
Abstract
Herein, we report compound KST9046, a new agent possessing quinazoline-urea scaffold. Preliminary biological evaluation done by the National Cancer Institute (NCI), USA, showed a great inhibitory effect of KST9046 over the 60 cell-line tumor panel. Accordingly, it was selected for a dose-response assay; a broad spectrum antiproliferative activity with GI50 ranging from 1.3 to 3.9 µM was exerted. To explore a potential kinase inhibitory effect, KST9046 was applied at a single dose of 10 µM against a kinase panel of 347 different enzymes representing >50% of the predicted human protein kinome. Interestingly, selective inhibition of 76% was observed on DDR1 kinase. Further, KST9046 showed an IC50 value of 4.38 µM for DDR1. A molecular docking model presented KST9046 as a potential type III inhibitor for DDR1 kinase with an allosteric mode of interaction, which may offer an explanation for its selectivity. As further investigation, CYP450 assay was carried out for KST9046, it showed a promising toxicity profile against four different isoforms. Based on these findings, KST9046 can be further evaluated as a promising safe new hit for the development of broad spectrum anticancer agents with a selectivity for DDR1 kinase.
Introduction
Cancer is thought to reflect a multi-step process, resulting from an accumulation of inherited and/or acquired defects in genes involved in cell survival and proliferation. The human genome encodes approximately 500 predicted protein kinases, many of them are participating in signal transduction pathways that regulate cell growth and survivalCitation1. Consequently, abnormal phosphorylation due to kinases overexpression can lead to numerous types of cancerCitation2–4. Currently, several protein kinase inhibitors have been approved by the FDA for the treatment of different types of both solid and non-solid tumors of various origins and types. However, many kinase inhibitors are in fact not selective for one particular kinase but target several kinases, which potentially increases the risk of unwanted effects and toxicity.
Achieving inhibitor selectivity for particular protein kinases often remains a significant challenge in the development of new small molecules as drugs or as tools for chemical biology research. Based on the mechanism of action, there are four types of kinase inhibitors; Type I which interacts directly with the ATP binding pocket via the hinge region. This type lacks the selectivity because the ATP-binding pocket is highly conserved among members of the kinase family. Type II binds to the hinge region as well as in a pocket created by the DFG residues of the activation loop in “DFG-out” conformation. As a result, they are able to achieve a higher level of selectivity compared to type I. The highest selectivity profile could be expected by both; type III (allosteric modulators exclusively bind in a pocket adjacent to the ATP site, without making any interactions with the hinge region) and type IV (allosteric modulators bind to a remote allosteric site on the kinase)Citation5. In this sense, kinase inhibitors possessing an allosteric mode of action could exhibit the highest degree of selectivity because they bind to less conserved allosteric sites outside the ATP pocket and exploit regulatory mechanisms that are unique to a specific kinaseCitation6.
Discoidin domain receptor 1 (DDR1), binds to several collagens and widely expressed in epithelial cells of different tissues. Recent reports indicated altered expression of DDR1 kinase in various human cancers, including brain, ovary, breast, lung and esophagous cancers, suggesting a potential role of DDR1 in tumor progressionCitation7–11. Although the exact mechanisms by which this receptor may contribute to oncogenesis is unknown, it has been demonstrated that it may act as a critical regulator of cell proliferation, adhesion, migration and subsequent tumor metastasisCitation12. Hence, inhibition of DDR1 function may provide a potential approach to selectively enhance treatment of such tumors.
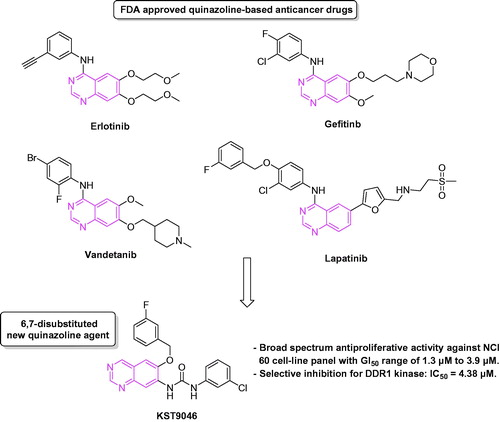
In a previous work reported by our group, novel quinazoline-urea derivatives were synthesized and biologically evaluated as modulators for Aβ-induced mitochondrial dysfunctionCitation13. Since several quinazoline derivatives have been approved by the FDA as kinase inhibitory anticancer drugs, such as Gefitinib, Erlotinib, Lapatinib and Vandetanib, we found that the novel quinazoline-urea chemical structure with its unique substitution pattern on positions C6 and C7 (), which has not been tested before for either antiproliferative or kinase inhibition effect, is worthy to be evaluated. Herein, we report the discovery of compound KST9046, a new agent with quinazoline-urea scaffold. The synthetic and screening protocols for the new agent are illustrated in details. Preliminary in vitro anticancer assay done by the National Cancer Institute (NCI), USA, showed a broad spectrum activity of the new compound in low micromolar range over all of the 60 NCI tumor cell-lines. Accordingly, the kinase inhibitory activity of the synthesized compound was tested over 347 different kinases representing >50% of the predicted human protein kinome, and it showed selectivity for DDR1 kinase.
Figure 1. Quinazoline-based anticancer drugs and KST9046 chemical structure.
Materials and methods
Chemistry
All reactions and manipulations were performed in nitrogen atmosphere using standard Schelenk techniques. Reagents and solvents were purchased from Aldrich Co. (St. Louis, MO) and Tokyo Chemical Industry Co. (Tokyo, Japan) and used without purification. Thin-layer chromatography was performed with Merck (White House Station, NJ) silica gel 60 F254 pre-coated glass sheets. Column chromatography was performed on Merck Silica Gel 60 (230–400 mesh) and the eluting solvents are noted as mixed solvent with given volume-to-volume ratios. 1H NMR 400 MHz was measured on Bruker Avance 400 (Billerica, MA), and chemical shifts and coupling constants are presented in parts per million relative to Me4Si and Hertz, respectively. Abbreviations are as follows: s, singlet; d, doublet; t, triplet; m, multiplet. High-resolution spectra were performed on Waters (Milford, MA) ACQUITY UPLC BEH C18 1.7µ–Q-TOF SYNAPT G2-Si High Definition Mass Spectrometry.
Preparation of the intermediates 2, 3, 4 and 5
The synthetic procedures and compounds’ characterization were carried out as reported by our research groupCitation13.
Preparation of compound 1-(3-chlorophenyl)-3-(6-(3-fluorobenzyloxy)quinazolin-7-yl)urea (KST9046)
A mixture of quinazolinamine 5 (1.0 mmol) and 3-chlorophenyl isocyanate (1.2 mmol) in anhydrous THF (10 mL) was stirred at 85 °C overnight. The mixture was evaporated under reduced pressure and the residue was purified by flash column chromatography (silica gel, methylene chloride/ethyl acetate 1:4 v/v). Yield 62%; mp: 147–148 °C, 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 9.96 (s, 1H), 9.26 (s, 1H), 9.08 (s, 1H), 8.85 (s, 1H), 8.75 (s, 1H), 7.79 (s, 1H), 7.66 (s, 1H), 7.53–7.43 (m, 3H), 7.37–7.29 (m, 2H), 7.22 (t, J = 8.2 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 5.50 (s, 2H). HRMS (ES+): m/z calculated for C22H16ClFN4O2: 445.0844 [M + Na]+. Found 445.0848.
NCI-cell line screening
Cell-line screening was applied at the National Cancer Institute (NCI), Bethesda, MC, USACitation14, by applying the following procedure. The human tumor cell lines of the cancer screening panel are grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM l-glutamine. For a typical screening experiment, cells are inoculated into 96-well microtiter plates in 100 μL at plating densities ranging from 5000 to 40 000 cells/well depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates are incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After 24 h, two plates of each cell line are fixed in situ with TCA, to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). Experimental drugs are solubilized in dimethyl sulfoxide at 400-fold of the desired final maximum test concentration and stored frozen prior to use. At the time of drug addition, an aliquot of frozen concentrate is thawed and diluted to twice the desired final maximum test concentration with a complete medium containing 50 μg/mL gentamicin. Additional four, 10-fold or 1/2 log serial dilutions are made to provide a total of five drug concentrations plus control. Aliquots of 100 μL of these different drug dilutions are added to the appropriate microtiter wells already containing 100 μL of medium, resulting in the required final drug concentrations. Following drug addition, the plates are incubated for an additional 48 h at 37 °C, 5% CO2, 95% air and 100% relative humidity. For adherent cells, the assay is terminated by the addition of cold TCA. Cells are fixed in situ by the gentle addition of 50 μL of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant is discarded, and the plates are washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 μL) at 0.4% (w/v) in 1% acetic acid is added to each well, and plates are incubated for 10 min at room temperature. After staining, unbound dye is removed by washing five times with 1% acetic acid and the plates are air dried. Bound stain is subsequently solubilized with 10 mM trizma base, and the absorbance is read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same except that the assay is terminated by fixing settled cells at the bottom of the wells by gently adding 50 μL of 80% TCA (final concentration, 16% TCA). Using the seven absorbance measurements [time zero (Tz), control growth (C) and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth is calculated at each of the drug concentrations levels. Percentage growth inhibition is calculated as:
Three dose–response parameters are calculated for each experimental agent. Growth inhibition of 50% (IC50) is calculated by using [(Ti − Tz)/(C − Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti = Tz. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti − Tz)/Tz] × 100 = −50. Values are calculated for each of these three parameters if the level of activity is reached; however, if the effect is not reached or is exceeded, the value for that parameter is expressed as greater or lesser than the maximum or minimum concentration tested.
Kinase screening
Kinase assays were performed at Reaction Biology Corporation (Malvern, PA) using the “HotSpot” assay platformCitation15. Kinase assay protocol; reaction buffer: base reaction buffer; 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO. Required cofactors were added individually (if needed) to each kinase reaction. Reaction procedure: To a freshly prepared buffer solution was added any required cofactor for the enzymatic reaction, followed by the addition of the selected kinase at a concentration of 20 μM. The contents were mixed gently, then the compound under test (KST9046) dissolved in DMSO was added to the reaction mixture in the appropriate concentration. 339-ATP (specific activity 500 μCi/μL) was added to the mixture in order to initiate the reaction, and the mixture was incubated at room temperature for 2 h. Initial screening over 347 kinases: KST9046 was tested by single dose duplicate made at a concentration of 10 μM. Staurosporine was used as a control compound in a 5-dose IC50 mode with 10-fold serial dilutions starting at 20 μM. Reaction was carried out at 10 μM ATP concentration. Testing against DDR1 kinase: KST9046 was tested in a 10-dose IC50 mode with three-fold serial dilutions starting at 20 μM. Staurosporine was used as a control compound in a 10-dose IC50 mode with five-fold serial dilutions starting at 20 μM. Reaction was carried out at 10 μM ATP concentration.
Molecular docking
We retrieve co-crystallized DDR1 kinase with Imatinib (PDB code: 4BKJ) from PDB bank. Compound was drawn with Chemdraw14 and then converted into 3D structure. The protein and ligand were prepared using ProteinPrepareWizard and Ligprep module in Maestro 9.7 (Schrödinger, LLC, New York, NY), respectively. After removing water and adding hydrogen, protein was neutralized and then optimized with energy minimization on only hydrogens. Ligand was prepared with protonation at pH 7.4 and energy minimization. InduceFit docking module was employed to predict binding mode of ligand into DDR1 kinase with XP-Gscore. The pose with the lowest XP-Gscore was selected as the best docking pose and used for the analysis of binding mode.
CYP 450 metabolic stability test
Cytochrome P-450 enzyme inhibition was tested using the P450-glo™ assay kit (Promega, Madison, WI) according to the protocols provided by the manufacturer. First, the compounds were diluted into 4× the final test concentrations. Each CYP membrane and the corresponding luciferin-tagged substrate were diluted with water. Equal volume (6.25 µL) of the 4× sample and 4× CYP were mixed in a 384-well white plate and pre-incubated at room temperature for 10 min. Then, the plate was stored for 20–30 min after adding 12.5 µL of a 2× NADPH generation system. Luminescence was detected in Fusion-alpha® (PerkinElmer, Waltham, MA) after 20 min stabilization with a luciferin detection reagent.
Results and discussion
Chemistry
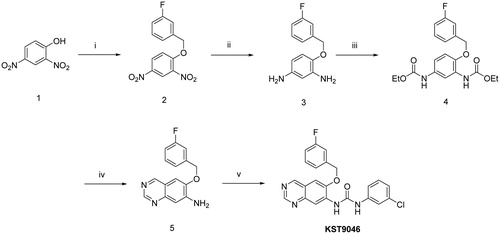
Synthesis of the target compound was achieved according to the sequence illustrated in . Heating 2,4-dinitrophenol 1 with 3-fluorobenzyl bromide in the presence of anhydrous K2CO3 and catalytic amount of potassium iodide in acetonitrile afforded the corresponding dinitrobenzene derivative 2 which by catalytic hydrogenation at room temperature using 10% Pt/C in methanol gave the diamine analogue 3. The dicarbamate derivative 4 was obtained by stirring compound 3 with ethyl chloroformate in the presence of triethylamine (TEA) in tetrahydrofuran (THF) at room temperature. Cyclization to quinazolineamine 5 was carried out by treatment of intermediate 4 with hexamethylenetetramine (HMTA) in the presence of trifluoroacetic acid (TFA) at room temperature, and subsequent heating with 10% aqueous-ethanolic KOH (1:1) and K3Fe(CN)6Citation13. Reaction of the amino group in compound 5 with 3-chlorophenyl isocyanate in THF at 85 °C provided the new quinazoline-urea derivative KST9046.
Full NCI 60 cell panel assay
The new agent was selected by the NCI, USA, for in vitro disease-oriented human cells screening panel assay for anticancer activity, under the Developmental Therapeutic Program (DTP). Preliminary in vitro assay was performed against a full panel of approximately 60 human tumor cell lines representing leukemia, melanoma and cancers of lung, colon, brain, breast, ovary, kidney and prostate in accordance with the protocol of the NCI. The compound was added at a single dose concentration of 10 µM, and the culture was incubated for 48 h. End point determinations were made with a protein binding dye, Sulforhodamine B (SRB). Results for the compound were reported as a mean graph of the percent growth inhibition (GI %) of the treated cells when compared to the untreated control cellsCitation14,Citation16,Citation17. As shown in , the compound exerted great inhibitions up to lethal effects over almost all of the 60 NCI cell-lines. Compound KST9046 (NCS: 777737) satisfied pre-determined threshold growth inhibition criteria and further selected for NCI full panel five-dose assay at 10-fold dilutions of five different concentrations (0.01, 0.1, 1, 10 and 100 µM). The result of tested compound is given by three response parameters (GI50, TGI and LC50) for each cell line from log concentration versus % growth inhibition curves on nine cancer disease (). The GI50 value (growth inhibitory activity) corresponds to the concentration of the compound causing 50% decrease in net cell growth, the TGI value (cytostatic activity) is the concentration of the compound resulting in total growth inhibition and LC50 value (cytotoxic activity) is the concentration of the compound causing net 50% loss of initial cells at the end of the incubation period of 48 h. Furthermore, a mean graph midpoint (MG-MID) is calculated giving an averaged activity parameter overall cell lines. The title compound under investigation exhibited remarkable anticancer activity against all the tested cell lines representing nine different subpanels with GI50 values between 1.3 and 3.9 µM ().
Figure 2. Dose-response curves (% growth versus sample concentration at NCI fixed protocol, µM) obtained from the NCI’s in vitro disease-oriented human tumor cells line of compound KST9046 on nine cancer disease. The different color and shape of NCI subpanel cell lines indicative of growth percentage inhibition with concentration of sample.
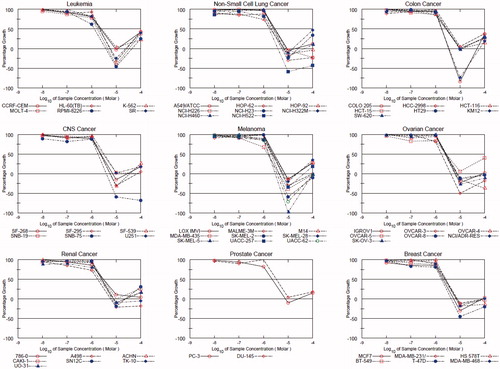
Table 1. % Growth inhibition at 10 µM, GI50, TGI and LC50 of compound KST9046 over the NCI cell-line panel.
In vitro kinase profile screening
In order to investigate a possible kinase inhibitory activity of the new compound, it was tested over a panel of 347 different kinases at Reaction Biology Corporation (Malvern, PA)Citation15. The screening results have revealed that a remarkable inhibitory activity was selectively shown at DDR1 kinase only. The compound was tested initially at a single dose concentration of 10 µM. At this concentration, 76% inhibition of the enzymatic activity of DDR1 kinase was observed, while the inhibition in activity was below 25% in all of the other kinases, and in the range of 41–53% in four kinases only (ARAF, FLT3, LIMK1 and MLK2) as summarized in (for full data screening results, see supplementary file).
Table 2. Summary of kinase inhibitory profile of the new agent KST9046 at 10 µM over a panel of 347 kinases.
Further, KST9046 was tested over DDR1 kinase in a 10-dose IC50 mode and showed an IC50 value of 4.38 µM. However, the potency of the compound over DDR1 kinase cannot justify for its strong and broad spectrum anticancer activity, and this suggests presence of other underlying mechanisms that may control the activity of this new hit against cancer.
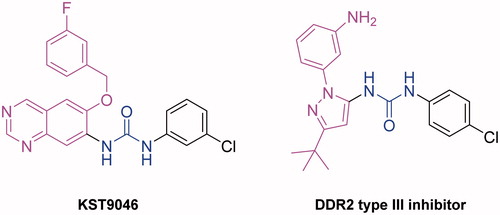
Since DDR1 and DDR2 have extensive homology, we have searched the literature for type III DRR1 and/or DDR2 inhibitors. Although only a few DDR2 inhibitors are known, a high structural similarity was noted between KSR9046 and a recently reported type III DDR2 inhibitor discovered through high-throughput screening as illustrated in Citation18. Both compounds possess a heterocyclic system, a hydrophobic chlorophenyl moiety and a hydrogen bond donor-acceptor pair (urea linker). This finding could support KST9046 as type III-like compound with possible allosteric modulation for DDR1.
Figure 3. Structure similarity between KST9046 and type III DDR2 allosteric modulator.
Molecular docking
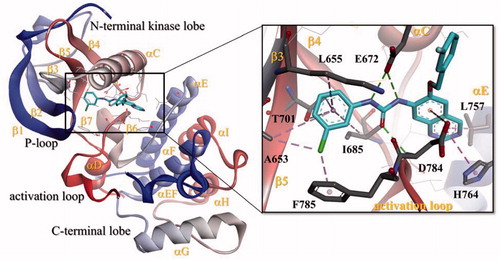
A molecular docking study using Maestro 9.7 (Schrödinger, LLC, New York) programCitation19 was performed in order to predict the binding mode of KST9046 with the catalytic domain of DDR1 enzyme. The docking model was carried out using X-ray cocrystal structure of DDR1 kinase with Imatinib (PDB: 4BKJ)Citation20, which is DFG-out conformation of DDR1 bound with type II inhibitor. In this study, induced Fit Docking XP-Gscore was used for analysis of the binding mode.
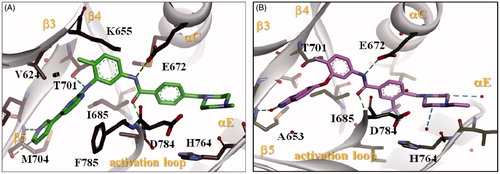
As shown in , different types of binding interactions have been achieved between KST9046 and DDR1 kinase catalytic domain immediately adjacent to the hinge region; the urea linker formed three H-bonds suggested to be the main keys responsible for the ligand-enzyme interaction; two H-bonds were formed between the urea NH groups and the carboxylic side chain of Glu672 residue in the αC helix, while another H-bond was noticed between the urea carboxylic group and NH moiety of Asp784 residue in DFG motif.
Figure 4. Docking of compound KST9046 in the binding site of DDR1 kinase catalytic domain (PDB ID: 4BKJ) in 3D style.
Further, we docked known type II DDR1 inhibitors; Imatinib () and DDR1-1 N-1 ()Citation21 to the catalytic domain of DDR1 and compared the binding modes with that of KST9046. As expected, Imatinib and DDR1-1 N-1 exhibited similar bonds to Glu672 and Asp784 residues via their amide linker as indicated in . Hence, it was clear that although KST9046 missed H-bonds formation with the hinge region, which was achieved by type II inhibitors, it could successfully fit and bound to the hydrophobic pocket in the DFG motif.
Figure 5. Docking of Imatinib (A) and DDR1-1N-1 (B) (type II DDR1 inhibitors) in the binding site of DDR1 kinase catalytic domain in 3D style.
Scheme 1. Reagents and conditions: (i) 3-fluorobenzyl bromide, K2CO3, KI, CH3CN, 75 °C, 8 h; (ii) H2, 10% Pt/C, CH3OH, rt, 6 h; (iii) Ethyl chloroformate, TEA, THF, rt, 2 h; (iv) (a) HMTA, TFA, rt, 1 h, (b) 10% KOH aqueous ethanolic (1:1), K3Fe(CN)6, 100 °C, 4 h; (v) 3-chlorophenyl isocyanate, THF, 85 °C, overnight.
It is worth pointing out that some other interactions with DDR1 binding pocket were exhibited by KST9046; the quinazoline core formed a hydrophobic binding with Leu757 and His764 residues in the αE helix, in addition to π-anion interaction with Asp784 residue in DFG motif. Moreover, the 3-chlorophenyl moiety attached to the urea linker also showed another hydrophobic interaction with Ala653 and Lys655 in β3 strand, Thr701 in β5 strand, and Ile685 residues in β4 strand.
Of special interest, the binding affinity of the illustrated docking pose of KST9046 has achieved XP-Gscore of −8.962 kcal/mol. From this model, type III kinase inhibitory allosteric mechanism of action is suggested to be a possible binding mode of KST9046 with DDR1 kinase which may offer an explanation of its selectivity.
CYP 450 metabolic stability test
Cytochrome P450 (CYP450) is an enzyme responsible for drug metabolism in human. Inhibition of CYP450 at a clinically relevant concentration causes drug–drug interactions as well as various side effects. Consequently, KST9046 was subjected to a metabolic stability test over four isoforms of CYP450 (2C9, 2C19, 2D6 and 3A4). The toxicity of KST9046 over CYP450 isoforms was tested and IC50 values are reported in . KST9046 exhibited very promising toxicity profile with high IC50 values compared to the positive control agents used in this experiment. The results indicated that KST9046 has no potential inhibitory effect over CYP450, which decreases the possibilities of drug–drug interactions as well as unwanted adverse side effects.
Table 3. IC50 values of compound KST9046 over four different isoforms of CYP450 comparing to positive control agents.
Conclusion
In conclusion, biological evaluation of KST9046 at NCI, aimed at screening its potential anticancer activity, resulted in discovery of a broad spectrum effect over almost all of the 60 NCI cell-lines. In addition, a selective kinase inhibition for DDR1 enzyme was disclosed. Currently, in cooperation with other research groups, detailed in vitro and in vivo pharmacological studies are carried out using compound KST9046 as a promising hit to develop new agents targeting glioblastoma multiforme (GBM), the most common and malignant type of human brain tumor. The results of the current work will be published as soon as done.
Supplementary material available online
Supplemental Material.pdf
Download PDF (393.6 KB)Acknowledgements
We would like to thank the National Cancer Institute (NCI), Bethesda, MD, USA, for performing the anticancer testing over the cell lines. We also appreciate Dr Haiching Ma from Reaction Biology Corporation for kinase profile screening.
Declaration of interest
The authors have declared no conflict of interest.
This research was supported by a grant (NRF-2011-0028676) from the creative/challenging research program of National Research Foundation of Korea.
References
- Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science 2002;298:1912–34
- Cohen P. The role of protein phosphorylation in human health and disease. Eur J Biochem 2001;268:5001–10
- Khadjavi A, Barbero G, Destefanis P, et al. Evidence of abnormal tyrosine phosphorylated proteins in the urine of patients with bladder cancer: the road toward a new diagnostic tool? J Urol 2011;185:1922–9
- Cicenas J, Urban P, Küng W, et al. Phosphorylation of tyrosine 1248-ERBB2 measured by chemiluminescence-linked immunoassay is an independent predictor of poor prognosis in primary breast cancer patients. Eur J Cancer (Oxford, England: 1990) 2006;42:636–45
- Gavrin LK, Saiah E. Approaches to discover non-ATP site kinase inhibitors. Med Chem Comm 2013;4:41–51
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Rev Cancer 2009;9:28–39
- Valiathan RR, Marco M, Leitinger B, et al. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metast Rev 2012;31:295–321
- Yamanaka R, Arao T, Yajima N, et al. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006;25:5994–6002
- Johansson FK, Goransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene 2005;24:3896–905
- Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, et al. Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin Cancer Res 2004;10:4427–36
- Weiner HL, Huang H, Zagzag D, et al. Consistent and selective expression of the discoidin domain receptor-1 tyrosine kinase in human brain tumors. Neurosurgery 2000;47:1400–9
- Ford CE, Lau SK, Zhu CQ, et al. Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Brit J Cancer 2007;96:808–14
- Elkamhawy A, Lee J, Park B-G, et al. Novel quinazoline-urea analogues as modulators for Aβ-induced mitochondrial dysfunction: design, synthesis, and molecular docking study. Eur J Med Chem 2014;84:466–75
- http://www.dtp.nci.nih.gov/
- http://www.reactionbiology.com/
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66
- Boyd MR, Paull KD. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Devel Res 1995;34:91–109
- Richters A, Nguyen HD, Phan T, et al. Identification of Type II and III DDR2 Inhibitors. J Med Chem 2014;57:4252–62
- http://www.schrodinger.com/Maestro
- Canning P, Tan L, Chu K, et al. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J Molec Biol 2014;426:2457–70
- Kim H-G, Tan L, Weisberg EL, et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol 2013;8:2145–50