
Abstract
Transthyretin (TTR) is a tetrameric protein, and its dissociation, aggregation, deposition, and misfolding are linked to several human amyloid diseases. As the main transporter for thyroxine (T4) in plasma and cerebrospinal fluid, TTR contains two T4-binding sites, which are docked with T4 and subsequently maintain the structural stability of TTR homotetramer. Affected by genetic disorders and detrimental environmental factors, TTR degrades to monomer and/or form amyloid fibrils. Reasonably, stabilization of TTR might be an efficient strategy for the treatment of TTR-related amyloidosis. However, only 10–25% of T4 in the plasma is bound to TTR under physiological conditions. Expectedly, T4 analogs with different structures aiming to bind to T4 pockets may displace the functions of T4. So far, a number of compounds including both natural and synthetic origin have been reported. In this paper, we summarized the potent inhibitors, including bisaryl structure-based compounds, flavonoids, crown ethers, and carboranes, for treating TTR-related amyloid diseases and the combination modes of some compounds binding to TTR protein.
Introduction
Transthyretin (TTR), functions as a mediator in transporting thyroxine (T4) in the cerebrospinal fluid and plasma,Citation1 is a 55 kDa homotetrameric protein composed of 127 amino acid residues in each monomer, which consists of two four-stranded β-sheets. Strands DAGH and CBEF are the β-sheets, and they are extended in the dimer to be DAGHH’G’A’D’ and CBEFF’E’B’C’, standing for their individual monomers. Hydrogen bonds between the main-chain atoms and strand H from different dimers promote the association of two dimers, forming a tetramer.Citation2 The contact areas in monomer-monomer and dimer-dimer are extensive in TTR. More than 120 TTR mutations have been associated with amyloidogenesis and diseases. Investigation on the roles of some specific amino acids, such as T119M,Citation2 V30M,Citation3 and R104H,Citation4 has been conducted. The A25T-TTR variant is among the most destabilized and fastest dissociating TTR tetramers.Citation5 Tetramer stability is mainly governed by hydrophobic interactions, and the subunits exchange among TTR may occur faster at 4°C than at 25°C or 37°C.Citation6
The tetramer structure of TTR contains two funnel-shaped T4-binding sites, and binding of T4 may maintain the stability of the TTR tetramer structure.Citation7,Citation8 Structurally, TTR is a homotetramer with an extensive β-sheet structure, and the two identical T4-binding sites locate at the dimer–dimer interfaces ().Citation9–Citation15 Specifically, two T4 molecules, with different orientations, bind to the unique binding sites between AC and BD dimers (), bridging these subunits. Within the binding cavity, some key amino acids (Glu54, Lys15, Leu17, Ala108, Thr119, Leu110, Ser117) () from the corresponding monomer are orchestrated to form halogen-binding pockets P1, P2, and P3 (). The amino- and carboxyl-terminal structures of T4 form hydrogen bonds by interacting with Glu54 and Lys15, respectively, in the P1 binding pocket.Citation16 Interaction of ligands with T4-binding sites is dominantly enforced by the hydrophobic effect and electrostatic interactions, which promote kinetic stabilization in the weaker dimer–dimer interface, stabilizing TTR tetramer, and increasing the kinetic barrier for dissociation.Citation16,Citation17
Figure 1 Crystal structure (PDB ID: 1IE4) of TTR tetramer with T4 interacting with the two T4-binding sits is shown. (A) The two T4-binding sites located at the dimer–dimer interfaces are framed by the white boxes. (B) The specific interaction between T4 and amino acids at the binding pockets is shown. The yellow rod structure is indicated as T4, and the green solid lines are hydrogen bonds. These pictures are prepared using the program UCSF Chimera developed by the University of California.
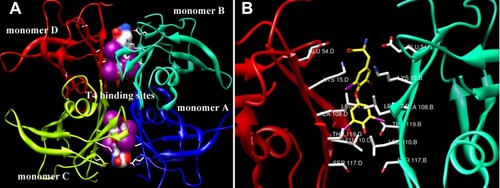
Table 1 The Structure–Activity Relationship of TTR Amyloidogenesis Inhibitors
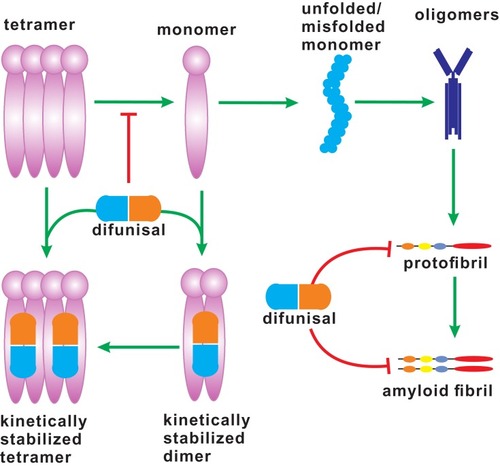
However, most of TTRs T4-binding cavities are unoccupied, only less than 25% of which in plasma is binding to T4. Therefore, under certain conditions (such as genetic mutation and induction by some chemical pollutants), TTR tetramer without T4 may become instability, dissociation into monomer, and misfolding, leading to initiation of oligomerization processes of monomeric TTR and formation of amyloid fibrils (), and induction of transthyretin amyloidogenesis (ATTR), and activation of NF-κB signaling pathway, inflammatory stress, and cell death.Citation18 A number of mutations in the gene encoding TTR protein have been identified in elderly individuals, and a conformational change in mutated TTR tetramer is observed, which results in the deposition of amyloid fibrils and induction of several diseases, such as familial amyloid polyneuropathies (FAP), familial amyloid cardiomyopathy (FAC), and senile systemic amyloidosis (SSA).Citation19 One of the possible environmentally etiological factors might be the inheritance from parents with TTR mutations. In addition, any pollutants may directly or indirectly affect the complex stability by, at least in partial, inducing genetic mutations. Under normal physiological conditions, clusterin is a plasma chaperone and may recognize exposed hydrophobic regions of misfolded protein, preventing them from aggregation (). Clusterin has been demonstrated to form a complex with a monomeric or oligomeric β-sheet rich structure of TTR in a stable manner, preventing TTR amyloid fibril formation.Citation20
The stabilization of TTR tetramer by small molecules like T4 and T4 analogs, which interact with the binding pockets, is a promising therapeutic strategy against ATTR.Citation21 This article will review the categories and structures of ATTR inhibitors.
Figure 2 The dissociation of TTR tetramer. TTR tetramer dissociates into monomers, which can be dimerized and further tetramerized by interacting with diflunisal. The unfolded/misfolded monomers of TTR aggregate to form amyloid fibrils, which may be inhibited by inhibitors, such as diflunisal.
Potent Inhibitors of ATTR
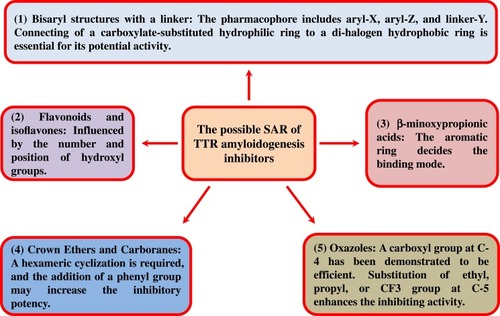
In the past few decades, a large number of small molecules with the activity of stabilizing the TTR tetramer structure have been reported. Most of these compounds contain a substructure with two aromatic rings by a linker. The bisaryl structures with a linker form the possible pharmacophoric elements, which have been indicated in . In addition, other compounds with different characteristic structures also show strong activity in the stabilization of TTR tetramer, such as flavonoids, crown ethers, and carboranes, although they bind to the T4-binding sites in different manners. Inhibitors with disaggregating TTR amyloid fibrils also play an important role for managing ATTR.
Bisaryl Structures with a Linker
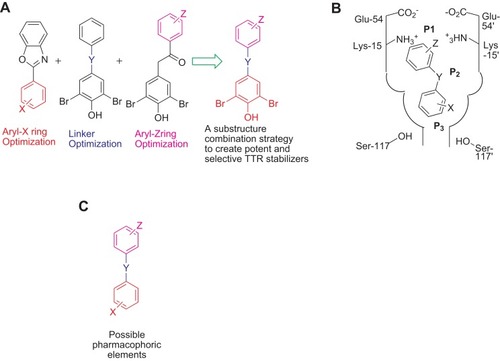
The substructure-combinational strategy has been used for producing potent and selective TTR stabilizers (). The groups including aryl-X and aryl-Z rings as well as the linker-Y can be varied in generating alternative ATTR inhibitors ().Citation22 Structure–activity relationship (SAR) study indicates that the Y linker optimization must be in concert with SAR data from aryl-X and aryl-Z optimization to predict potential structures for selective TTR amyloidogenesis inhibition. The linker (Y) can be formed by variable chemical structures with different lengths, and both the two aryl rings (X and Z) bear a combination of substituents, including alkyl, carboxyl, halide, trifluoromethyl or hydroxyl groups ().Citation23,Citation24 Structural optimization for better activities is focusing on aryl-X, -Z, and Y. Thyroid hormone-like substitution (3,5-X-4-OH, where X=CH3, F, Cl, Br, and I) produces potent selective TTR amyloidogenesis inhibitors. The linker Y structure is designed as non-polar E-olefin or –CH2CH2- group also generates highly selective inhibitors, and the hydrophobic effect of linker Y contributes to binding energy. Similar to that of T4, the binding mode of these compounds to the binding pockets has been indicated that two aromatic rings occupy P1 and P3 pockets, respectively, and the linkers occupy the P2 pocket.Citation25 A series of ATTR small molecular inhibitors with two aromatic rings and the linkers have been designed and synthesized, which will be described below.
Figure 3 The substructure-combinational strategy is used for producing potent and selective ATTR inhibitors (A). The binding model is indicated within the T4-binding pockets (B). The indicated structure may be considered as the possible pharmacophoric elements (C), and the alternative substitutions may be showed as Z, Y, and X.
Under physiological conditions, T4 (1, ) with a diphenyl ether structure maintains the stability of TTR tetramer by binding to the T4-binding sites within TTR protein. Fenoprofen (2, ), a non-steroidal anti–inflammatory drug (NSAID), is also a diphenyl ether structure compound. The activity of Fenoprofen in inhibiting amyloid fibril formation has been evaluated with a maximal therapeutic concentration of 96 μM ().Citation26 Diflunisal reaches the maximal therapeutic concentration of 224 μM with 0.85 equiv of drug bound to TTR. Similarly, flufenamic acid reaches 54 μM with 0.59 equiv. Increasing amount of molecules with diphenyl ether structure has been explored (3–12, ). All of these compounds show a high affinity to TTR protein and prevent against fibrillogenesis by stabilizing the tetramer structure of TTR.Citation18
Figure 4 Biphenyl ethers act as the potent ATTR inhibitors.
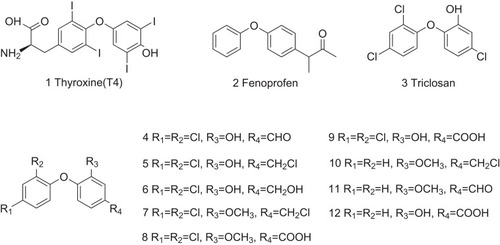
Similarly, diphenyl compounds can also bind to T4-binding sites, stabilizing TTR tetramer. A variety of NSAIDs with diphenyl groups, such as diflunisal (13, ) and flurbiprofen (14, ), have been identified to contribute to the stability of TTR tetramer ().Citation27,Citation28 Specifically, the biaryl moiety of flurbiprofen is flanked on both sides by the hydrophobic side chains of Lys15, Leu17, Ala108, Leu110, Ser117, Thr119, and Val121. The substituted phenyl ring forms van der Waals interactions with Val17 and Ala108. The carboxylate group of the CH3CHCOOH substituent is observed to electrostatically interact with Lys15. Furthermore, a number of diphenyl structure-related derivatives have been synthesized (15–65, ) and tested as potent inhibitors against amyloid fibril formation. High inhibitory activity has been observed for about half of these analogs, and eight of them greatly promote tetramer dissociation into monomer.Citation27 Structure–activity relationship study shows that connecting of a carboxylate-substituted hydrophilic ring to a di-halogen hydrophobic ring is essential for its potential activity in inhibiting amyloid fibril formation.
Figure 5 Continued.
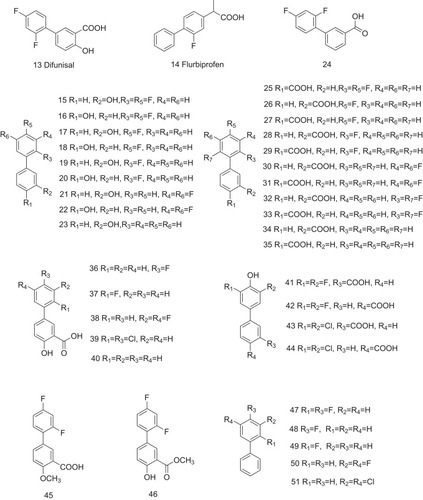
Figure 5 Diphenyl structure-related derivatives act as the potent inhibitors against ATTR.
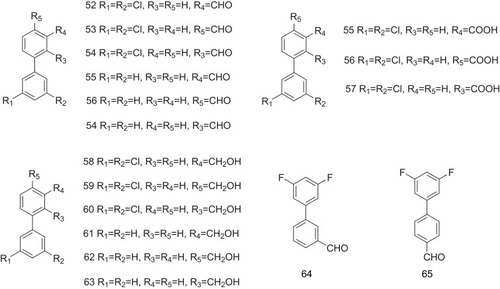
The TTR binding affinity of bromine- and iodine-substituted diphenyl compounds iododiflunisal and bromodiflunisal (66–67, ) has also been investigated that both bromine and iodine substitutions increase the binding potencies with values of 0.85 and 0.53, respectively, calculated by EC50 T4/EC50 tested compound, compared with diflunisal with a value of 0.04 ().Citation29 A series of iodine-substituted biphenyls, including iododiflunisal (68–89, ), have been produced. Data from the T4 competition assay show that the ability of these iodine-substituted biphenyls to stabilize TTR has been improved significantly than that of those inhibitors without iodine atoms ().Citation30 The iodine atom in the iododiflunisal complex produces hydrophobic interactions with Leu17, Thr106, Ala108, Thr119, and Val121, occupying the P1 pocket. Studies have showed that methyl and chloro groups in the phenyl ring and carboxyl group are the necessary requirement for flurbiprofen to create high affinity and stabilization effects on binding to TTR. Flurbiprofen analogs CHF5074 (90, ), CHF5075 (91, ), CHF4795 (92, ), and CHF4802 (93, ) are also involved in the investigation of their effects on stabilizing TTR. CHF5075 (91) and CHF4802 (93) exhibit greater activities than flurbiprofen on TTR stabilization. CHF4795 (92) shows a similar effect, and CHF5074 (90) indicates a worse effect on TTR stabilization.Citation31
Figure 6 Bromine- and iodine-substituted diphenyls act as the potent inhibitors against ATTR.
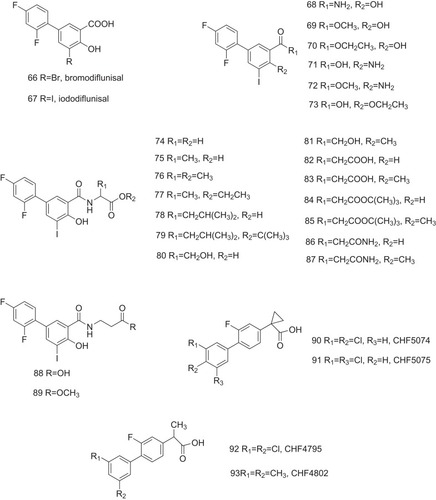
Polychlorinated biphenyls (PCBs) and hydroxylated polychlorinated biphenyls (OH-PCBs), due to their slow degradation, are known for their pollution to the environment and toxicity to humans.Citation32–Citation35 Both PCBs and OH-PCBs bind to TTR tetramer with a high affinity with Ki values of 10–140 nM, similar to the natural ligand T4 (a Ki value of 62 nM) ().Citation36,Citation37 Expectedly, several PCBs and OH-PCBs are reported (94–123, ), and their inhibitory activities against ATTR are also measured.Citation38,Citation39 Only 12–50% of the normal amount of fibril formation is observed after a period of 72 h incubation (PCB 3.6 μM, TTR 3.6 μM). Studies on the binding selectivity of PCBs and OH-PCBs for TTR in human blood plasma have been shown that compounds 102–109 bind to TTR with IC50 values of less than 50 nM.Citation37 In addition, OH-PCBs (compounds 110–123) competitively bind to the T4-binding sites of TTR and lower T4 levels in mice or rats.Citation26,Citation40,Citation41
Figure 7 PCBs and OH-PCBs act as the potent inhibitors against ATTR.
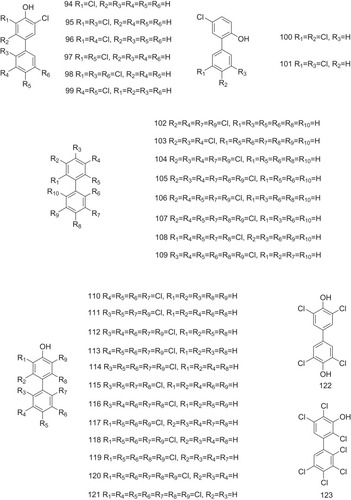
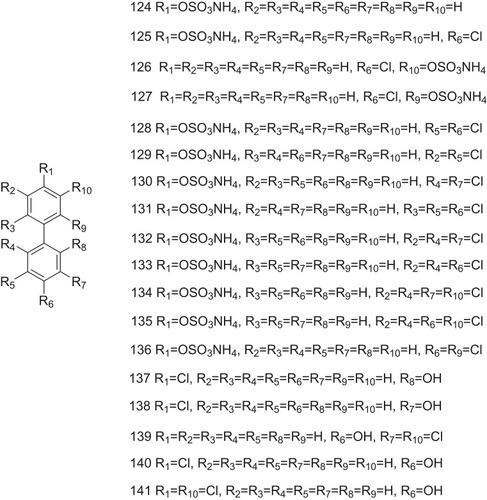
However, due to bioaccumulation and toxicity to the human body, PCBs and OH-PCBs are exclusively used in industry, such as lubricants and coolants.Citation42 In contrast to the higher-chlorinated congeners, the lower-chlorinated PCB congeners (LC-PCBs) are more susceptible to metabolic conversion, producing less toxicity to the human body.Citation43 Specifically, LC-PCBs may undergo oxidative metabolism and become hydroxylated LC-PCBs (OH-LC-PCBs) catalyzed by cytochrome P-450 enzymes.Citation44 LC-PCB sulfates, the further metabolites of LC-PCBs, are also artificially synthesized (124–141, ).Citation45,Citation46 The binding potentials of several LC-PCB sulfates (compounds 125–129) and OH-LC-PCBs (compounds 137–141) to TTR have been investigated that PCB sulfates non-covalently act as the ligands with higher affinity to TTR than their respective OH-PCB precursors. The equilibrium dissociation constants of LC-PCB sulfates in binding to TTR are in a low nanomolar range (4.8–16.8 nM), similar to that observed for T4 (4.7 nM) ().Citation46 Docking simulations provide multiple high-affinity model of binding. The lowest-energy binding conformations indicate an orientation that produces hydrogen bonding interactions between the sulfate groups and Lys15. The ortho-sulfate group appears to form hydrogen bonding with Leu110.
Figure 8 OH-LC-PCBs and PCB sulfates act as the potent inhibitors against ATTR.
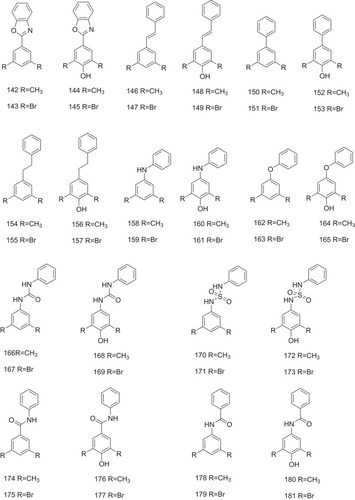
In addition to diphenyl ethers and biphenyls, 40 binary molecules based on 10 unique linker substructure (142–181, ) have been developed and evaluated as potent and highly selective ATTR inhibitors.Citation23 Of the 40 tested compounds, 20 compounds exhibit potent inhibitory activity against TTR aggregation with percent fibril formation (% F.F.) values of <20% (). TTR binding stoichiometry (P.S.) values are obtained by the plasma selectivity assay for the active candidate compounds. The average values of the % F.F. and P.S. are calculated and indicated as % F.F.ave and P.S.ave, respectively.Citation26,Citation47,Citation48 The two factors were then input into the equation below to afford an “Efficacy Score” for each linker, as indicated that higher efficacy scores correspond to more potent and selective linkers.
Table 2 The Efficacy Scores of Compounds 142–181
The results show that inhibitors with connection of two aryls by non-polar E-olefin or –CH2-CH2- exhibit higher efficacy scores () and more potent and selective activity in inhibiting ATTR ().Citation23
Figure 9 Compounds 142–181 with the different linkers as the potent inhibitors against ATTR are indicated.
Furthermore, the effects of Aryl-Z with different substitution positions and substituted groups in binding to TTR using a library of 80 benzamides (compounds 182–262, ) have been investigated. Of which, 56 compounds are involved in the evaluation of the amyloid inhibition potency, and 41 display increased potency, compared with their parent compound 182. The capacity of 41 compounds in selectively binding to TTR in blood plasma using ex vivo plasma TTR binding selectivity assay is also studied, as indicated in .Citation24,Citation49,Citation50 Kinetic stabilizer binding to TTR needs to adopt similar out-of-plane aryl conformation to achieve optimal receptor–ligand interactions. Optimal TTR-ligands interactions can also be affected by the differences in structure and energy between the preferred conformations of the free versus bound ligands. The rotational energy barriers for aryls adjacent to a linking NH group are dramatically higher than when adjacent to carbon or oxygen atoms. It is likely that other factors substantially affect ligand binding affinity and inhibitor potency, notably desolvation energies, which are difficult to predict.
Figure 10 Compounds 182–262 with different substitution positions of benzamides as the potent inhibitors against ATTR are indicated.
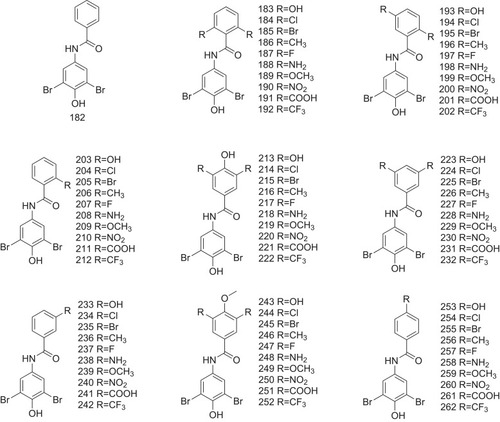
Table 3 The Efficacy Scores of Compounds 178–258
Inhibitors with position 2,6; 2,5; 2; 3,4,5 and 3,5 substitutions generate excellent potency and selectivity, and the efficacy scores of which are 0.789, 0.748, 0.734, 0.697, and 0.538, respectively, higher than that of their parent compound 182 (P.S. value of 0.41). It has been shown that substitution by -OH, -Cl, -Br, -CH3, -F, -NH2, and -OCH3 groups in Aryl-Z generates significant potency in interacting with TTR. The efficacy scores are 0.706, 0.667, 0.599, 0.597, 0.552, 0.525, and 0.456, respectively, and higher than that of the parent compound 182 (P.S. value of 0.41).Citation24
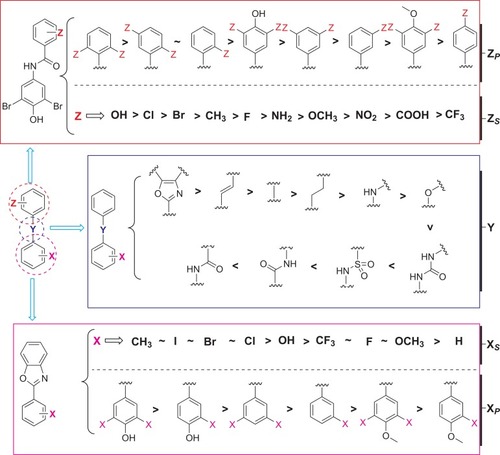
Another study reports some TTR-related amyloidogenesis inhibitors composed of two aryl rings and linkers of variable chemical composition ().Citation16,Citation51-61 The biological activity of these bisaryl molecules in inhibiting ATTR is significant, and the relative summary on different linker connections, substitution positions, and substituted groups in the two aryl rings has also been reported (). Two semi-quantitative models for predicting the optimal structures of potent and selective TTR kinetic stabilizers have been established. By comparing the efficacy scores, connection of two aryls with linkage through non-polar E-olefin or –CH2-CH2- generates more potency in interacting with TTR. The activities of Aryl-Z inhibitors with position 2.6, 2.5, and 2 substitutions are indicated as the most potency. Hydroxyl substitution on Aryl-Z inhibitors increases the most in activity. In contrast, the activity of Aryl-Z inhibitors with trifluoromethyl substitution is lowest. In addition, the substitution of 4ʹ-hydroxyl groups on Aryl-X can improve the inhibitory activity.Citation62
Figure 11 Summary of the structure–activity relationships of small molecule ATTR inhibitors composed of two aryl rings and variable linkers.
Flavonoids and Isoflavones
Flavonoid is considered as a representative compound that exhibits inhibitory activity against TTR-related amyloid fibril formation. Nine flavonoids (263–271, ) with different amounts of phenolic hydroxyl groups have been screened for their inhibitory activity against TTR-related amyloid fibril formation. Conversion from normally folded TTR to amyloid fibrils shows that the more hydroxyl groups are substituted on flavonoids, the lower the conversion degree to amyloid fibrils is observed. As indicated in , compounds 267–271 exhibit the percent conversion less than 10%. Apigenin (270) has been identified as the best inhibitor in all the tested flavones, exhibiting the conversion value of 6% at the concentration of 10.8 μM and completely inhibiting fibril formation at the concentration of 36 μM ().Citation60
Several natural flavonoids (272–278, ), including luteolin (272), have been tested to probe the influence of the number and position of hydroxyl groups in flavonoid scaffold on the interactions between flavonoids and TTR. The results indicated that changing the number and position of hydroxyl groups attached to the flavonoid core strongly influences flavonoid recognition by TTR. Hydrophobic interaction with Lys15 may be favorable to binding to TTR. The Lys15 side chains at the entrance of TTR orients the position of flavonoid AC rings on the center and bottom of the binding sites. The flexibility of the B ring also contributes to the binding entropy. Luteolin, being considered as a potent ATTR inhibitor, has a highest inhibitory potency against TTR disaggregation in vitro with an IC50 value of 5.68 ± 1.10 μM, lower than that of the natural ligand T4 with an IC50 value of 7.14 ± 1.08 μM ().Citation63,Citation64
Figure 12 Flavonoids act as small potent inhibitors against ATTR.
Genistein (279, ) and its O-glucoside conjugate genistin (280, ), which are isoflavones found in soybeans, are excellent acid-mediated ATTR inhibitors and reduce acid-mediated fibril formation to <10%. Additionally, these two natural products exhibit high selectivity in binding to TTR in plasma. Another two natural isoflavones from soybeans, daidzein (281, ) and its O-glucoside conjugate (282, ), exhibit lower potency in fibril aggregation inhibition as they lack the hydroxyl groups at position 5 on genistein and genistin. This suggests that the hydroxyl group at position 5 on flavonoids plays an important role in fibril aggregation inhibition.Citation65
Figure 13 Isoflavones act as small potent inhibitors against ATTR.
β-Aminoxypropionic Acids
A series of β-aminoxypropionic acids (283–299, ) and their derivatives (300–310, ) have been synthesized and reported as TTR-related amyloid formation inhibitors.Citation66–Citation68 These compounds contain a flexible oxime-based tether between the aromatic (aryl or fluorenyl) and acidic moieties, which are structurally distinct from the native ligand thyroxine and typical halogenated biaryl NSAID-like inhibitors and avoid off-target hormonal or anti–inflammatory activity. The aromatic ring is docked into P3 and plays a role in deciding the binding mode for this series of TTR inhibitors, and the β-aminoxyethyl chain directs the carboxylic group to Lys15. The ortho-CF3 substitution on the phenyl ring is engaged in interaction with P3 residues Ala108, Leu110, Ser117, and Thr119. However, substitutions in the meta- and para-position do not interact with P3 to the same extent. Of the 28 tested compounds, six compounds (291, 293, 294, 302, 304, and 305, ) at a concentration of 3.6 μM show significant inhibition in TTR amyloidogenesis (fibril formation inhibition >50%) by using an in vitro fibril formation assay. Notably, fluorenyl compounds 293, 294 and 304 display stronger inhibitory activities in TTR-related fibril formation (69% inhibition for 293 and 294 and 65% for 304) than the well-known TTR amyloid inhibitor diflunisal (63% for diflunisal) (). Citation27,Citation68 Compound 293, the most potent inhibitor, also avoids anti–inflammatory activity and shows good selectivity in contrast to NSAID-based TTR inhibitors, determined by an in vivo carrageenan-induced paw edema assay in rats.Citation68
Figure 14 β-aminoxypropionic acids act as small potent inhibitors against ATTR.
Crown Ethers and Carboranes
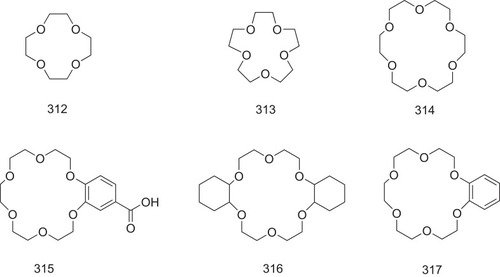
Of the seven lysine residues in the human transthyretin monomer, only Lys15 is located at the T4-binding site, providing a protonated amino group to form a salt bridge with the TTR stabilizers that have a ring structure.Citation28,Citation69-71 The cyclic hexamer 18-crown-6 (315) is well known as a receptor of protonated amino groups of lysine residues due to its reasonably sized cavity ().Citation70 CLR01, a Lys-specific molecular tweezer with a circular three-dimensional structure (311, ), has been reported to inhibit the fibrillization by binding to lysine residues in TTR protein and indicate an IC50 value of 50 μM in inhibiting the toxicity of amyloidogenic proteins in PC12 cells.Citation72,Citation73 However, due to its high molecular weight, CLR01 may not be a favorable TTR stabilizer. Therefore, small cyclic molecules are promising to be developed as TTR-related fibrosis inhibitors.
Figure 15 Lys-specific molecular tweezer CLR01 interacts with Lys by forming a salt bridge.
Six crown ethers (312–317, ) containing a ring structure with different sizes are tested and identified as TTR-related amyloidogenesis inhibitors using fluorescence probes and chemical cross-linking assays. Unlike crown ethers 312 and 313 that do not produce any effects on amyloid fibril formation, the hexameric cyclization crown ether 314 significantly suppresses amyloid fibril formation at a concentration of 20 nM. In addition, crown ethers 315 and 317 are found to be the most potent candidates among the selected crown ethers, inhibiting the formation of TTR-related amyloid fibril by 58% (at a concentration of 2 mM) and 47% (at a concentration of 10 mM), respectively ().Citation15 It indicates that a hexameric cyclization is required for crown ethers in the inhibition of TTR-related amyloid fibril formation, and the addition of a phenyl group may increase the inhibitory potency. However, the carboxyl phenyl group is more important for crown ethers 315 to bind to the allosteric sites of TTR than the cyclic polyether moiety. In addition, crown ethers 315 and 317 stabilize the TTR tetramer in a dose-dependent manner, compared with other crown ethers.Citation74
Carboranes, being rich and untapped, have been considered as a novel class of inorganic pharmacophores, which contain a rigid skeleton of dicarba-closo-dodecaboranes. This rigid scaffolding structure affords carboranes extraordinary chemical properties, such as strong hydrophobicity, resistance to catabolism, and especially high selectivity. Furthermore, carboranes share approximately the same volume as the rotated three-dimensional aromatics.Citation75 A study on the structure and properties of carboranes shows that replacing a benzene ring in NSAIDs with the carborane moiety can improve their selectivity for TTR protein and decrease their anti-COX activity, indicating that the carborane-substituted compounds may have the potentials to be developed as ATTR inhibitors.
Figure 16 Crown ethers act as small potent inhibitors against ATTR.
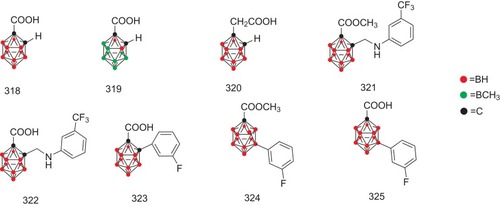
A group of carborane-based compounds (318–325, ) have been recognized as the promising inhibitors against TTR-related amyloid formation.Citation52,Citation60,Citation75 In TTR assays, compound 318 has been proven to be a moderate inhibitor against TTR-related amyloid formation with the percent fibril formation by 46% at a concentration of 3.6 μM. Consistently, compounds 322 and 325 have been identified as the most potent inhibitors with the percent fibril formation by 22% and 15%, respectively.Citation76
Figure 17 Carboranes act as small potent inhibitors against ATTR.
Oxazoles
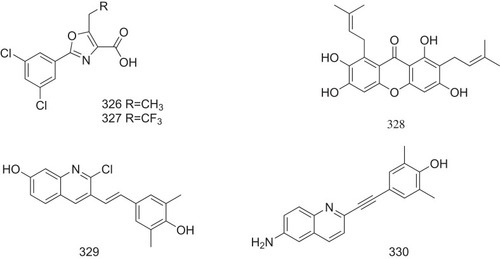
Aryl oxazoles bearing a carboxyl group at C-4 have been demonstrated to be an efficient class in inhibiting TTR amyloidogenesis. Substitution of ethyl, propyl, or CF3 group at C-5 may significantly enhance the inhibiting activity. In contrast, the substitution of a 3.5-dichlorophenyl at C-2 on the oxazole ring dramatically reduces its activity. Compounds 326 and 327 () have been showed to inhibit TTR activity and fibril formation at the dose of 3.6 μM and 7.2 μM, respectively ().Citation77
γ-Mangostin
γ-Mangostin (328, ), a xanthone derivative isolated from mangosteen, has been reported to bind to T4-binding sites directly, stabilize TTR tetramer, and inhibit the amyloid fibril formation of V30M amyloidogenic TTR with EC50 value of 7 ± 0.6 μM ().Citation78
Figure 18 Structures of oxazoles, γ-Mangostin, and quinolone derivatives.
Quinoline Derivatives
A library of quinolone derivatives has been synthesized. Among these quinolone derivatives, compound 329 has been screened to dock into the T4-binding sites of TTR and be the most potent in inhibiting TTR fibril formation with an IC50 value of 1.49 μM against wild-type TTR and 1.63 μM against V30M TTR variant. Intravenous injection of 2 mg/kg compound 329 and 330 () produces the peak plasma concentration (Cmax) in rats as 3.107 and 2.381 μg/mL, respectively, and half-life (T1/2) as 2.710 and 2.745 h, respectively ().Citation79
TTR-Targeting RNAi
RNA interference (RNAi) is a clinically validated technology, being a promising approach for managing ATTR amyloidosis. TTR-targeting siRNAs have been evaluated in hTTR V30M HSF1± mice. Knockdown of TTR expression by RNAi significantly inhibits the deposition of TTR and promotes regression of existing TTR deposits in pathological tissues, showing a greater activity than that of tafamidis and highlighting the potential of RNAi.Citation80 TTR siRNA promotes clearance of TTR deposition in the extracellular matrix of meninges and brain blood vessels. However, the cerebrospinal fluid TTR concentration is unchanged, although it is declined significantly in the blood. This indicates that TTR-targeting siRNA may not affect the neuroprotective activity of TTR in the central nervous system.Citation81
ATTR Inhibitors by Disaggregating TTR-Related Amyloid fibrils
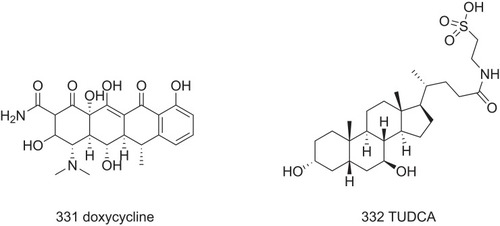
In addition to stabilizer of TTR tetramers, small molecules that directly disaggregate amyloid fibrils have been developed to manage ATTR disease. Animal experiments show that doxycycline (331, ) inhibits the formation of amyloid fibers and destroys the deposited fibers, exhibiting good effects on FAP and light-chain amyloidosis.Citation82,Citation83 Tauroursodeoxycholic acid (TUDCA) (332, ) can reduce the deposition of TTR precursor fibers, which is often combined with doxycycline to significantly promote the deposition of amyloid substances and non-fibrous TTR oligomers.Citation84,Citation85
Figure 19 Structures of doxycycline and TUDCA.
Several natural products have been analyzed for their activity in inducing amyloid fibril disaggregation, as revealed that gossypol, rottlerin, and hematoxylin (333–335, ) disrupt the pre-formed amyloid fibril. The competitive assays are involved to judge that, except hematoxylin, both gossypol and rottlerin competitively bind to the T4-binding site. Collectively, hematoxylin is an amyloid fibril disrupter, and gossypol and rottlerin are dual inhibitors.Citation7 Indeed, some biphenyl ether compounds have been shown to exhibit TTR stabilization and fibril disruption activities.Citation18
Figure 20 Structures of gossypol, rottlerin, and hematoxylin.
Clinical Perspectives
The familiar NSAID diflunisal (13, ), which may bind to the T4-binding site of TTR, stabilize the tetramer, and inhibit the amyloidogenesis, has already been approved as a prescription drug in more than 40 countries.Citation27,Citation28,Citation86-88 In addition, tafamidis (336, ), an oral small molecule inhibitor, has been developed for the treatment of FAP by stabilizing the TTR tetramer.Citation89–Citation91 However, tafamidis is exclusively effective in the early stage of FAP and currently available in Europe, Japan, Mexico, and Argentina till 2018.Citation92 In the management of TTR amyloid cardiomyopathy, tafamidis has been showed to be associated with lower all-cause mortality (29.5%) than placebo (42.9%). At month 30, tafamidis is indicated with a lower rate of decline in distance for the 6-min walk test and in KCCQ-OS score, reducing cardiovascular-related hospitalizations and the decline in functional capacity and life quality.Citation93 Pfizer is now working on the application for a US listing of tafamidis, which are expected to be approved by the end of 2019. Inotersen, an antisense oligonucleotide, has been recently demonstrated to improve disease course and life quality in the early hereditary transthyretin amyloidosis polyneuropathy by reducing the production of transthyretin.Citation94 Inotersen has been clinically approved in the European Union for the management of adult patients with hATTR-PN on August 20, 2018.Citation95 And now it is accepted by USA, European Union, and Canada.Citation94
Epigallocatechin-3-gallate (EGCG) (337, ) and resveratrol (338, ), two polyphenols isolated from green tea and grape skins, respectively, have been shown to stabilize TTR tetramers.Citation96,Citation97 EGCG may inhibit the deposition of TTR pre-fiber by up to 50% and decompose amyloid.Citation98 In addition, EGCG stabilizes myocardial mass in wtATTR-CM patients with good tolerance and no major safety concerns.Citation99,Citation100 Therefore, EGCG has been expected to be developed for the treatment of amyloid cardiomyopathy. In vitro, studies have shown that resveratrol may promote free monomers to form a tetramer and decrease the toxicity of pre-fiber TTR molecules.Citation97 However, the clinical application of resveratrol is limited, due to its poor drug-like physicochemical and ADMET properties.Citation101,Citation102
Figure 21 Structures of tafamidis, EGCG, and resveratrol.
Reduction or stop of endogenous TTR protein production is also an effective strategy to prevent the further progression of ATTR disease, as TTR has been shown to be a non-essential protein transporter of T4.Citation103 Oligonucleotides, such as patisiran, revusiran, and ASOs, are molecules that reduce the production of mutated proteins, thus modifying ATTR disease outcome.Citation104 Patisiran (ALN-TTR02) and revusiran (ALN-TTRsc), the two siRNA molecules currently under clinical investigation, contain a double-stranded RNA that targets wild-type and mutated TTR proteins, inhibit the synthesis of TTR, and promote the degradation of surrounding amyloid.Citation105–Citation107 Specifically, patisiran is a siRNA encapsulated in a lipid nanoparticle and binds to the mutant and wild-type TTR mRNA, reducing the level of TTR in the blood.Citation108 The results from Phase III clinical trials show that intravenous administration of patisiran significantly reduced blood TTR level at an optimal dose of 0.3 mg/kg of body weight for every 3 weeks.Citation109 Promisingly, patisiran has been approved by the FDA.Citation94 Revusiran, another one siRNA developed by Alnylam Company, is conjugated with N-acetylgalactosamine.Citation106 Unfortunately, revusiran was halted at phase III clinical trials, due to more deaths in the treatment group.Citation110 ASOs, an antisense oligonucleotide developed by Isis pharmaceuticals, have been used to treat amyloid cardiomyopathy. Combination of ASOs and TTR mRNA inhibits the production of mutant TTR and wild-type TTR. After 4 weeks of subcutaneous injection, the inhibitory rate may reach over 90%, indicating that this strategy can be applied for the treatment of hereditary ATTR and SSA.Citation111 In addition, the neuro-energy turbulence and living quality in FAP patients may be improved by ASOs in phase III clinical trials.Citation112 Tolcapone (SOM0266), a catechol-O-methyltransferase inhibitor in Phase II–III in the clinical trial, has been used as an adjunct to levodopa/carbidopa for management of Parkinson’s diseases. Tolcapone, better than tafamidis, may dock into the T4-binding sites of wild-type TTR and the V122I cardiomyopathy-associated TTR variant with enthalpically driven optimization, inhibiting their aggregation with EC50 values of 1.50 ± 0.12 μM and 4.72 ± 0.45 μM, respectively ().Citation113 In addition, tolcapone also inhibits the aggregation of the leptomeningeal-associated A25T-TTR variant. Tolcapone increases 52% of TTR stabilization at the dose of 200 mg within 2 h in phase A and increases 38.8% at the dose of 100 mg.Citation114 In vitro, tolcapone partially ameliorates the activation of caspase-3 induced by Y78F-TTR oligomers in RN22 cells.Citation115
Figure 22 The possible structure–activity relationship of TTR amyloidogenesis inhibitors, including bisayl structures with a linker, flavonoids and isoflavones, β-minoxypropionic acids, crown ethers and carboranes, and oxazoles.
Conclusion
In this article, the different types of ATTR inhibitors and a summary of the structure–activity relationship of some compounds have been reviewed (). Bisaryl structures with a linker, such as diphenyl ethers and biphenyl compounds, functions as the largest class of ATTR inhibitors. This class of compounds is mainly docked into the two T4 active pockets of TTR, maintaining the stability of the TTR tetramer, and the spatial distance between the two active pockets determines the length of the biphenyl linker. In addition, the types and position of substituents on the two aryl rings also affect the activity. Inhibitors that inhibit TTR amyloid fibril formation play an important role in the treatment of ATTR. Flavonoids and isoflavones, represented by luteolin and genistein, have been considered as the potential inhibitors against TTR protein fiber formation. β-aminoxypropionic acid, a class of well-known amyloid fibrinogen formation inhibitors, has the distinct advantage of avoiding off-target and producing incoherent anti–inflammatory activity. Crown ethers and carboranes also show good selectivity contrast to NSAID-based TTR inhibitors. Amyloid fibril disrupters are the novel therapeutic compounds, providing a new strategy for the management of ATTR. Some molecules that directly disaggregate amyloid fibrils have been developed. For example, antibiotic doxycycline and natural product hematoxylin have been shown the surprising disaggregation activity against amyloid fibrils.
Data Sharing Statement
The experimental data used to support the findings of this study are included within the article.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (81660371, 81860388, 81860261, and 81960883), The Youth Jinggang Scholar Program in Jiangxi Province, and Innovative Teamwork Project of Gannan Medical University (TD201707).
References
- SaraivaMJ, MagalhaesJ, FerreiraN, AlmeidaMR. Transthyretin deposition in familial amyloidotic polyneuropathy. Curr Med Chem. 2012;19(15):2304–2311. doi:10.2174/09298671280026923622471982
- SebastiaoMP, LamzinV, SaraivaMJ, DamasAM. Transthyretin stability as a key factor in amyloidogenesis: X-ray analysis at atomic resolution. J Mol Biol. 2001;306(4):733–744. doi:10.1006/jmbi.2000.441511243784
- Longo AlvesI, HaysMT, SaraivaMJ. Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur J Biochem. 1997;249(3):662–668. doi:10.1111/j.1432-1033.1997.00662.x9395311
- TerazakiH, AndoY, MisumiS, et al. A novel compound heterozygote (FAP ATTR Arg104His/ATTR Val30Met) with high serum transthyretin (TTR) and retinol binding protein (RBP) levels. Biochem Biophys Res Commun. 1999;264(2):365–370. doi:10.1006/bbrc.1999.151410529370
- SekijimaY, HammarstromP, MatsumuraM, et al. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83(3):409–417. doi:10.1097/01.LAB.0000059937.11023.1F12649341
- RobinsonLZ, ReixachN. Quantification of quaternary structure stability in aggregation-prone proteins under physiological conditions: the transthyretin case. Biochemistry. 2014;53(41):6496–6510. doi:10.1021/bi500739q25245430
- YokoyamaT, MizuguchiM. Inhibition of the amyloidogenesis of transthyretin by natural products and synthetic compounds. Biol Pharm Bull. 2018;41(7):979–984. doi:10.1248/bpb.b18-0016629962408
- BlakeCC, GeisowMJ, OatleySJ, RératB, RératC. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121(3):339–356. doi:10.1016/0022-2836(78)90368-6671542
- NaylorHM, NewcomerME. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38(9):2647–2653. doi:10.1021/bi982291i10052934
- ChoiS, ConnellyS, ReixachN, WilsonIA, KellyJW. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat Chem Biol. 2010;6(2):133–139. doi:10.1038/nchembio.28120081815
- CicconeL, Fruchart-gaillardC, MourierG, et al. Copper mediated amyloid-β binding to transthyretin. Sci Rep. 2018;8(1):13744. doi:10.1038/s41598-018-31808-530213975
- YeeAW, AldeghiM, BlakeleyMP, et al. A molecular mechanism for transthyretin amyloidogenesis. Nat Commun. 2019;10(1):925. doi:10.1038/s41467-019-08609-z30804345
- SaelicesL, SieversSA, SawayaMR, EisenbergDS. Crystal structures of amyloidogenic segments of human transthyretin. Protein Sci. 2018;27(7):1295–1303. doi:10.1002/pro.342029626847
- ZhangJ, GrundströmC, BrännströmK, et al. Interspecies variation between fish and human transthyretins in their binding of thyroid-disrupting chemicals. Environ Sci Technol. 2018;52(20):11865–11874.30226982
- YokoyamaT, MizuguchiM. Crown ethers as transthyretin amyloidogenesis inhibitors. J Med Chem. 2019;62(4):2076–2082. doi:10.1021/acs.jmedchem.8b0170030688456
- PetrassiHM, KlabundeT, SacchettiniJ, KellyJW. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J Am Chem Soc. 2000;122(10):2178–2192. doi:10.1021/ja993309v
- HammarstromP, WisemanRL, PowersET, KellyJW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299(5607):713–716. doi:10.1126/science.107958912560553
- GuptaS, ChhibberM, SinhaS, SuroliaA. Design of mechanism-based inhibitors of transthyretin amyloidosis: studies with biphenyl ethers and new structural templates. J Med Chem. 2007;50(23):5589–5599. doi:10.1021/jm070015917948976
- OrtoreG, MartinelliA. Identification of transthyretin fibril formation inhibitors using structure-based virtual screening. ChemMedChem. 2017;12(16):1327–1334. doi:10.1002/cmdc.20170005128422428
- GreeneMJ, KlimtchukES, SeldinDC, BerkJL, ConnorsLH. Cooperative stabilization of transthyretin by clusterin and diflunisal. Biochemistry. 2015;54(2):268–278. doi:10.1021/bi501124925478940
- UedaM, AndoY. Recent advances in transthyretin amyloidosis therapy. Transl Neurodegener. 2014;3:19. doi:10.1186/2047-9158-3-1925228988
- ChoiS, ReixachN, ConnellyS, JohnsonSM, WilsonIA, KellyJW. A substructure combination strategy to create potent and selective transthyretin kinetic stabilizers that prevent amyloidogenesis and cytotoxicity. J Am Chem Soc. 2010;132(4):1359–1370. doi:10.1021/ja908562q20043671
- JohnsonSM, ConnellyS, WilsonIA, KellyJW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51(20):6348–6358. doi:10.1021/jm800435s18811132
- JohnsonSM, ConnellyS, WilsonIA, KellyJW. Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2009;52(4):1115–1125. doi:10.1021/jm801347s19191553
- WojtczakA, CodyV, LuftJ, PangbornW. Structure of rat transthyretin (rTTR) complex with thyroxine at 2.5 Å resolution: first non-biased insight into thyroxine binding reveals different hormone orientation in two binding sites. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt8):1061–1070. doi:10.1107/S090744490100723511468389
- PurkeyHE, DorrellMI, KellyJW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98(10):5566–5571. doi:10.1073/pnas.09143179811344299
- Adamski-WernerSL, PalaninathanSK, SacchettiniJC, KellyJW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355–374. doi:10.1021/jm030347n14711308
- KlabundeT, PetrassiHM, OzaVB, RamanP, KellyJW, SacchettiniJC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7(4):312–321. doi:10.1038/7408210742177
- AlmeidaMR, MacedoB, CardosoI, et al. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem J. 2004;381(Pt 2):351–356. doi:10.1042/BJ2004001115080795
- MairalT, NietoJ, PintoM, et al. Iodine atoms: a new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS One. 2009;4(1):e4124. doi:10.1371/journal.pone.000412419125186
- LoconteV, MenozziI, FerrariA, et al. Structure-activity relationships of flurbiprofen analogues as stabilizers of the amyloidogenic protein transthyretin. J Struct Biol. 2019;208(2):165–173. doi:10.1016/j.jsb.2019.08.01131473362
- SafeS. Toxicology, structure-function relationship, and human and environmental health impacts of polychlorinated biphenyls: progress and problems. Environ Health Perspect. 1993;100:259–268. doi:10.1289/ehp.931002598354174
- SafeSH. Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol. 1994;24(2):87–149.8037844
- SafeS. Human toxicology of chlorinated organic micropollutants In: HesterRE, HarrisonRM, editors. Chlorinated Organic Micropollutants. Vol. 6 The Royal Society of Chemistry; 1996:73–88.
- KossG, WölfleD. Chapter 29 - Dioxin and dioxin-like polychlorinated hydrocarbons and biphenyls In: MarquardtH, SchäferSG, McClellanR, WelschF, editors. Toxicology. San Diego: Academic Press; 1999:699–728.
- CheekAO, KowK, ChenJ, McLachlanJA. Potential mechanisms of thyroid disruption in humans: interaction of organochlorine compounds with thyroid receptor, transthyretin, and thyroid-binding globulin. Environ Health Perspect. 1999;107(4):273–278. doi:10.1289/ehp.99107273
- ChauhanKR, KodavantiPRS, McKinneyJD. Assessing the role of ortho-substitution on polychlorinated biphenyl binding to transthyretin, a thyroxine transport protein. Toxicol Appl Pharmacol. 2000;162(1):10–21. doi:10.1006/taap.1999.882610631123
- LehmlerHJ, RobertsonLW. Synthesis of hydroxylated PCB metabolites with the Suzuki-coupling. Chemosphere. 2001;45(8):1119–1127. doi:10.1016/S0045-6535(01)00052-211695625
- PurkeyHE, PalaninathanSK, KentKC, et al. Hydroxylated polychlorinated biphenyls selectively bind transthyretin in blood and inhibit amyloidogenesis: rationalizing rodent PCB toxicity. Chem Biol. 2004;11(12):1719–1728. doi:10.1016/j.chembiol.2004.10.00915610856
- SandauCD, MeertsIATM, LetcherRJ, et al. Identification of 4-hydroxyheptachlorostyrene in polar bear plasma and its binding affinity to transthyretin: a metabolite of octachlorostyrene? Environ Sci Technol. 2000;34(18):3871–3877. doi:10.1021/es001134f
- MorseDC, WehlerEK, WesselingW, KoemanJH, BrouwerA. Alterations in rat brain thyroid hormone status following pre- and postnatal exposure to polychlorinated biphenyls (Aroclor 1254). Toxicol Appl Pharmacol. 1996;136(2):269–279. doi:10.1006/taap.1996.00348619235
- MargaretO. James. Polychlorinated Biphenyls: Metabolism and Metabolites. In: Robertson LW, Hansen LG, editors. Recent Advances in Environmental Toxicology and Health Effects. University Press of Kentucky; 2001:35–46.
- RobertsonLW, LudewigG. Polychlorinated biphenyl (PCB) carcinogenicity with special emphasis on airborne PCBs. Air Qual Control. 2011;71(1–2):25–32.
- LetcherRJ, Klasson-wehlerE, BergmanA. Methyl sulfone and hydroxylated metabolites of polychlorinated biphenyls In: HutzingerO, PaasivirtaJ, editors. Volume 3 Anthropogenic Compounds Part K. Berlin, Heidelberg: Springer Berlin Heidelberg; 2000:315–359.
- LiX, ParkinS, DuffelMW, RobertsonLW, LehmlerH-J. An efficient approach to sulfate metabolites of polychlorinated biphenyls. Environ Int. 2010;36(8):843–848. doi:10.1016/j.envint.2009.02.00519345419
- GrimmFA, LehmlerH-J, HeX, RobertsonLW, DuffelMW. Sulfated metabolites of polychlorinated biphenyls are high-affinity ligands for the thyroid hormone transport protein transthyretin. Environ Health Perspect. 2013;121(6):657–662. doi:10.1289/ehp.120619823584369
- LashuelHA, WurthC, WooL, KellyJW. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry. 1999;38(41):13560–13573. doi:10.1021/bi991021c10521263
- JohnsonSM, ConnellyS, WilsonIA, KellyJW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem. 2008;51(2):260–270. doi:10.1021/jm070873518095641
- GlaserK, SungML, O’neillK, et al. Etodolac selectively inhibits human prostaglandin G/H synthase 2 (PGHS-2) versus human PGHS-1. Eur J Pharmacol. 1995;281(1):107–111. doi:10.1016/0014-2999(95)00302-28566109
- InoueA, YamakawaJ, YukiokaM, MorisawaS. Filter-binding assay procedure for thyroid hormone receptors. Anal Biochem. 1983;134(1):176–183. doi:10.1016/0003-2697(83)90280-46318596
- OzaVB, PetrassiHM, PurkeyHE, KellyJW. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 1999;9(1):1–6. doi:10.1016/S0960-894X(98)00696-99990446
- BauresPW, OzaVB, PetersonSA, KellyJW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Med Chem. 1999;7(7):1339–1347. doi:10.1016/S0968-0896(99)00066-810465408
- MillerSR, SekijimaY, KellyJW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84(5):545–552. doi:10.1038/labinvest.370005914968122
- JohnsonSM, WisemanRL, SekijimaY, GreenNS, Adamski-wernerSL, KellyJW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38(12):911–921. doi:10.1021/ar020073i16359163
- GreenNS, PalaninathanSK, SacchettiniJC, KellyJW. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J Am Chem Soc. 2003;125(44):13404–13414. doi:10.1021/ja030294z14583036
- WisemanRL, JohnsonSM, KelkerMS, FossT, WilsonIA, KellyJW. Kinetic stabilization of an oligomeric protein by a single ligand binding event. J Am Chem Soc. 2005;127(15):5540–5551. doi:10.1021/ja042929f15826192
- PetrassiHM, JohnsonSM, PurkeyHE, et al. Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloidogenesis: kinetic stabilization of the native state. J Am Chem Soc. 2005;127(18):6662–6671. doi:10.1021/ja044351f15869287
- JohnsonSM, PetrassiHM, PalaninathanSK, et al. Bisaryloxime ethers as potent inhibitors of transthyretin amyloid fibril formation. J Med Chem. 2005;48(5):1576–1587. doi:10.1021/jm049274d15743199
- RazaviH, PalaninathanSK, PowersET, et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed Engl. 2003;42(24):2758–2761. doi:10.1002/anie.20035117912820260
- BauresPW, PetersonSA, KellyJW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6(8):1389–1401. doi:10.1016/S0968-0896(98)00130-89784876
- FerreiraN, SaraivaMJ, AlmeidaMR. Uncovering the neuroprotective mechanisms of curcumin on transthyretin amyloidosis. Int J Mol Sci. 2019;20:6. doi:10.3390/ijms20061287
- ConnellyS, MortensonDE, ChoiS, et al. Semi-quantitative models for identifying potent and selective transthyretin amyloidogenesis inhibitors. Bioorg Med Chem Lett. 2017;27(15):3441–3449. doi:10.1016/j.bmcl.2017.05.08028625364
- TrivellaDBB, Dos ReisCV, LimaLMTR, FoguelD, PolikarpovI. Flavonoid interactions with human transthyretin: combined structural and thermodynamic analysis. J Struct Biol. 2012;180(1):143–153. doi:10.1016/j.jsb.2012.07.00822842046
- IakovlevaI, BegumA, PokrzywaM, WalfridssonM, Sauer-erikssonAE, OlofssonA. The flavonoid luteolin, but not luteolin-7-O-glucoside, prevents a transthyretin mediated toxic response. PLoS One. 2015;10(5):e0128222. doi:10.1371/journal.pone.012822226020516
- GreenNS, FossTR, KellyJW. Genistein, a natural product from soy, is a potent inhibitor of transthyretin amyloidosis. Proc Natl Acad Sci U S A. 2005;102(41):14545–14550. doi:10.1073/pnas.050160910216195386
- MacchiaB, BalsamoA, LapucciA, et al. Molecular design, synthesis, and antiinflammatory activity of a series of beta-aminoxypropionic acids. J Med Chem. 1990;33(5):1423–1430. doi:10.1021/jm00167a0232329564
- BalsamoA, BertiniS, GervasiG, et al. Enantiopure 3-(arylmethylidene)aminoxy-2-methylpropionic acids: synthesis and antiinflammatory properties. Eur J Med Chem. 2001;36(10):799–807. doi:10.1016/S0223-5234(01)01275-211738487
- PalaninathanSK, MohamedmohaideenNN, OrlandiniE, et al. Novel transthyretin amyloid fibril formation inhibitors: synthesis, biological evaluation, and X-ray structural analysis. PLoS One. 2009;4(7):e6290. doi:10.1371/journal.pone.000629019621084
- OshimaT, SuetsuguA, BabaY. Extraction and separation of a lysine-rich protein by formation of supramolecule between crown ether and protein in aqueous two-phase system. Anal Chim Acta. 2010;674(2):211–219. doi:10.1016/j.aca.2010.06.03920678632
- Lee-C-C, Maestre-reynaM, HsuK-C, et al. Crowning proteins: modulating the protein surface properties using crown ethers. Angew Chem Int Ed Engl. 2014;53(48):13054–13058. doi:10.1002/anie.20140566425287606
- TomarD, KhanT, SinghRR, et al. Crystallographic study of novel transthyretin ligands exhibiting negative-cooperativity between two thyroxine binding sites. PLoS One. 2012;7(9):e43522. doi:10.1371/journal.pone.004352222973437
- SinhaS, LopesDHJ, DuZ, et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc. 2011;133(42):16958–16969. doi:10.1021/ja206279b21916458
- FerreiraN, Pereira-henriquesA, AttarA, et al. Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics. 2014;11(2):450–461. doi:10.1007/s13311-013-0256-824459092
- ColonW, KellyJW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31(36):8654–8660. doi:10.1021/bi00151a0361390650
- JuliusRL, FarhaOK, ChiangJ, PerryLJ, HawthorneMF. Synthesis and evaluation of transthyretin amyloidosis inhibitors containing carborane pharmacophores. Proc Natl Acad Sci U S A. 2007;104(12):4808–4813. doi:10.1073/pnas.070031610417360344
- MiroyGJ, LaiZ, LashuelHA, PetersonSA, StrangC, KellyJW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93(26):15051–15056. doi:10.1073/pnas.93.26.150518986762
- RazaviH, PowersET, PurkeyHE, et al. Design, synthesis, and evaluation of oxazole transthyretin amyloidogenesis inhibitors. Bioorg Med Chem Lett. 2005;15(4):1075–1078. doi:10.1016/j.bmcl.2004.12.02215686915
- YokoyamaT, UedaM, AndoY, MizuguchiM. Discovery of γ-mangostin as an amyloidogenesis inhibitor. Sci Rep. 2015;5:13570. doi:10.1038/srep1357026310724
- KimB, ParkH, LeeSK, et al. Systemic optimization and structural evaluation of quinoline derivatives as transthyretin amyloidogenesis inhibitors. Eur J Med Chem. 2016;123:777–787. doi:10.1016/j.ejmech.2016.08.00327541261
- ButlerJS, ChanA, CostelhaS, et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid. 2016;23(2):109–118. doi:10.3109/13506129.2016.116088227033334
- GoncalvesP, MartinsH, CostelhaS, MaiaLF, SaraivaMJ. Efficiency of silencing RNA for removal of transthyretin V30M in a TTR leptomeningeal animal model. Amyloid. 2016;23(4):249–253. doi:10.1080/13506129.2016.125628227884058
- CardosoI, SaraivaMJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20(2):234–239. doi:10.1096/fj.05-4509com16449795
- WardJE, RenR, ToraldoG, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118(25):6610–6617. doi:10.1182/blood-2011-04-35164321998211
- CardosoI, MartinsD, RibeiroT, MerliniG, SaraivaMJ. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74. doi:10.1186/1479-5876-8-7420673327
- ObiciL, CorteseA, LozzaA, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a Phase II study. Amyloid. 2012;19(Suppl 1):34–36. doi:10.3109/13506129.2012.67850822551192
- SekijimaY, DendleMA, KellyJW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–249. doi:10.1080/1350612060096088217107884
- TojoK, SekijimaY, KellyJW, IkedaS-I. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56(4):441–449. doi:10.1016/j.neures.2006.08.01417028027
- SekijimaY. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther. 2014;39(3):225–233. doi:10.1111/jcpt.2014.39.issue-324749898
- CoelhoT, MaiaLF, da SilvaAM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–2814. doi:10.1007/s00415-013-7051-723974642
- CorteseA, VitaG, LuigettiM, et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–924. doi:10.1007/s00415-016-8064-926984605
- BulawaCE, ConnellyS, DevitM, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi:10.1073/pnas.112100510922645360
- Waddington CruzM, BensonMD. A review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4(2):61–79. doi:10.1007/s40120-015-0031-326662359
- MaurerMS, SchwartzJH, GundapaneniB, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa180568930145929
- MathewV, WangAK. Inotersen: new promise for the treatment of hereditary transthyretin amyloidosis. Drug Des Devel Ther. 2019;13:1515–1525. doi:10.2147/DDDT.S162913
- KeamSJ. Inotersen: first global approval. Drugs. 2018;78(13):1371–1376. doi:10.1007/s40265-018-0968-530120737
- MerelesD, BussSJ, HardtSE, HunsteinW, KatusHA. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin Res Cardiol. 2010;99(8):483–490. doi:10.1007/s00392-010-0142-x20221615
- BourgaultS, ChoiS, BuxbaumJN, KellyJW, PriceJL, ReixachN. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410(4):707–713. doi:10.1016/j.bbrc.2011.04.13321557933
- FerreiraN, SaraivaMJ, AlmeidaMR. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “In vivo” evidence from FAP mice models. PLoS One. 2012;7(1):e29933. doi:10.1371/journal.pone.002993322253829
- Aus Dem SiepenF, BauerR, AurichM, et al. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: an observational study. Drug Des Devel Ther. 2015;9:6319––6325.
- CappelliF, MartoneR, TaborchiG, et al. Epigallocatechin-3-gallate tolerability and impact on survival in a cohort of patients with transthyretin-related cardiac amyloidosis. A single-center retrospective study. Intern Emerg Med. 2018;13(6):873–880. doi:10.1007/s11739-018-1887-x29882023
- AndradeS, RamalhoMJ, PereiraM, LoureiroJA. Resveratrol brain delivery for neurological disorders prevention and treatment. Front Pharmacol. 2018;9:1261. doi:10.3389/fphar.2018.0126130524273
- SaqibU, KelleyTT, PanguluriSK, et al. Polypharmacology or promiscuity? Structural interactions of resveratrol with its bandwagon of targets. Front Pharmacol. 2018;9:1201. doi:10.3389/fphar.2018.0120130405416
- PalhaJA, HaysMT. Morreale de EscobarG, EpiskopouV, GottesmanME, SaraivaMJ. Transthyretin is not essential for thyroxine to reach the brain and other tissues in transthyretin-null mice. Am J Physiol. 1997;272(3 Pt 1):E485–E493. doi:10.1152/ajpendo.1997.272.3.E4859124556
- NiemietzC, ChandhokG, SchmidtH. Therapeutic oligonucleotides targeting liver disease: TTR amyloidosis. Molecules. 2015;20(10):17944–17975. doi:10.3390/molecules20101794426437390
- CrookeST, WitztumJL, BennettCF, BakerBF. RNA-targeted therapeutics. Cell Metab. 2018;27(4):714–739. doi:10.1016/j.cmet.2018.03.00429617640
- ShenX, CoreyDR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–1600. doi:10.1093/nar/gkx123929240946
- RossorAM, ReillyMM, SleighJN. Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol. 2018;18(2):126–131. doi:10.1136/practneurol-2017-00176429455156
- CoelhoT, AdamsD, SilvaA, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi:10.1056/NEJMoa120876023984729
- AdamsD, Gonzalez-duarteA, O’riordanWD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa171615329972753
- GarberK. Alnylam terminates revusiran program, stock plunges. Nat Biotechnol. 2016;34(12):1213–1214. doi:10.1038/nbt1216-121327926717
- AckermannEJ, GuoS, BootenS, et al. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19(Suppl 1):43–44. doi:10.3109/13506129.2012.67314022494066
- HayashiY, JonoH. Recent advances in oligonucleotide-based therapy for transthyretin amyloidosis: clinical impact and future prospects. Biol Pharm Bull. 2018;41(12):1737–1744. doi:10.1248/bpb.b18-0062530504675
- Sant’annaR, GallegoP, RobinsonLZ, et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. 2016;7:10787. doi:10.1038/ncomms1078726902880
- GamezJ, SalvadoM, ReigN, et al. Transthyretin stabilization activity of the catechol-O-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: proof-of-concept study. Amyloid. 2019;26(2):74–84. doi:10.1080/13506129.2019.159770231119947
- FerreiraN, SaraivaMJ, AlmeidaMR. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett. 2011;585(15):2424–2430. doi:10.1016/j.febslet.2011.06.03021740906