Abstract
Context: Triosephosphate isomerase (TIM) is a ubiquitous enzyme that has been targeted for the discovery of small molecular weight compounds with potential use against Trypanosoma cruzi, the causative agent of Chagas disease. We have identified a new selective inhibitor chemotype of TIM from T. cruzi (TcTIM), 1,2,4-thiadiazol-5(4H)-one.
Objective: Study the mechanism of TcTIM inhibition by a 1,2,4-thiadiazol derivative.
Methods: We performed the biochemical characterization of the interaction of the 1,2,4-thiadiazol derivative with the wild-type and mutant TcTIMs, using DOSY-NMR and MS experiments. Studies of T. cruzi growth inhibition were additionally carried out.
Results and conclusion: At low micromolar concentrations, the compound induces highly selective irreversible inactivation of TcTIM through non-covalent binding. Our studies indicate that it interferes with the association of the two monomers of the dimeric enzyme. We also show that it inhibits T. cruzi growth in culture.
| Abbreviations: | ||
| TIM, | = | triosephosphate isomerase; |
| TcTIM, | = | triosephosphate isomerase from Trypanosoma cruzi; |
| TbTIM, | = | triosephosphate isomerase from Trypanosoma brucei; |
| LmTIM, | = | triosephosphate isomerase from Leishmania mexicana; |
| HsTIM, | = | triosephosphate isomerase from Homo sapiens; |
| DOSY-NMR, | = | diffusion-ordered nuclear magnetic resonance experiment; |
| EDTA, | = | ethylenediaminetetraacetic acid; |
| GAP, | = | glyceraldehyde 3-phosphate; |
| αGPDH, | = | α-glycerol phosphate dehydrogenase; |
| NS-nanoLC-MS, | = | nano spray ionization liquid chromatography-mass spectrometry analysis; |
| PL, | = | phospholipids; |
| BHI, | = | brain–heart infusion; |
| ID, | = | inhibitory dosis; |
| IC, | = | inhibitory concentration; |
| GdnHCl, | = | guanidine chloride; |
| HOMO, | = | highest energy occupied molecular orbital; |
Introduction
Trypanosoma cruzi (T. cruzi) has been identified for years as the causative agent of Chagas disease, an endemic disease widely disseminated in the American continent that affects 9.8–11.0 million people, leaving 60 million people at riskCitation1. Currently available therapies have shown to be inadequate concerning safety, efficacy, resistance, toxicity, difficulty of administration in impoverished conditions and cost issues. Despite the urgent need for new drugs, most pharmaceutical companies have neglected this disease, largely because of market policies.
T. cruzi dependence on glycolysis makes enzymes in this pathway excellent drug targets. Particularly, triosephosphate isomerase from Trypanosoma cruzi (TcTIM), widely investigated for the search of small molecular weight compounds that could selectively inhibit it, killing the parasiteCitation2–4.
TIM catalyzes the isomerization of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate in the fifth step of the glycolytic pathway. Most of the described TIMs are homodimers and each monomer consists of eight parallel β-strands, surrounded by eight α-helices forming the classical α/β-barrel structure. In TcTIMCitation5, the interface between monomers represents a significant portion of the molecular surface area of each polypeptide chain: approximately 1496 Å2 in TcTIMCitation5. In most organisms, including T. cruzi, only the homodimeric form of the enzyme is activeCitation6,Citation7.
We examined the interface of TIM from three different trypanosomatids, T. cruzi, Trypanosoma brucei (TbTIM) and Leishmania mexicana (LmTIM) and five other species, yeast, chicken, Homo sapiens (HsTIM), Plasmodium falciparum and Entamoeba histolytica. Our data shows that the structure of this portion of the interface is highly conserved in the enzymes from the trypanosomatids (TcTIM, LmTIM and TbTIM), differing from the other analyzed speciesCitation8. In fact, the identity of the 32 interfacial residues of TcTIM, TbTIM and LmTIM with the homology of HsTIM is approximately 52 %. TIMs from T. cruzi, T. brucei and L. mexicana have a high degree of sequence identity; being of 73.9 % for the trypanosomal TIMs, with a sequence similarity of 92.4 %Citation9. In fact, the potential use of protein–protein interfaces of homodimeric enzymes as targets for drug discovery has recently been reviewedCitation10.
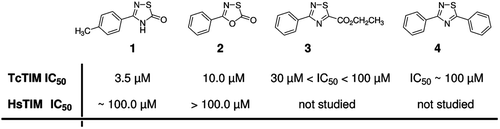
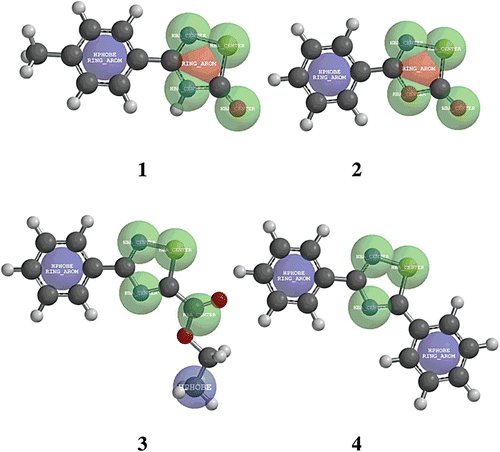
As part of our ongoing program in the search of molecules that could provide leads in the design of a new drug for the treatment of Chagas diseaseCitation8,Citation9,Citation11, we undertook a massive screening for inhibitors of TcTIM, using nearly 230 compounds from an in-house libraryCitation12. We identified two compounds (1 and 2, ), belonging to the 1,2,4-thiadiazol and 1,3,4-oxathiazol chemotypes, that displayed selectivity for TcTIM over HsTIM. Other 1,2,4-thiadiazol derivatives (i.e. 3 and 4, ) exerted very low inhibition.
Figure 1. Chemical structure of 1,2,4-thiadiazol derivatives and the previously studiedCitation12 1,3,4-oxathiazol. Derivatives 1 and 2 were also analyzed against HsTIM showing at least 10-fold selectivity for TcTIM.
In this work, we characterize the mechanism through which compound 1 causes TcTIM inactivation. Our data shows that it is a noncompetitive inhibitor that induces the dissociation of the two monomers and inhibits their association after they were unfolded by high guanidine chloride concentrations. In order to determine the residues or amino acids sequences of the enzyme that are essential for the action of compound 1, we studied its effect on some TcTIM mutants. We further show that, although compound 1 binds non-covalently to the enzyme, the binding character is strong as revealed by diffusion nuclear magnetic resonance (DOSY-NMR) experiments, and it is an effective inhibitor of T. cruzi growth.
Methods
Expression and purification of proteins
TcTIMCitation13, TbTIMCitation14, LmTIMCitation15, TcTIM 2,3,5-8 (a chimeral protein that contains amino acids 1 to 35 and 92 to 119 of TbTIM and the rest is identical to TcTIM)Citation16 and TcTIMC15A (a mutant protein, that contains an Ala-residue at position 14 of the sequence instead of a Cys present in wild-type TcTIM)Citation17, were expressed in Escherichia coli and purified as described in the referred literature. After purification, the enzymes, dissolved in 100 mM triethanolamine, 10 mM EDTA and 1 mM dithiothreitol (pH 8), were precipitated with ammonium sulfate (75% saturation) and stored at 4 °C. Before use, extensive dialysis against 100 mM triethanolamine/10 mM EDTA (pH 7.4) was performed. Protein concentration was determined by absorbance at 280 nmCitation18.
Activity assays
Activity was determined following the conversion of glyceraldehyde 3-phosphate into dihydroxyacetone phosphateCitation2. The decrease in absorbance at 340 nm was followed in a multicell Hewlett-Packard spectrophotometer at 25°C. The reaction mixture (1 mL, pH 7.4) contained 100 mM triethanolamine, 10 mM EDTA, 0.2 mM NADH, 1 mM glyceraldehyde 3-phosphate (GAP), and 0.9 units of α-glycerol phosphate dehydrogenase (α-GPDH). The reaction was initiated by addition of 5 ng/mL of the corresponding TIM. To calculate the kinetic parameters, we used increasing GAP concentrations from 0.05 to 10 mM. The data were adjusted according to the Michaelis-Menten model and the values of Km and Vmax were calculated by non-linear regression.
Effect of compound 1 on the stability of TcTIM dimers
The enzyme was incubated at concentrations between 6 × 10−4 and 6 × 10−1 µM for 2 h at 37°C with and without of compound 1 (3.5 µM), and the remaining activity was determined.
Folding/reactivation assay
TIM (TcTIM, HsTIM, or TbTIM, 0.5 mg/mL) was denatured with guanidine chloride (6 M) in a final volume of 200 µL for 1 h. A portion (10 µL) of this solution was diluted to a final volume of 1 mL containing increasing concentrations of compound 1. After 15 and 60 min, 1 µL of mixture was diluted to 1 mL and the activity was determined as previously described (see Activity assays).
DOSY-NMR experiments
NMR experiments were recorded on a Varian VNMR500 (499.8 MHz) with a z-axis gradient coil. The experiments were done with a spectral window of 8012.8 Hz (14.0 to −2.0 ppm) at 298 K. Compounds 1 and 2 were dissolved in deuterated 20 mM phosphate buffer, pH 7.4, at a final dose of 11 mM in 500 µL. In the different experiments, 70 µL of TcTIM (7.4 mg/mL 137 µM), prepared in the same buffer, were added to the compound solutions to reach a ratio compound:TcTIM of 650:1. Diffusion experiments: For each 2D-DOSY experiment 32 acquisitions were carried out, in which the gradient strength G went from 1.83 to 40.3 G cm−1, the diffusion time was 50 ms and the gradient pulse time 2 ms. Each experiment was 36 min long and the signal of H2O was suppressed by presaturation.
Mass spectrometry analysis
For the molecular mass determinations of the whole enzyme, native and TIM treated with compound 1 (using the same conditions used for enzyme activity assays) were bound on a C(4) Ziptip (P10, Millipore Corp., Bedford, MA), washed with H2O-0.1% formic acid and directly eluted into the ESI-ion trap with 30% AcCN and 0.1% formic acidCitation19. The deconvoluted spectra were obtained by using the Promass Deconvolution for Xcalibur Software (Thermo Electron Corp.).
Peptide mapping and nanospray ionization liquid chromatography-mass spectrometry analysis
Native and TIM treated with compound 1 (1 µmol) were digested with sequencing grade trypsin in 50 mM bicarbonate buffer, pH 8.0 at 37 °C for 15 h using an enzyme:substrate ratio of approximately 1:50 (w/w). In some cases, protein was reduced with dithiothreitol (25 mM) and alkylated with iodoacetamide (100 mM) before digestion. Peptide samples were analyzed by nanospray ionization liquid chromatography-mass spectrometry analysis (NSI-nanoLC-MS) using a LTQ Velos ion trap mass spectrometer (Thermo Electron Corp., San Jose, CA) in line with an Easy nLC (formerly Proxeon, Thermo Electron Corp., San Jose, CA). A reversed phase pre-column (EASY-Column, 2 cm, ID100 µm, 5 µm) in line with an analytical reverse phase column (EASY-Column, 10 cm, ID 75 µm, 3 µm) from Thermo (Thermo Electron Corp., San Jose, CA) were used at 300 nL/min, using a gradient from 0–45% of solvent B in 70 min (solvent A: 0.1% formic acid; solvent B: acetonitrile, 0.1% formic acid). The nanospray voltage was 1.4 kV, and the temperature of the capillary was 230°C. Peptides were detected in the positive ion mode using a mass range of 300–2000 m/z. The top 5 MS/MS peptide analyses were performed by data dependent acquisition. The data was analyzed by using Proteome Discoverer software (Thermo Electron Corp., San Jose, CA), using Sequest and/or Mascot as search engines, and allowing the following dynamic modifications: dehydro-Cys, carboxamidomethyl-Cys, Met oxidation.
Theoretical calculations
The molecular structures of compounds 1–4, were subjected to complete geometry optimization, in gas phase, using the PC SPARTAN 04 packageCitation20 as follows: Conformational search using the MMFF conformer module, the structure of the most stable conformer was optimized by applying density functional B3LYP-6-31G*. Structural, geometrical and electronic descriptors were extracted from comparisons.
Anti-T. cruzi activity of compound 1 encapsulated in liposomes
The liposomes were prepared by hydrating thin lipid films by shaking them with biological culture milieu at room temperature. The lipid films were obtained by evaporation at reduced pressure in a rotary evaporatorCitation21, from a chloroformic solution of phospholipid (PL) (P3556L-α-Phosphatidylcholine (100 mg), Aldrich) containing different concentrations of compound 1. The biological evaluation was performed as previously describedCitation22. Briefly, T. cruzi epimastigotes (Tulahuen 2 strain) were grown axenically at 28°C in BHI-Tryptose, complemented with 5% fetal calf serum. Cells were harvested in the late log phase, suspended in fresh culture medium, counted in a Neubauer chamber and placed in 24-well plates (2 × 106 cells/mL). Cell growth was measured by counting trypanosomes in the Neubauer chamber. Before inoculation, the medium was supplemented with the indicated amount of liposomes containing the compound to be analyzed. No effect on the growth of epimastigotes was observed at the maximum concentration of PL (1000 µM) without compound. The percent of growth inhibition was calculated as follows: {1 − [(Ap − A0p) / (Ac − A0c)]} × 100, where Ap = amount of parasites containing the compound to be analyzed on day 5; A0p = amount of parasites containing the compound to be analyzed, just after adding the compound (day 0); Ac = amount of parasites in the absence of compound (control) on day 5; A0c = amount of parasites in the absence of compound on day 0. To determine the ID50, parasite growth was followed in the absence (control), and the presence, of increasing concentrations of the corresponding compound. The IC50 values were considered as the drug concentrations required to decrease the number of parasites by half of those measured for untreated controls. Nifurtimox (Nfx) and Benznidazole (Bnz) were used as reference drugs.
Results
Effect of compounds 1 and 2 on the activity of different TIMs
We studied the effect of different concentrations of compounds 1 and 2 on the activity of TcTIM. The results show that although both compounds are effective inhibitors of TcTIM (), compound 1 (with an IC50 of 3.5 µM) is about three-times more effective than compound 2. The effect of different concentrations of compound 1 on the activity of TcTIM is shown in . It is important to denote that the activity of HsTIM is not affected by 100 µM of compound 2, and that the IC50 of compound 1 on the activity of HsTIM is around 100 µM (). To further examine the selectivity of these compounds for TcTIM, we assessed their effect on the activity of LmTIM and TbTIM. Our data shows that compounds 1 and 2, at a concentration of 100 µM, induced a rather small inhibition of the latter enzymes (). Thus, the compounds seem to be highly selective for TcTIM.
Table 1. Classic inhibition assay of Tc, Hs, Lm and Tb TIMs to evaluate the selectivity of the inhibitor compounds.
Figure 2. Effect of compounds 1 on the activity of TcTIM. (a) Lineweaver–Burk plot of TcTIM alone and TcTIM+compound 1 (3.5 µM). (b) TcTIM activity dependence on compound 1 concentration.
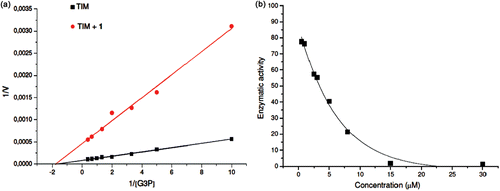
The kinetic studies of TcTIM with and without compound 1, using different concentrations of glyceraldehyde 3-phosphate, indicated that this compound is a noncompetitive inhibitor of the enzyme. At a concentration of 3.5 µM, compound 1 reduced the Vmax six-fold without significantly affecting the Km ().
Some of the most powerful inhibitors of TcTIM act by lowering the association between the two monomers of the enzymeCitation23. Accordingly, we explored if compound 1 exerts its action through the perturbation of monomer-monomer association. TcTIM was incubated with different concentrations of the compound for 2 h and the activity was measured (). The data shown in suggest that compound 1 exerts its action by favouring the dissociation over the association reaction of TIM monomers; the presence of compound 1 increased 15-fold the concentration of TIM needed to measure the same enzyme activity detected in the absence of the compound.
Figure 3. Effect of compound 1 on TcTIM stability. (a) Effect of compound 1 on TcTIM stability. The enzyme was incubated at the indicated concentrations for 2 h at 37°C with (▪) and without (•;) 3.5 µM compound 1 and the activity of each of the samples was measured. (b) Effect of compound 1 on the formation of active TcTIM from GdnHCl unfolded monomers. The experiment was performed as described in the Methods section. Activity was measured 15 (•) and 60 (▪) min after the denaturing mixture was diluted one hundred fold.
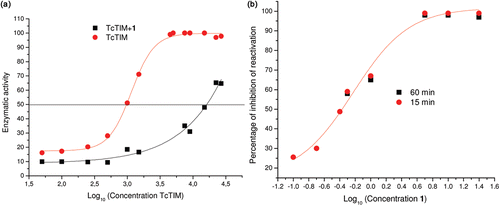
To examine if the disruption of the dimeric form of TIM is due to the association of compound 1 with its monomers, TcTIM was denatured in 6 M guanidine chloride (GdnHCl). In a second step of the experiment, the denaturing mixture was diluted in buffer containing different concentrations of the compound. As previously shown byCitation24, the removal of the denaturant by dilution leads to the refolding and association of the monomers to form the catalytically active dimer. This reaction can be followed by measuring the appearance of activity through time. shows that compound 1 effectively inhibits the formation of active TcTIM dimers from its corresponding unfolded monomers. The IC50 of compound 1 for the inhibition of dimer formation was 0.6 µM and the IC90 was 3.0 µM.
NMR experiments
The proton signals for compounds 1 and 2 incubated with, or without, TcTIM (ratio compound:TcTIM=260:1) displayed the same diffusion coefficient values in both conditions. The results obtained in this experiment suggest that these compounds could be establishing covalent or strong interactions with TcTIM, preventing us to evidence any weak compound-biomolecule interaction. In this conditions, compounds 1 and 2, strongly bound to TcTIM, displayed a diffusion coefficient similar to the protein alone and the fact that the new entity was at a lower concentration (free compound:bound compound = 650:1) led to the absence of new signals.
Mass spectrometry analysis
Native TIM and TIM treated with compound 1 were analyzed by mass spectrometry (see Materials and Methods) to ascertain if adducts of the inhibitor and the enzyme were formed. The deconvoluted spectra of native versus treated enzyme, in the same experimental conditions where inhibition was observed, showed no differences in the molecular mass of the enzyme. These results strongly suggest that the inhibitor is not forming a covalent bond with the enzyme (data not shown). To confirm the observation, we analyzed the peptide map of native versus treated TcTIM. In both cases, 98% of the sequence was covered, including all the Cys residues. No evidence of covalent binding of the inhibitor to TcTIM was detected (data not shown).
Theoretical calculations
Theoretical calculations were made in order to assess the contribution of the electrophilic centre of compound 1 to the inactivation of TcTIM; its molecular similarity to the less active or inert compounds 2–4 was also determined. Compound 1 possesses a very different electronic distribution when compared to derivatives 2–4 (); its nitrogen 2 (N2) is more negative and, consequently, may undergo protonation, promoting a heterocyclic opening process that may be related to its inactivating properties. Furthermore, the higher S1-N2 bond length for compound 1 () clearly reinforced the idea that this compound possesses the lowest bond order and consequently the highest capability to be opened by a biological nucleophile in the bond S1-N2. Additionally, we studied the covalent interaction between compound 1 and a model of low molecular mass nucleophile-thiol, β-mercaptoethanol, in order to probe these theoretical observations (Figure S1, Supplementary Material). The mass spectrometry analysis confirmed the reactive site (S1-N2) for compound 1 (Figures S2 and S3, Supplementary Material).
Figure 4. Calculated electronic distribution for compounds 1–4. (a) Potential electrostatic molecular maps. The solid arrow shows the reaction centre (sulfur 1, S1) and the dotted arrow points the protonated centre (nitrogen 2, N2). (b) HOMO maps (red-blue) superimposed to electron density bond maps (silver). The solid arrows show the contribution of S1 and N2 to the HOMO only in derivative 1. The S1-N2 bond lengths for each compound are also shown.
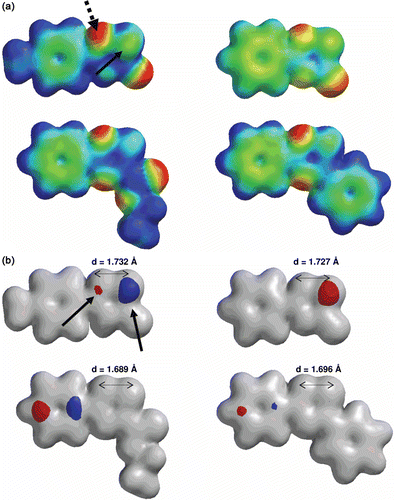
We also analyzed compounds 1–4, considering their stereo-electronic convergences, using the structural similarities-module implemented in SPARTAN package. The two more effective inhibitors, compounds 1 and 2, share common structural features and differ in some stereo-electronic features compared to the inactive thiadiazol molecules, compounds 3 and 4 ( and for details). The good inhibitors, 1 and 2, share the presence of a ring aromatic system (1,2,4-thiadiazol-5(4H)-one and 1,3,4-oxathiazol-2-one, red region, RING_AROM in ) with different stereo-electronic features than the inactive derivatives (3 and 4, absence of these red regions, ). Additionally, the weak inhibitors share the presence of a hydrophobic substituent, an ethyl group in compound 3 and a phenyl moiety in compound 4 (blue regions, HPHOBE and HPHOBE RING_AROM, respectively, ) that are not present in the structures of compounds 1 and 2. Both groups of structural characteristics could contribute to the adequate accessibility to TcTIM explaining the marked differences in the potency of compounds 1, 2, 3 and 4 in the inactivation of TcTIM.
Figure 5. Structural similarities of the analyzed compounds (1–4).
Attempts to identify the amino acid or regions of TcTIM that make it susceptible to the action of compound 1
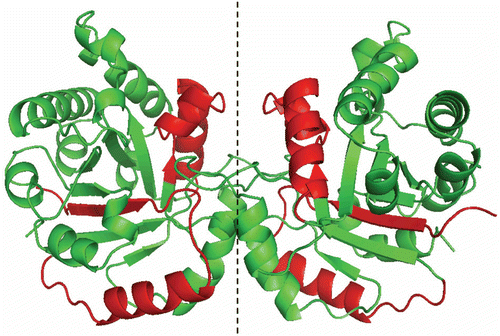
It has been reported that the integrity of the interface Cys of TcTIM is central in maintaining the stability of dimeric TcTIMCitation10. Therefore, we examined the effect of compound 1 on a C15A mutant of TcTIM (). The mutant was inactivated by compound 1 and was slightly more sensitive than the wild type. Recently, we reportedCitation16 that it is possible to use chimeras of TcTIM and TbTIM in order to identify the amino acids or amino acid sequences that are involved in a given property of the enzymes. Thus, we examined the effect of compound 1 on a TcTIM in which regions 1 (residues 1–35) and 4 (residues 92–119) were replaced by the corresponding regions of TbTIM, namely TcTIM 2,3,5-8 (). The chimera showed to be largely insensitive to both compounds 1 and 2 ().
Table 2. Classic inhibition assay, using modified TcTIMs, to evaluate the selectivity of the inhibitor 1.
Figure 6. Stereoview of TcTIM showing the regions of TbTIM (in red) that were grafted in TcTIM to generate the chimera studied herein.
Proof of concept
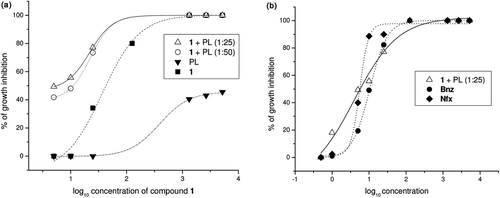
In order to test the hypothesis that TcTIM inhibition could affect the life of T. cruzi, we examined the effect of compound 1 on epimastigotes in culture. It is noted that we have previously evaluated the effect of compound 1 on epimastigotesCitation12, and observed that it displayed very low activity against the whole parasite. We hypothesized that the poor effect of compound 1 could be explained, by its relatively high hydrophilicity that could be affecting its penetration into the parasite. Accordingly, we encapsulated compound 1 into liposomes with different proportions of phospholipids (PL), and tested their effect on T. cruzi epimastigotes (Tulahuen 2 strain). In these conditions, compound 1 was an excellent inhibitor of growth for T. cruzi () with an IC50 of 5.8 µM, similar to the value found for inhibition of TcTIM activity in vitro.
Figure 7. T. cruzi growth inhibition assays. (a) Dose-response curves showing growth inhibition of T. cruzi by compound 1 incorporated into phospholipid (PL). (b) Dose-response curves showing growth inhibition of T. cruzi by compound 1 incorporated into PL and the reference drugs, Nifurtimox (Nfx) and Benznidazole (Bnz).
Discussion
Using a massive screening assay that monitors the inactivation of TcTIM, we identified the 1,2,4-thiadiazol derivative 1 as a small molecule with potential anti-T. cruzi activityCitation12. We found that this compound, together with its oxa-analogue compound 2, selectively inhibits the T. cruzi enzyme, with rather meager effects on TIMs from H. sapiens, Leishmania sp. and T. brucei. As shown here, compound 1 diminishes the stability of the TcTIM dimer and prevents its formation from unfolded monomers; furthermore, our data indicates that the inactivating effect of compound 1 on dimeric TcTIM is highly specific. Nonetheless, at submicromolar concentrations, compound 1 inhibits the formation of TbTIM and HsTIM dimers from their respective unfolded monomers (not shown), suggesting that all the tested TIMs have a binding site for compound 1. However, in TcTIM the union site for the compound seems to be accessible in the whole dimer, whereas in the other TIMs, compound 1 is able to bind to the inhibitory site only when the dimeric structure is exposed to the solvent. Thus, it would appear that the high selectivity of TcTIM to compound 1 is related to a higher solvent accessibility of the binding site for the molecule. It is important to denote that the DOSY-NMR experiments of compounds 1 and 2 after incubation with TcTIM (ratio compound:TcTIM = 650:1), indicated that these compounds establish strong interactions with the protein. However, it is important to consider that we could not detect covalent interactions between the enzyme and compounds 1 and 2, even though these may function as nucleophilic acceptors with the potential to bind covalently to some amino acid residues. Since we have not been able to obtain crystals of TcTIM in complex with compound 1, we could not elucidate its precise binding site. However, the data with the chimeras have given insight into the residues that contribute to the binding of the compounds to TcTIM.
In summary, we have found a compound that, in the low micromolar range, specifically inhibits the activity of TcTIM and inhibits T. cruzi epimastigotes growth.
Declaration of interest
The authors thank the financial supports from Cooperación Mexicana para el Desarrollo, from the Instituto de Ciencia y Tecnología del Distrito Federal No. 304/2009 (R. P-M.), from the Dirección General de Asuntos del Personal Académico, U.N.A.M. No. IN221812-3 (R. P-M.), from the Consejo Nacional de Ciencia y Tecnología, México No. 167823 (R.P-M.), and from RIDIMEDCHAG-CYTED. The authors also thank ANII for the scholarships to GA and the Biochemistry Lia Randall for reviewing the language. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Schofield CJ, Jannin J, Salvatella R. The future of Chagas disease control. Trends Parasitol 2006;22:583–588.
- Gómez-Puyou A, Saavedra-Lira E, Becker I, Zubillaga RA, Rojo-Domínguez A, Pérez-Montfort R. Using evolutionary changes to achieve species-specific inhibition of enzyme action–studies with triosephosphate isomerase. Chem Biol 1995;2:847–855.
- Velanker SS, Ray SS, Gokhale RS, Suma S, Balaram H, Balaram P et al. Triosephosphate isomerase from Plasmodium falciparum: the crystal structure provides insights into antimalarial drug design. Structure 1997;5:751–761.
- Cortés-Figueroa AA, Pérez-Torres A, Salaiza N, Cabrera N, Escalona-Montaño A, Rondán A et al. A monoclonal antibody that inhibits Trypanosoma cruzi growth in vitro and its reaction with intracellular triosephosphate isomerase. Parasitol Res 2008;102:635–643.
- Maldonado E, Soriano-García M, Moreno A, Cabrera N, Garza-Ramos G, de Gómez-Puyou M et al. Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. J Mol Biol 1998;283:193–203.
- Waley SG. Refolding of triose phosphate isomerase. Biochem J 1973;135:165–172.
- Zabori S, Rudolph R, Jaenicke R. Folding and association of triose phosphate isomerase from rabbit muscle. Z Naturforsch, C, Biosci 1980;35:999–1004.
- Olivares-Illana V, Pérez-Montfort R, López-Calahorra F, Costas M, Rodríguez-Romero A, Tuena de Gómez-Puyou M et al. Structural differences in triosephosphate isomerase from different species and discovery of a multitrypanosomatid inhibitor. Biochemistry 2006;45:2556–2560.
- Olivares-Illana V, Rodríguez-Romero A, Becker I, Berzunza M, García J, Pérez-Montfort R et al. Perturbation of the dimer interface of triosephosphate isomerase and its effect on Trypanosoma cruzi. PLoS Negl Trop Dis 2007;1:e1.
- Cardinale D, Salo-Ahen OM, Ferrari S, Ponterini G, Cruciani G, Carosati E et al. Homodimeric enzymes as drug targets. Curr Med Chem 2010;17:826–846.
- Cerecetto H, González M. Anti-T. cruzi agents: our experience in the evaluation of more than five hundred compounds. Mini Rev Med Chem 2008;8:1355–1383.
- Alvarez G, Aguirre-López B, Varela J, Cabrera M, Merlino A, López GV et al. Massive screening yields novel and selective Trypanosoma cruzi triosephosphate isomerase dimer-interface-irreversible inhibitors with anti-trypanosomal activity. Eur J Med Chem 2010;45:5767–5772.
- Ostoa-Saloma P, Garza-Ramos G, Ramírez J, Becker I, Berzunza M, Landa A et al. Cloning, expression, purification and characterization of triosephosphate isomerase from Trypanosoma cruzi. Eur J Biochem 1997;244:700–705.
- Borchert TV, Pratt K, Zeelen JP, Callens M, Noble ME, Opperdoes FR et al. Overexpression of trypanosomal triosephosphate isomerase in Escherichia coli and characterisation of a dimer-interface mutant. Eur J Biochem 1993;211:703–710.
- Kohl L, Callens M, Wierenga RK, Opperdoes FR, Michels PA. Triose-phosphate isomerase of Leishmania mexicana mexicana. Cloning and characterization of the gene, overexpression in Escherichia coli and analysis of the protein. Eur J Biochem 1994;220:331–338.
- García-Torres I, Cabrera N, Torres-Larios A, Rodríguez-Bolaños M, Díaz-Mazariegos S, Gómez-Puyou A et al. Identification of amino acids that account for long-range interactions in two triosephosphate isomerases from pathogenic trypanosomes. PLoS ONE 2011;6:e18791.
- Zomosa-Signoret V, Aguirre-López B, Hernández-Alcántara G, Pérez-Montfort R, de Gómez-Puyou MT, Gómez-Puyou A. Crosstalk between the subunits of the homodimeric enzyme triosephosphate isomerase. Proteins 2007;67:75–83.
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 1995;4:2411–2423.
- Rhoads TW, Lopez NI, Zollinger DR, Morré JT, Arbogast BL, Maier CS et al. Measuring copper and zinc superoxide dismutase from spinal cord tissue using electrospray mass spectrometry. Anal Biochem 2011;415:52–58.
- Spartan’04; Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370. Irvine, California 92612 USA.
- Crommelin DJA, Schreier H. (1994) Liposomes. In: Kreuter J, ed. Colloidal Drug Delivery Systems. New York: Marcel Dekker, 73–190.
- Benitez D, Cabrera M, Hernández P, Boiani L, Lavaggi ML, Di Maio R et al. 3-Trifluoromethylquinoxaline N,N’-dioxides as anti-trypanosomatid agents. Identification of optimal anti-T. cruzi agents and mechanism of action studies. J Med Chem 2011;54:3624–3636.
- Téllez-Valencia A, Avila-Ríos S, Pérez-Montfort R, Rodríguez-Romero A, Tuena de Gómez-Puyou M, López-Calahorra F et al. Highly specific inactivation of triosephosphate isomerase from Trypanosoma cruzi. Biochem Biophys Res Commun 2002;295:958–963.
- Zomosa-Signoret V, Hernández-Alcántara G, Reyes-Vivas H, Martínez-Martínez E, Garza-Ramos G, Pérez-Montfort R et al. Control of the reactivation kinetics of homodimeric triosephosphate isomerase from unfolded monomers. Biochemistry 2003;42:3311–3318.