
Abstract
In this article, 62−α-maltotriosyl-maltotriose was consecutively prepared by hydrolysis and reverse synthesis action of Bacillus Acidopullulyticus pullulanase using pullulan as a substrate. Maltotriose syrup was obtained by hydrolyzing of pullulan with pullulanase. The optimal reverse synthesis conditions were then investigated and the optimum conditions were as follows: maltotriose syrup concentration, 70–75%; time, 40 h; pH, 4.0; temperature, 65°C; amount of pullulanase, 120 U. Under these conditions, the relative conversion rate of reverse synthesis reached up to 26%. The products were further isolated by preparative high performance liquid chromatography on Lichrospher NH2 column. 62−α-Maltotriosyl-maltotriose was purified and maltotriose syrup was collected and recycled as the substrate for reverse synthesis. Structure of 62−α-maltotriosyl-maltotriose was elucidated by electrospray ionization mass spectrometry, fourier transform infrared spectroscopy, 1H nuclear magnetic resonance, 13C nuclear magnetic resonance, and heteronuclear multiple bond correlation. These results suggested that this was a promising way of preparation of 62−α-maltotriosyl-maltotriose.
INTRODUCTION
With the development of carbohydrate chemistry, oligosaccharides demand more and more attention due to their important biological functionality and molecular diversity.[Citation1−Citation3] However, chemical synthesis of oligosaccharides is more laborious than that of other biological macromolecules. Multifarious protections and de-protection strategies are essential for the achievement of the desired oligosaccharides.[Citation4Citation5] These complicated processes cause limitations to the application and research of oligosaccharides. Enzymatic synthesis of oligosaccharides is a promising alternative to the chemical method. The panose and isopanose were prepared in good yield from hydrolyzates of pullulan.[Citation6] Li et al.[Citation7] reported a simple method of preparing diverse neoagaro-oligosaccharides with β-agarase. 6ʹ-α-Maltosyl-maltotriose and 6″-α-glucosyl-maltotriose were prepared from Novamyl degradation of branched cylodextrins.[Citation8]
Pullulan is an exocellular polysaccharide containing a succession of maltotriose linked by α-(1, 6) glycosidic bonds and 7% maltotetraose.[Citation9Citation10] The distinctive physical traits of pullulan are attributable to the regular linkages pattern.[Citation11] Pullulanase (EC 3.2.1.41) is a member of the α-amylase family and specifically cleaves α-(1, 6) glycosidic bonds in starch, amylopectin, and pullulan. Maltotriose syrup has been prepared by hydrolyzing of pullulan with pullulanase.[Citation12Citation13] Reversible action of pullulanase was verified through condensation reactions, and the smallest known substrate for the reverse action of pullulanase was maltose.[Citation14] The reversibility of hydrolysis for pullulanase has been employed to produce branched cyclodextrins, such as maltosyl-β-cyclodextrin.[Citation15] The specific synthesis of 62−α-maltotriosyl-maltotriose by chemical process is complicated and difficult. Therefore, it is of interest to investigate the way for producing 62−α-maltotriosyl-maltotriose by hydrolysis and reverse synthesis action of pullulanase consecutively.
MATERIALS AND METHOD
Materials
Pullulan (purity >98%) was purchased from Threewin Trading Co., Ltd (Shanghai). Pullulanase from Bacillus acidopullulyticus was purchased from Genencor Inc. (Wuxi, China). All other chemicals and reagents were of reagent grade.
Assay of Pullulanase Activity
Pullulanase activity was determined by measuring the amount of enzyme to release reducing sugars during the incubation with pullulan. In this assay, the reaction mixture containing 0.5 mL of 1% (w/v) pullulan in 20 mM acetate buffer (pH 5.0) and 0.5 mL of enzyme sample was incubated at 50°C for 30 min. The reducing sugar released was determined by dinitrosalisyilic acid method (DNS).[Citation16] One unit of pullulan activity was defined as the amount of enzyme that catalyzed the formation of 1 μmol of reducing sugars per minute. A V-1800 spectrophotometer (Mapada, Shanghai, China) was used for all assays.
General Procedures for the Preparation of 62−α-Maltotriosyl-Maltotriose
Preparation of maltotriose syrup was performed through a modification of the method of Wu et al.[Citation12] and Singh et al.[Citation13] Pullulan was dissolved in 50 mM acetate buffer to make a solution of a concentration of 6% (w/v). Then, 100 ml of pullulan solution was incubated with pullulanase (8 U/ml) under agitation of 100 rpm at 45°C for 8 h. To investigate the optimal reverse synthesis condition, the resulting maltotriose syrup was concentrated in vacuo. Pullulanase was added into the maltotriose syrup and the reaction mixture was stirred with a speed of 100 rpm at 60°C for 48 h. After the reaction, the mixture was heated at 100°C for 15 min to inactivate the enzyme and centrifuged to remove the precipitate. The relative conversion rate (RC) was used for evaluating the degree of reverse synthesis:
The peak area of 62−α-maltotriosyl-maltotriose and the peak area of maltotriose were detected and calculated by high performance liquid chromatography (HPLC).
Purification of 62−α-Maltotriosyl-Maltotriose
62−α-Maltotriosyl-maltotriose was isolated by preparative HPLC from the reaction supernatant. The conditions of this process are as follows: column, Lichrospher NH2 (5 μm 10 × 300 mm); solvent system, acetonitrile-water (63:37, v/v); column temperature, 35°C; flow rate, 3 ml/min; detector, refractive index detector. The unreacted maltotriose syrup were collected and recycled as substrate for reverse systhesis.
HPLC, Mass Spectrometry (MS), and Nuclear Magnetic Resonance (NMR) Analysis
The product was analyzed by HPLC under the following conditions: column, Lichrospher NH2 (5 μm 4.6 × 250 mm); solvent system, acetonitrile-water (70:30, v/v); column temperature, 35°C; flow rate, 1 ml/min; detector, refractive index detector. Electrospray Ionization mass spectrometry (ESIMS) analysis of 62−α-maltotriosyl-maltotriose was performed using a Waters Platform ZMD 4000 mass spectrometer with electro spray ionization. Citation1H nuclear magnetic resonance (1H NMR), Citation13C nuclear magnetic resonance (13C NMR), and heteronuclear multiple bond correlation (HMBC) spectra were carried out on a Bruker AV400 MHz (Bruker, Rneinstetten, Germany). Samples were solved in D2O with tetramethylsilane (TMS) as an internal standard.
Statistical Analysis
The data were expressed as means of triplicate. Statistical significance was assessed with one-way analysis of variance (ANOVA) using ORIGIN 7.5 (OriginLab Inc., USA). A value of p < 0.05 was considered to be statistically significant.
RESULTS AND DISCUSSION
Effect of Maltotriose Syrup Concentration on the Preparation of 62−α-Maltotriosyl-Maltotriose
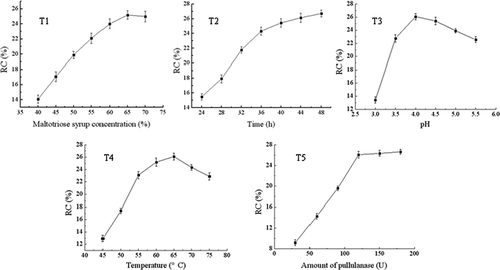
Effects of maltotriose syrup concentration on the reverse synthesis of pullulanase were shown in (T1). RC was found to increase with increasing concentration of maltotriose syrup. The optimum concentration for preparation of 62−α-maltotriosyl-maltotriose was 65% (w/w). This indicated that the reverse synthesis activity of pullulanase depended on the concentration and tended to be enhanced with the increase of maltotriose syrup. The result was confirmed with previous reports. Little tetrasaccharide were synthesized at a maltose concentration of under 5%.[Citation14] Maltosyl cyclodextrins were prepared through the reverse reaction of pullulanase incubated with concentration of 70–75%.[Citation15]
FIGURE 1 T1: Effect of maltotriose syrup concentration on the preparation of 62−α-maltotriosyl-maltotriose; T2: Effect of time on the preparation of 62−α-maltotriosyl-maltotriose; T3: Effect of pH on the preparation of 62−α-maltotriosyl-maltotriose: T4: Effect of temperature on the preparation of 62−α-maltotriosyl-maltotriose; and T5: Effect of pullulanase amount on the preparation of 62−α-maltotriosyl-maltotriose.
Effect of Time on the Preparation of 62−α-Maltotriosyl-Maltotriose
The time course experiment for reverse synthesis of 62−α-maltotriosyl-maltotriose was presented in (T2). The results showed that RC increased with the proceeding of reaction and had an obvious increase within 40 h. Thereafter, RC increased slowly with the proceeding of reaction. This indicated that the consumption of maltotriose syrup decreased the reverse synthesis activity of pullulanase. This was in accordance with the preceding results that the concentration was crucial for reverse synthesis activity of pullulanase.
Effect of pH on the Preparation of 62−α-Maltotriosyl-Maltotriose
(T3) presents the influence of reaction pH on RC. A wide pH range from 3.5 to 5.5 was applicable to the reverse synthesis reaction. The maximum RC reached 26% at a pH of 4.0. This characteristic was in agreement with the operational pH (4.0–4.5) for formation of maltosyl cyclodextrins.[Citation15] Compared with the optimum pH 5.0 for hydrolysis,[Citation12] it shifted to a more acidic value.
Effect of Temperature on the Preparation of 62−α-Maltotriosyl-Maltotriose
The influence of the reaction temperature on RC was evaluated as shown in (T4). It was evident that RC was optimal at 65°C. In contrast, optimal temperatures for hydrolysis of pullulanase were obtained at 45°C[Citation13] and 50°C.[Citation12] The result showed that higher temperature promoted the reverse synthesis activity of pullulanase and more activation energy was required. Moreover, higher temperature accelerated the diffusion between substrate and pullulanase. These results agree well with previous reports claiming the crucial role of temperature in improving reaction rate.[Citation17Citation18]
Effect of Pullulanase Amount on the Preparation of 62−α-Maltotriosyl-Maltotriose
As shown in (T5), the increase of pullulanase loading from 30 to 120 U resulted in linear increases of RC. The conversion of maltotriose maintained 26% at high levels of pullulanase (120 U). Compared with hydrolysis, more pullulanase was required in the reverse synthesis reaction. This characteristic was in good agreement with the preparation of branched cyclodextrins.[Citation15]
Identification of 62−α-Maltotriosyl-Maltotriose
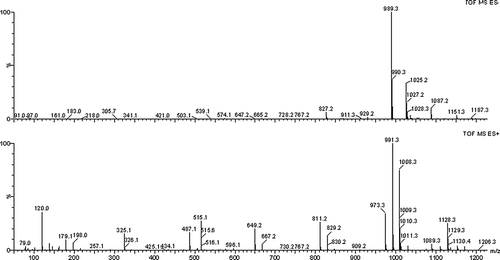
The FTIR spectrum of 62−α-maltotriosyl-maltotriose is shown in . The product exhibited an absorption peak at 3400 cm−Citation1, which was the characteristic of the –OH group. There was an apparent absorption at 2927 cm−Citation1 corresponding to the –CH– group. The FTIR absorption of the product at 1150 cm−Citation1 was the absorption of the –CO– group. The characteristic bands at 847 cm−Citation1 were attributed to α-glycosidic linkage and there was no characteristic absorption of β-glycosidic linkage in FTIR spectrum. From the FTIR spectrum of the product it was shown that the structure of the product was similar to that of maltotriose. The ESI-MS spectrum of 62−α-maltotriosyl-maltotriose showed [M-H]+ at m/z 989.3 and [M-H]− at m/z 991.3 (). The molecular weight was corresponding to six glucose moleculars.
FIGURE 2 FTIR spectrum of 62−α-maltotriosyl-maltotriose.
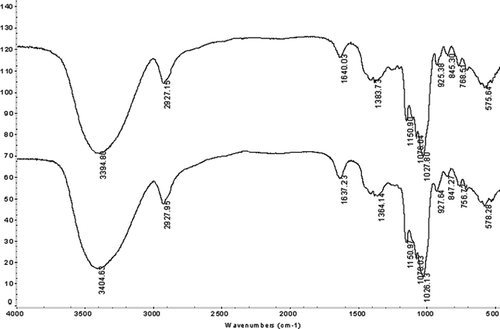
FIGURE 3 ESIMS spectra of 62−α-maltotriosyl-maltotriose in the negative and positive mode.
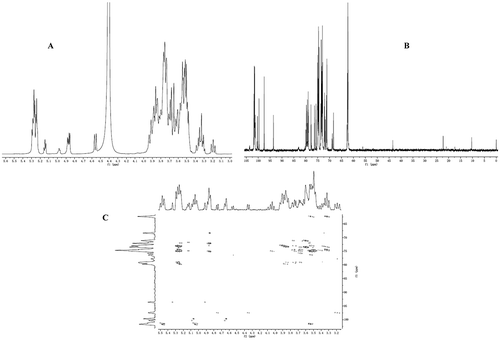
The Citation1H NMR spectrum of 62−α-maltotriosyl-maltotriose is shown in . These signals were assigned on the basis of comparison with other spectra of branched oligosaccharides.[Citation8Citation19−Citation21] The spectrum comprised four kinds of anomeric proton assigned to H-1 of internal anomeric (δ 5.34–5.20), H-1 of branched point (δ 4.87), and H-1 of the reducing end included α and β forms (δ 5.15 and δ 4.56). The structure of 62−α-maltotriosyl-maltotriose was elucidated by these data and guidelines.[Citation22] It was deduced that there were four internal α1,4-linkages, one α1,6-branch, and one reducing end (α + β form). The Citation13C NMR spectrum of 62−α-maltotriosyl-maltotriose is shown in . Compared with other Citation13C NMR spectra of branched oligosaccharides,[Citation19−Citation22] the resonance peaks in the Citation13C NMR spectrum were assigned to glucose residue. Four kinds of C-1 signals were observed: C-1s of internal anomeric (δ 101.85–101.21), C-1 of branched point (δ 100.31), and C-1 of the reducing end included α and β forms (δ 93.61 and δ 97.51). The C-6 signals of glucopyranose residues normally appeared at the highest field. The peaks at δ 62.67–62.29 could be attributed to C-6 of internal anomeric. The C-6 resonance of the branched point was shifted to the lower field at δ 68.38. The large downfield shifts indicated that branched maltotriose residues were linked to the respective residues. Two kinds of C-4 signals appeared at δ 71.24 and δ 79.88–78.99. The peak at δ 71.24 was due to the C-4 of the reducing end and the shift of C-4 resonance was caused by the glycosylation of hydroxyl group. This downfield shift of C resonance was confirmed by previous research.[Citation4Citation20] In addition, the HMBC spectra allowed for determination of the order of the branched point. As shown in c, C-4 and H-1 of the product had an α1,4-linkage at the inner residues (δ 79.23, 5.26); C-1 and H-4 of the product was found to couple to signals (δ 101.58, 3.24); C-6 and H-1 of the product had an α1,4-linkage at branched points (δ 68.38, 4.85). These results suggest that the product was 62−α-maltotriosyl-maltotriose.
FIGURE 4 (A) Citation1H NMR spectrum of 62−α-maltotriosyl-maltotriose; (B) Citation13C NMR spectrum of 62−α-maltotriosyl-maltotriose; and (C) HMBC spectrum of 62−α-maltotriosyl-maltotrios.
CONCLUSION
Preparation of maltotriose syrup was performed through the hydrolysis of pullulan with a commercial pullulanase. 62−α-Maltotriosyl-maltotriose can be prepared through the reverse synthesis of the same enzyme. The optimum reverse synthesis conditions were as follows: maltotriose syrup concentration, 70–75%; time, 40 h; pH, 4.0; temperature, 65°C; amount of pullulanase, 120 U. The product could be isolated directly through Lichrospher NH2 column by preparative HPLC. The structural identities of the product were determined by the analysis of ESIMS, FTIR, Citation1H NMR, Citation13C NMR, and HMBC. The result showed that the oligosaccharide was a 62−α-maltotriosyl-maltotriose.
FUNDING
This study was also supported by the National Natural Science Foundation of China (Nos. 31230057 and 31071490), the National ‘Twelfth Five-Year’ Plan for Science & Technology Support of China (2012BAD37B02), and The Youth Foundation of Hubei Province Education (Q20132602).
REFERENCES
- Sigulinsky, C.; Bab, P.; Victor, X.V.; Kuberan, B. Preparation and characterization of 15N-enriched, size-defined heparan sulfate precursor oligosaccharides. Carbohydrate Research 2010, 345 (2), 250–256.
- Li, S.; Li, T.; Zhu, R.; Wang, N.; Song, Y.; Wang, S.; Guo, M. Antibacterial action of haw pectic oligosaccharides. International Journal of Food Properties 2013, 16 (3), 706–712.
- Sodhi, N.S.; Chang, Y.; Midha, S.; Kohyama, K. Molecular structure and physicochemical properties of acid-methanol-treated chickpea starch. International Journal of Food Properties 2013, 16 (1), 125–138.
- Motawia, M.S.; Olsen, C.E.; Enevoldsen, K.; Marcussen, J.; Møller, B.L. Chemical synthesis of 6ʹ-α-maltosyl-maltotriose, a branched oligosaccharide representing the branch point of starch. Carbohydrate Research 1995, 277 (1), 109–123.
- Wang, C.C.; Lee, K.L.; Hung, S.C. Regioselective one-pot protection of carbohydrates. Nature 2007, 446 (7138), 896–899.
- Sakano, Y.; Kogure, M.; Kobayashi, T.; Tamura, M.; Suekane, M. Enzymic preparation of panose and isopanose from pullulan. Carbohydrate Research 1978, 61 (1), 175–179.
- Li, J.; Han, F.; Lu, X.; Fu, X.; Ma, C.; Chu, Y.; Yu, W. A simple method of preparing diverse neoagaro-oligosaccharides with β-agarase. Carbohydrate Research 2007, 342 (8), 1030–1033.
- Jørgensen, C.T.; Svendsen, A.; Brask, J. Enzymatic synthesis of oligosaccharides from branched cyclodextrins. Carbohydtrate Research 2005, 340 (6), 1233–1237.
- Wallenfels, K.; Keilich, G.; Bechtler, G.; Freudenberger, D. Investigations on pullulan. IV. Resolution of structural problems using physical, chemical and enzymatic methods. Biochemische Zeitschrift 1965, 341, 433–450.
- Catley, B.J.; Ramsay, A.; Servis, C. Observations on the structure of the fungal extracellular polysaccharide, pullulan. Carbohydrate Research 1986, 153 (1), 79–86.
- Leathers, T.D. Biotechnological production and applications of pullulan. Applied Microbiology and Biotechnology 2003, 62 (5–6), 468–473.
- Wu, S.; Chen, H.; Tong, Q.; Xu, X.; Jin, Z. Preparation of maltotriose by hydrolyzing of pullulan with pullulanase. European Food Research and Technology 2009, 229 (5), 821–824.
- Singh, R.S.; Saini, G.K.; Kennedy, J.F. Maltotriose syrup preparation from pullulan using pullulanase. Carbohydrate Polymers 2010, 80 (2), 401–407.
- Abdullah, M.; French, D. Reversible action of pullulanase. Nature 1966, 210 (5032), 200.
- Shiraishi, T.; Kusano, S.; Tsumuraya, Y.; Sakano, Y. Synthesis of maltosyl(α1→6)cyclodextrins through the reverse reaction of thermostable bacillus acidopullulyticus pullulanase. Agricultural and Biological Chemistry 1989, 53 (8), 2181–2188.
- Miller, G.L. Use of dinitrosalicyclic acid reagent for determination of reducing sugar. Analytical Chemistry 1959, 31 (3), 426–428.
- Guo, Z.; Sun, Y. Solvent-free production of 1,3-diglyceride of CLA: Strategy consideration and protocol design. Food Chemistry 2007, 100 (3), 1076–1084.
- Watanabe, T.; Shimizu, M.; Sugiura, M.; Sato, M.; Kohori, J.; Yamada, N.; Nakanishi, K. Optimization of reaction conditions for the production of DAG using immobilized 1,3-regiospecific lipase Lipozyme RM IM. Journal of the American Oil Chemists’ Society 2003, 80 (12), 1201–1207.
- Ishizuka, Y.; Nemoto, T.; Kanazawa, K.; Nakanishi, H. 1H NMR spectra of branched-chain cyclomaltohexaoses (α-cyclodextrins). Carbohydrate Research 2004, 339 (4), 777–785.
- Okada, Y.; Semma, M.; Ichikawa, A. Physicochemical and biological properties of 61,63,65−tri-O-α-maltosyl-cyclomaltoheptaose (61,63,65−tri-O-α-maltosyl-β-cycylodextrin). Carbohydrate Research 2007, 342 (10), 1315–1322.
- Cui, B.; Jin, Z. Enzymatic synthesis and identification of maltosyl(α-1→6) β-cycylodextrin. Chemical Journal of Chinese University 2007, 28 (2), 283–285.
- Bock, K. Carbohydrate-protein interactions: Substrate specificity of enzymes used in the degradation of oligosaccharides related to starch and cellulose. Pure and Applied Chemistry 1987, 59 (11), 1447–1456