangDChatterjeeDJCo-administration of liraglutide with insulin detemir demonstrates additive pharmacodynamic effects with no pharmacokinetic interactionDiabetes Obes Metab2011131758021114606
- AnsellJHirshJHylekEJacobsonACrowtherMPalaretiGPharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition)Chest20081336 Suppl160S19818574265
- JobskiKBehrSGarbeEDrug interactions with phenprocoumon and the risk of serious haemorrhage: a nested case-control study in a large population-based German databaseEur J Clin Pharmacol201167994195121452031
- TallaridaRJDrug synergism: its detection and applicationsJ Pharmacol Exp Ther2001298386587211504778
- BaughRCalvertRTThe effect of diphenhydramine alone and in combination with ethanol on histamine skin response and mental performanceEur J Clin Pharmacol197712320120422437
- LoboIAHarrisRAGABA(A) receptors and alcoholPharmacol Biochem Behav2008901909418423561
- LiuHFarleyJMEffects of first and second generation antihistamines on muscarinic induced mucus gland cell ion transportBMC Pharmacol20055815790419
- FilitzJIhmsenHGüntherWSupra-additive effects of tramadol and acetaminophen in a human pain modelPain2008136326227017709207
- WyllieDJChenPETaking the time to study competitive antagonismBr J Pharmacol2007150554155117245371
- MaggiCABevanSWalpoleCSRangHPGiulianiSA comparison of capsazepine and ruthenium red as capsaicin antagonists in the rat isolated urinary bladder and vas deferensBr J Pharmacol199310838018057682139
- WermelingDPReview of naloxone safety for opioid overdose: practical considerations for new technology and expanded public accessTher Adv Drug Saf201561203125642320
- KrimmerECBarryHMeasuring naloxone antagonism of discriminative opioid stimulusFed Proc1982417231923227042395
- PrestonKLBigelowGELiebsonIABuprenorphine and naloxone alone and in combination in opioid-dependent humansPsychopharmacology19889444844902453895
- PingleSCMattaJAAhernGPCapsaicin receptor: TRPV1 a promiscuous TRP channelHdb Exp Pharmacol2007179155171
- MassoomiFSavageJDestacheCJOmeprazole: a comprehensive reviewPharmacotherapy199313146598437967
- ShinJMSachsGPharmacology of proton pump inhibitorsCurr Gastroenterol Rep200810652853419006606
- JohnsonJWKotermanskiSEMechanism of action of memantineCurr Opin Pharmacol200661616716368266
- ThomasSJGrossbergGTMemantine: a review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementiasClin Interv Aging2009436737719851512
- GujralTSPeshkinLKirschnerMWExploiting polypharmacology for drug target deconvolutionProc Natl Acad Sci U S A2014111135048505324707051
- ParkKKimDHaSLeeDPredicting pharmacodynamic drug-drug interactions through signaling propagation interference on protein-protein interaction networksPLoS One20151010e014081626469276
- LuoHZhangPHuangHDDI-CPI, a server that predicts drug-drug interactions through implementing the chemical-protein interactomeNucleic Acids Res201442Web Server issueW46W5224875476
- TakedaTHaoMChengTBryantSHWangYPredicting drug-drug interactions through drug structural similarities and interaction networks incorporating pharmacokinetics and pharmacodynamics knowledgeJ Cheminform201791628316654
- Rubinchik-SternMEyalSDrug interactions at the human placenta: what is the evidence?Front Pharmacol2012312622787449