
ABSTRACT
Introduction: Diseases of the Central Nervous System (CNS) affect millions of people worldwide, with the number of people affected quickly growing. Unfortunately, the successful development of CNS-acting drugs is less than 10%, and this is attributed to the complexity of the CNS, unexpected side effects, difficulties in penetrating the blood-brain barrier and lack of biomarkers.
Areas covered: Herein, the authors first review how pharmacokinetic/pharmacodynamic (PK/PD) models are designed to predict the dose-dependent time course of effect, and how they are used to translate drug effects from animal to man. Then, the authors discuss how pharmacometabolomics gives insight into system-wide pharmacological effects and why it is a promising method to study interspecies differences. Finally, the authors advocate the application of PK/PD-metabolomics modeling to advance translational CNS drug development by discussing its opportunities and challenges.
Expert opinion: It is envisioned that PK/PD-metabolomics will increase our understanding of CNS drug effects and improve translational CNS drug development, thereby increasing success rates. The successful future development of this concept will require multi-level and longitudinal biomarker evaluation over a large dose range, multi-tissue biomarker evaluation, and the generation of a proof of principle by application to multiple CNS drugs in multiple species.
1. Introduction
Central nervous system (CNS) diseases affect millions of people worldwide, and the number of people with such disease is quickly growing [Citation1]. They are characterized by their high complexity as multiple neurotransmitter systems and biochemical pathways are involved [Citation2–Citation4]. It is therefore not surprising that CNS drug development suffers from low success rates (<10%) and long duration (~12.6 years) [Citation5,Citation6]. Moreover, it is hampered by CNS-mediated side effects (e.g. nausea, dizziness), the presence of the blood–brain barrier (BBB), lack of effective animal models and/or lack of integrative investigations in animals to investigate the mechanisms of CNS pathology and pharmacology, and the lack of biomarkers representing these mechanisms [Citation6–Citation9]. In particular, the translation from preclinical to early clinical studies is difficult.
Clearly, there is a need to improve the current methodologies within CNS drug development. Two promising methods in this regard are pharmacokinetic/pharmacodynamic (PK/PD) modeling and pharmacometabolomics [Citation10–Citation12]. PK/PD modeling allows to ‘characterize and predict the time course of drug effects under (patho)physiological conditions’ [Citation13]. Pharmacometabolomics involves the ‘determination of the metabolic state to define signatures before and after drug exposure that might inform treatment outcomes’ [Citation14]. This review discusses how translational CNS drug development can be improved by the integrated application of PK/PD modeling and pharmacometabolomics. An overview will be provided of the role of both fields in translational CNS drug development, after which the opportunities and challenges of an integrated approach will be discussed.
2. Biomarker-driven development of CNS drugs
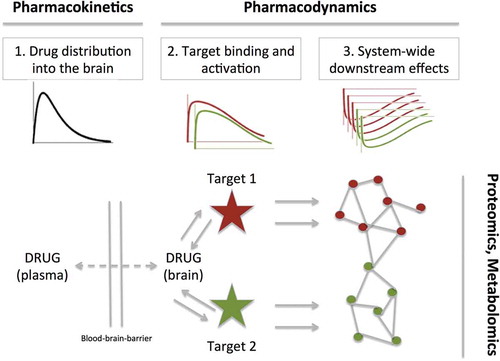
Current translational CNS drug development highly relies on behavioral endpoints, such as the 5-choice serial reaction time task. While these end points may provide reasonable construct validity, their predictive validity is low [Citation15,Citation16]. Predictive validity, which includes a mechanistic rationale between the drug effect and the end point, is important to translate the preclinical to the clinical pharmacology [Citation17]. It is therefore that biomarkers are increasingly recognized as an essential element of CNS drug development [Citation7,Citation18–Citation20]. Indeed, biomarkers have been defined as indicators of specific pharmacological or physiological processes [Citation21,Citation22]. Current biomarker strategies include receptor occupancy [Citation23–Citation25], functional imaging [Citation26,Citation27], biochemical measures in CSF [Citation20], EEG [Citation28,Citation29], or physiological measures such as hormone release [Citation30]. Biomarkers have been classified into multiple pharmacological levels following the causal relation of the drug dose to the clinical effect [Citation31]. These are (i) genotype or phenotype, (ii) drug exposure, (iii) target occupancy, (iv) target activation, (v) physiological/laboratory measures, (vi) disease processes, (vii) clinical scales. Such classification provides a framework for rational drug development. In particular, as depicted in , confidence in the drug exposure, target binding, and target activation are key components to guarantee successful translational drug development [Citation12].
Figure 1. The conceptualization of an integrative approach. The plasma and brain drug exposure profile are determined by the pharmacokinetics, to drive the target binding and activation of potentially multiple targets. The activation (or inhibition) of these targets elicits multiple downstream biochemical effects, which can be evaluated by proteomics or metabolomics. These processes are described by mathematical expressions as developed in the field of PK/PD modeling.
3. PK/PD modeling in biomarker-driven CNS drug development
Not only the measurement of biomarkers is important for prediction of the dose–effect relation. It is also important to quantify the nonlinear and time-dependent relations between the biomarkers to obtain insight into the dynamics of the pharmacological processes. PK/PD modeling is used to mathematically describe these processes in terms of PK and PD parameters, for example, clearance, volume of distribution, maximal drug effect or in vivo potency. Biomarkers thus enable the quantitative characterization of the processes that are on the causal path between dose and effect. More specifically, biomarker data give insight into pharmacokinetic (PK) parameters such as clearance and volume of distribution, or pharmacodynamic (PD) parameters such as maximal effect and in vivo potency. As such, PK/PD parameters provide a quantitative and scalable perspective on interspecies differences, thereby allowing the prediction of the first-in-human dose [Citation17,Citation30,Citation32]. The components of a PK/PD model are the (i) PK model that describes the exposure of the drug in the body; (ii) the PD model that captures the relation between the drug concentration and the effect; and (iii) the link model that accounts for the possible delay between the concentration-time and the effect-time profile [Citation13]. These components are further described in the next section.
3.1. PK/PD models
3.1.1. PK models
A crucial aspect of successful CNS drug development is the understanding of the distribution of the drug into the brain [Citation33–Citation35]. The intensity, onset, and duration of CNS drug effects depend on the concentration-time profile at the site of drug action. This brain is separated from the plasma by the BBB, which often influences the rate and extent of drug distribution into the brain. The transport over this barrier may be passive (driven by concentration gradient) and active (driven by transporters). In addition to the BBB, other factors, such as plasma protein binding, brain tissue binding, cellular uptake, brain metabolism, CSF flow, and physicochemical properties of the drug influence the drug exposure profile in the brain (for reviews and key research on this topic see references [Citation36–Citation41]). Although classical PK modeling still often is used, physiology-based PK (PBPK) modeling is increasingly applied to predict the time course of drug concentrations at the site of drug action.
3.1.2. PD models
Whereas the understanding of the drug exposure at the target site is a crucial aspect in CNS drug development, the subsequent linkage to the PD (i.e. target binding and activation, and downstream physiological responses) is equally important for understanding drug effects [Citation11,Citation12]. Among others, receptor occupancy [Citation23,Citation25], EEG measures [Citation28,Citation42], hormone release [Citation30] have been used to characterize the pharmacodynamic response of CNS drugs. The mathematical linkage of PD responses to the drug exposure has been extensively reviewed by Danhof et al. [Citation43]. Still, in practice, an integrative approach including PK and PD in one study is often lacking. A widely used equation to link PK to PD is the empirical sigmoid Emax equation:
where Emax is the maximal observed drug effect, EC50 is the in vivo potency and C is the concentration around the target (e.g. brainECF).
3.1.3. Link models
The effect-time profile is often delayed as compared to the drug concentration-time profile. If only plasma drug concentrations are known, one may assume that the delay is caused by slow distribution from plasma to the site of drug action. In such case, an effect compartment model is used to account for the delay [Citation44]. Slow target-binding kinetics may also cause a delay between PK and PD, and in such case, these can be explicitly included in the model [Citation45]. Finally, downstream signal transduction may be relatively slow compared to the plasma PK, drug distribution, and the target binding kinetics, being responsible for the delay of the effect-time profile. This is often accounted for by a turnover model [Citation46]. It assumes a continuous process of production and degradation (turnover) that drives the basal biomarker levels. The drug effect influences either the production or the degradation rates through inhibition or stimulation, thereby causing an increase or a decrease of the biomarkers levels.
3.2. Interspecies scaling
PK/PD modeling enables the rational extrapolation of drug effects between animal and men [Citation47]. It does so by explicitly distinguishing the drug- and system-specific parameters [Citation17,Citation32]. Typical drug-specific parameters are plasma protein binding, target-binding affinity, and intrinsic efficacy, while examples of system-specific parameters are tissue volumes, clearances, receptor expression, and turnover rate constants. While drug-specific parameters can be obtained from in vitro experiments, system-specific parameters can only be estimated from in vivo data and may be species-dependent. The interspecies scaling of these parameters follows two principles: allometric scaling and physiology-based scaling. With allometric scaling, it is assumed that the parameters are dependent on bodyweight following a power function [Citation48,Citation49]:
where P is the mathematical model parameter, BW is the bodyweight, and b is the species-independent scaling exponent. Typically, allometric scaling is applied to clearance, volume of distribution, and turnover rate constants. The scaling exponent generally is 0.75 for the clearance, 1 for the drug distribution volume and −0.25 for the turnover rate constant [Citation50,Citation51]. As an illustrative example, the acetaminophen clearance extrapolates over a large range of species, including zebrafish larvae, rat and human, using allometric scaling [Citation52]. In another study, the prolactin effects of remoxipride were successfully scaled from rat to man, by applying allometric scaling on the turnover rate of prolactin in plasma [Citation30].
The principle of physiology-based scaling is to replace the animal parameters by the human parameters [Citation53]. While the physiology-based scaling of CNS PK is well developed, for example to predict the human CSF drug concentrations of acetaminophen and morphine [Citation37,Citation39,Citation54,Citation55], it has only started to emerge for PD. Some studies have shown that PD parameters such as Emax and EC50 may be similar across species for a series of drugs, for example for opioids and their effect on electrocardiogram output [Citation29,Citation56]. In contrast, other studies showed species-dependent PD parameters. A recent evaluation of a Transient Receptor Potential Melastatin-8 blocker showed threefold cross-species (mouse versus dog) differences in its potency, resulting in clinically important differences in core body temperature predictions [Citation57]. In another study, the Emax and EC50 for prolonging the QT-interval were found to differ between humans and dogs [Citation58]. A third publication showed that the affinity of psychoactive drugs differed significantly between, for example, the D1rat and 5HT2rat, and D1human and 5HT2human receptors [Citation9]. Also, the Emax and the EC50 of prolactin to control its own release was found different between rats and humans [Citation30].
Overall, these examples show that the interspecies translation of CNS drug effects needs to be driven by the mechanistic understanding of drug- and system-specific properties at the level of PK and PD. Both allometric scaling and physiology-based scaling of PK/PD parameters can be used to support interspecies translation on basis of in vitro (drug-specific) and in vivo (system-specific) parameters. If clinical data are not available from same-in-class drugs, multiple species can be evaluated for these properties and simulations of worst-to-best case scenarios can be used to guide the dosing strategies during early clinical development [Citation57].
4. Pharmacometabolomics in biomarker-driven CNS drug development
Although PK/PD modeling aims to predict single biomarker time courses, it appears that CNS drugs typically affect multiple biochemical pathways [Citation59,Citation60]. For example, risperidone affected multiple pathways including energy metabolism, antioxidant defense systems, neurotransmitter metabolism, fatty acid biosynthesis, and phospholipid metabolism [Citation61]. In fact, many successful CNS drugs were identified by serendipity on basis of phenotypic changes in vivo [Citation62]. Indeed, the efficacy of neurological drugs is associated with multi-target affinity [Citation63–Citation65]. As an example, antipsychotics typically have interactions with multiple targets (up to 26 for clozapine and quetiapine) [Citation63,Citation64]. A comparison of haloperidol and clozapine showed that they caused a different biochemical phenotype, that of clozapine close to that of the 5-HT2A antagonist M100907 [Citation66]. However, although multi-target pharmacology may be related to the efficacy of e.g. clozapine, it is also associated with unwanted effects, for example cardiovascular disease [Citation63]. Good insight into the systems behavior of multi-target drugs is essential to anticipate the (post-)clinical benefit-risk balance of drugs during early development. As such, pharmacometabolomics is suggested as an important method in drug development to biochemically understand in vivo neuropharmacological effects [Citation67–Citation69]. For example, using lipidomics, the underlying pathways were identified that may explain antipsychotic-induced weight gain [Citation70]. Metabolomics analyzes hundreds of biochemical molecules in biological samples, and as such, it can provide system-wide pharmacological biomarkers [Citation14]. By measuring the biochemical end-products of cellular reactions, it provides an intermediate metabolic phenotype between gene expression and drug effects on one hand, and clinical outcome on the other hand. In other words, it fulfills the definition of a type 4 biomarker [Citation31] and can provide insights into the pharmacological pathways relevant to the clinical outcome. For example, a urinary metabolomics fingerprint could be used to predict the Kellgren–Lawrence grade as a clinical end point for osteoarthritis [Citation71]. As compared to other biomarker types, such as functional imaging, pharmacometabolomics is relatively cheap and easy to apply in preclinical and early clinical studies. Moreover, biochemical pathways are relatively similar across mammalian species, suggesting potential for applying pharmacometabolomics in translational drug development [Citation72,Citation73]. The main analytical tools that are used for metabolomics are nuclear magnetic resonance (NMR) technology and liquid chromatography-tandem mass spectrometry (LC-MS/MS). Both technologies have the advantage that they can identify a wide range of small molecules, providing a comprehensive picture of the metabolome. The metabolome contains more than 40,000 molecules, which typically have a molecular weight below 2 kD [Citation74].
Of interest for the CNS-pharmacology are the energy substrates, neurotransmitters, amino acids, and structural lipids, all of which are involved in cell viability, signaling, and cell membrane function [Citation2]. It was specifically observed that the corresponding pathways were overlapping among CNS drugs and diseases, indicating that multi-biomarker approaches are important for the evaluation of drug effects [Citation2,Citation59]. Several clinical studies have been performed utilizing pharmacometabolomics for the study of CNS drug effects, although the main focus has been on the disease rather than on the treatment [Citation75]. These studies showed that pharmacometabolomics has the potential to reveal new insights into lipid-related side effects of antipsychotics [Citation61,Citation70], enable the early prediction of antidepressant effects on multiple biochemical pathways [Citation76], or identify systems biomarkers of motor neuron disease treatment [Citation77] and anti-Parkinson drugs [Citation78].
4.1. Multivariate analysis of pharmacometabolomics data
The endogenous metabolites are members of biologically highly connected pathways. Pharmacometabolomics data are therefore often evaluated by multivariate data analysis, which takes into account the connectivity among the individual metabolites. The purpose is to identify biomarkers that classify subgroups (e.g. treated vs. nontreated), and to elucidate the biochemical pathways that are perturbed with drug treatment. There are roughly three types of multivariate analyses: descriptive analyses (e.g. correlations), unsupervised methods, and supervised methods (for review, see [Citation79]). An example of descriptive analysis is correlations between metabolite levels. These can be used to define a network with metabolites as nodes, while edges are drawn if the correlation coefficient exceeds a certain threshold (e.g. 0.8). In addition to correlation-based networks, more sophisticated methods have been developed, such as Gaussian graphical networks. These networks eliminate the direct correlations that are explained by indirect correlations, providing a much cleaner network [Citation80,Citation81]. The power of network analysis is that it shows a clear picture of the multifactorial changes under particular conditions, for example, treated vs. nontreated. In particular, it can identify the key metabolite pathways that underlie the pharmacological effects [Citation82], as well as their synergistic or resilient characteristics [Citation83]. A network approach was, for example, used to understand the systems-wide effects of sertraline, showing that the tricarboxylic acid and the urea cycle, fatty acids and intermediates of lipid biosynthesis, amino acids, sugars and gut-derived metabolites were changed with four-week treatment [Citation76]. A well-known unsupervised method is cluster analysis, which classifies samples or metabolites on basis of the proximity to each other with regard to, for example, the metabolite levels or the chemical similarity. This can reveal interesting patterns in the data, such as clusters of genes or metabolites that have similar biological functions [Citation84]. Another well-known unsupervised method is principal component analysis (PCA), which identifies the latent variables (principal components) underlying pharmacometabolomics data [Citation85]. These latent variables then represent the ‘overall’ effect of a treatment in case of a pharmacometabolomics study. Closely related to PCA is the supervised partial least squares regression (PLS). This method optimizes a model to predict a certain output variable, for example, disease status or dose [Citation77]. Both PCA and PLS elucidate which metabolites are most influential in explaining the variation between the subgroups.
4.2. Translational pharmacometabolomics to study CNS drug effects
The specific application of metabolomics in translational drug development has gained attention more than 10 years ago [Citation72]. Metabolomics has an advantage over other ‘omics’ approaches with regard to interspecies translation. Indeed, endogenous metabolite pathways are highly identical among mammalian species. A recent study thoroughly compared the biochemical reaction network of rat and human, showing a strong overlap [Citation73]. There are, however, only a few studies that applied metabolomics in vivo to compare different species. Some studies showed how the metabolic phenotype of animal disease models for osteoarthritis and multiple sclerosis overlapped with the patients’ metabolic phenotype, indicating the potential of metabolomics for interspecies translation [Citation71,Citation72,Citation86,Citation87]. Although no efforts have yet been made to compare the animal and human metabolic phenotypes after drug treatment in vivo, the rat and mouse metabolic phenotypes were compared to study their differential sensitivity to cocaine [Citation88]. It was found that the aryl hydroxylation pathway was dominant in rats, causing increased excretion of cocaine, which was not the case in mice. Interestingly, when comparing microsomes of humans versus these two species, the human cocaine metabolism showed a closer resemblance to the mice cocaine metabolism, indicating that the mouse is a better animal model for evaluation of cocaine sensitivity in humans. This study shows how pharmacometabolomics could be used to guide interspecies translation of CNS drug effects. Nevertheless, care should be taken with regard to the assumption that the biochemical reaction networks are species independent. The bile acid, carbohydrate, glycine-serine-threonine, purine and ascorbic acid pathways were found to have reactions specific for rats, while the glycan and sphingolipid pathways included human-specific reactions, as measured in hepatic cells. These species differences may result in large differences in even opposite effects on certain endogenous metabolites [Citation73]. In such case, further information on the pathway is important to extrapolate the preclinical findings. The ascorbic acid change in rats, for example, reflects a change in the glucuronic acid metabolism, which is also present in humans [Citation89]. This information can then be used for the interspecies translation.
5. The integration of pharmacometabolomics and PK/PD modeling in translational CNS drug development
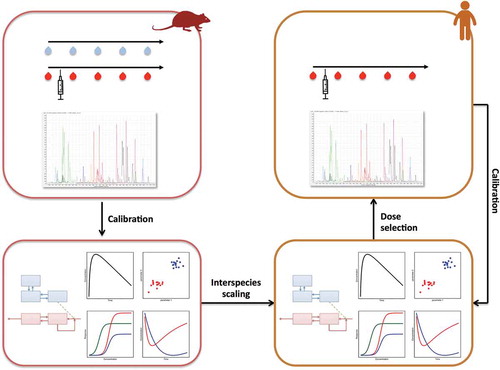
Translational CNS drug development can thus potentially profit from both PK/PD modeling and pharmacometabolomics; both are envisioned to contribute to biomarker-driven development. An integration of PK/PD modeling and pharmacometabolomics is envisioned to provide scalable system-specific parameters for multiple biochemical pathways that are potentially relevant for the clinical drug effects. A conceptual workflow of such translational approach is depicted in . Recent suggestions have been made to use pharmacometabolomics in PK/PD frameworks as static or dynamic markers [Citation90,Citation91]. While static metabolic phenotypes can be used as a predictor for treatment responses, dynamic metabolic phenotypes allow to follow the treatment effect over time to evaluate the system-wide dynamics [Citation90,Citation92,Citation93].
Figure 2. The integrative approach of metabolomics and PK/PD modeling as applied to interspecies scaling in CNS drug development. Such approach starts with animal experiments to collect longitudinal brainECF and plasma samples during treatment with a CNS drug. These samples are analyzed for drug concentrations and metabolomics to subsequently develop a multivariate PK/PD model. By applying the principles of interspecies scaling a humanized model is defined to select doses for the clinical study. Plasma drug concentrations and metabolomics data of the clinical study will be used to recalibrate the model and increase the understanding of interspecies differences.
5.1. Longitudinal analysis of pharmacometabolomics responses
A longitudinal multivariate evaluation of pharmacometabolomics data was performed by Rasmussen and colleagues, who were one of the firsts doing that in the field of clinical pharmacology [Citation94]. This multivariate fingerprint was suitable for guiding dose selection of recombinant interleukin-21 in patients with metastatic melanoma.
5.2. PK/PD-based analysis of pharmacometabolomics responses
In addition to longitudinal evaluation of the pharmacometabolomics response, the integration with PK/PD modeling has been shown in a few studies. Clustering of longitudinal transcriptomics data formed the basis for the 6 turnover models in one study. Together, these turnover models formed a complex PK/PD model that described the gene-expression signaling cascade in the rat liver after corticosteroid treatment [Citation84]. In another study, clustering was applied to the PK/PD parameters identified from pharmacometabolomics data in rats after remoxipride treatment [Citation95]. This analysis revealed 6 unique PK/PD relations, 18 potential biomarkers and two perturbed pathways (). It has the potential to define a therapeutic window on basis of multiple biomarkers, provides a list of biomarkers to take into account in additional studies, and gives insight into biological effects of remoxipride. The application of such analysis in multiple species will give insights into species differences on the PK/PD parameters that describe the longitudinal pharmacometabolomics response. Depending on the differences in parameters, dosing strategies can be defined following simulation of worst-to-best case scenarios as was performed by Gosset et al. [Citation57] for the effect of a Transient Receptor Potential Melastatin-8 (TRPM8) blocker on a single marker (core body temperature). Eventually, pharmacometabolomics data analysis methods can aid the development of quantitative systems pharmacology (QSP) models which aim to mathematically describe the interactions between multiple elements of the biological system (e.g. biomolecules, cells, tissues) in order to understand the impact of drugs on the system as a whole [Citation91,Citation96,Citation97]. Quantitative metabolic networks can provide a topological basis of QSP models to be integrated with organ-level networks, receptor binding kinetics and PK [Citation91,Citation97]. QSP models are promising for interspecies translation by humanizing the animal-based model parameters [Citation9,Citation98,Citation99].
Figure 3. A metabolomics study combined with multivariate PK/PD modeling revealed 6 diverse response patterns (middle) for remoxipride in rats. These response patterns were represented by 18 metabolites that could potentially function as biomarker (right), rendering further validation. The response clusters were associated with 2 known biological pathways (left). Modified from reference [Citation95] with permission of Elsevier.
![Figure 3. A metabolomics study combined with multivariate PK/PD modeling revealed 6 diverse response patterns (middle) for remoxipride in rats. These response patterns were represented by 18 metabolites that could potentially function as biomarker (right), rendering further validation. The response clusters were associated with 2 known biological pathways (left). Modified from reference [Citation95] with permission of Elsevier.](/cms/asset/5f7b7d5a-67c3-4c0d-ab85-da1234a46874/iedc_a_1446935_f0003_oc.jpg)
5.3. Prediction of the human brain pharmacometabolomics responses
In vivo pharmacometabolomics studies typically use plasma samples to characterize the system-wide drug effects. The plasma metabolic phenotype is a composite extraction of all individual tissue metabolic phenotypes. Although this provides the opportunity to evaluate whole-body treatment effects in an easily accessible body fluid, it can limit the quantitative interpretation of the treatment response that originates in a specific tissue. This is particularly true for CNS treatments, for which the metabolic biomarkers have to distribute over the BBB (). This was illustrated by the fact that plasma monoamine levels were decreased with CNS drug treatment, whereas CSF levels were not affected [Citation20,Citation78]. Likely, the effects were caused in the periphery, and did not provide information on the central brain effects of these drugs.
Figure 4. Brain metabolic phenotypes are reflected in the periphery via three mechanisms: i) individual metabolites distribute to CSF, plasma and urine, and become integrated in the peripheral metabolic phenotype; ii) the brain metabolic phenotype affects the peripheral nervous signaling, thereby controlling the release of peripheral metabolites, such as acetylcholine or norepinephrine; iii) the brain metabolic phenotype influences the neuroendocrine system via the hypothalamus, modifying the pituitary hormone release. Modified from reference [Citation59] with permission of Springer.
![Figure 4. Brain metabolic phenotypes are reflected in the periphery via three mechanisms: i) individual metabolites distribute to CSF, plasma and urine, and become integrated in the peripheral metabolic phenotype; ii) the brain metabolic phenotype affects the peripheral nervous signaling, thereby controlling the release of peripheral metabolites, such as acetylcholine or norepinephrine; iii) the brain metabolic phenotype influences the neuroendocrine system via the hypothalamus, modifying the pituitary hormone release. Modified from reference [Citation59] with permission of Springer.](/cms/asset/02f425ae-24d4-4819-be56-ed29ac254b97/iedc_a_1446935_f0004_oc.jpg)
A useful technique that has been used to study CNS drug PK and PD is intracerebral microdialysis [Citation100–Citation103]. It allows longitudinal sampling within a single individual to follow the treatment response over time. Moreover, since microdialysis allows the collection of molecules with a molecular weight below 20 kD, it is highly suitable for pharmacometabolomics analysis [Citation104,Citation105]. Notably, microdialysis, for ethical reasons, is limited in humans. Animals are therefore typically used to characterize the relation between the brain-the CSF-and the plasma metabolic phenotypes. Following the translation PK/PD-metabolomics workflow depicted in , the human brain metabolic phenotype can subsequently be predicted using the principles of interspecies scaling and calibrated with the human plasma and CSF metabolic phenotypes.
5.4. Disease-dependent PK/PD metabolomics approach
This review has mainly focused on the treatment, rather than on the disease. Here, we would like to spend a few words on the influence of pathology on the pharmacology; a patient may respond differently to a treatment than a healthy individual. Both the CNS drug PK and PD can be affected by the disease, and this influence is drug specific. For example, the morphine PK changed with traumatic brain injury [Citation39], and the rate of dopamine metabolism was higher in a rotenone rat model of Parkinson’s Disease as compared to control [Citation103]. Thus, the understanding of the two-way interaction between pathology and pharmacology in the context of translational CNS drug development is important. Metabolomics was found useful to understand species differences with regard to pathology [Citation72]. As such, it has potential to translate the pathology-dependent pharmacology from animal to men [Citation106].
6. Conclusion
This review discussed the merits of PK/PD modeling and pharmacometabolomics in the field of translational CNS drug development. PK/PD models can predict human biomarker time courses on the basis of animal data using the principles of interspecies scaling. Pharmacometabolomics can measure the biochemical responses to evaluate the system-wide CNS drug effects among species. The integration of PK/PD modeling and pharmacometabolomics studies is envisioned to enable the prediction of longitudinal, dose-dependent system-wide responses, and has begun to receive attention [Citation90,Citation91,Citation95]. The opportunities and challenges of such integration were discussed with regard to translational CNS drug development. Although we are still at the stage of early conceptual development, such integration is envisioned to increase understanding of system-wide pharmacology and to improve the interspecies translation of CNS drug effects.
7. Expert opinion
7.1 The potential of integrated PK/PD and pharmacometabolomics in translational CNS drug development
CNS drug development is suffering from low success rates, which, for a large part, can be attributed to the empirical approach in translational development [Citation6,Citation12]. This led to the realization to shift toward mechanism-based prediction of clinical on basis of preclinical pharmacology. In particular, PBPK and PK/PD modeling are increasingly applied in drug development to guide dosing strategies for early clinical studies [Citation39,Citation47,Citation57,Citation107]. The strength of these models is that they describe the dynamics of pharmacological processes, which can be scaled from animals to humans. While the PD models typically describe the drug effect on a single biological pathway, pharmacometabolomics provides a means to evaluate multiple pathways obtaining a comprehensive insight into the system-wide pharmacology of a CNS drug [Citation69,Citation108]. Interestingly, the metabolome is structurally very similar among mammalian species, enabling a direct comparison of their metabolic phenotypes, although there are a few differences that need caution (e.g. ascorbic acid production in rats, but not in humans) [Citation72,Citation73]. At this moment, only very few studies have been performed to investigate the interspecies correlations of metabolic phenotypes. Moreover, pharmacometabolomics is mostly applied in a static manner, although dynamic approaches are emerging [Citation90,Citation94,Citation95].
PK/PD modeling and pharmacometabolomics are thus complementary to each other. Since both fields have a potential for translational CNS drug development, their integration is promising. It has the potential to identify the pharmacologically relevant parameters of the system-wide drug effects [Citation95]. Using the principles of interspecies scaling, these parameters can be humanized, and predict the clinical on basis of the preclinical pharmacology. The model can subsequently be validated on basis of the clinical metabolic phenotype ().
7.2. Challenges and recommendations for the integration of PK/PD modeling and pharmacometabolomics
Several aspects of study design and data analysis need consideration to achieve an integration of PK/PD modeling and pharmacometabolomics.
7.2.1. Multilevel biomarker evaluation
To achieve an integrative understanding of the pharmacological action, multilevel biomarker data need to be collected, for example, plasma drug concentrations, brain drug concentrations, (multiple) target occupancies, biochemical biomarkers. Eventually, these biomarkers will be linked to physiological measures and clinical outcome during clinical development.
7.2.2. Longitudinal sampling over a large dose range
To capture the dynamics of the PK/PD response, longitudinal data are essential. Serial plasma sampling and intracerebral microdialysis are useful methods to obtain time courses of CNS drug concentrations, as well as biochemical markers, in plasma and brain. Of interest, isotope-labeling-based metabolomics (also called flux-based metabolomics) is an emerging discipline that enables the capturing of network dynamics when applied in combination with longitudinal sampling [Citation109,Citation110]. Additionally, a large drug concentration range is needed to have information on all parts of the nonlinear concentration-response curve. This is particularly important with a comprehensive pharmacometabolomics evaluation, since individual metabolites may have a different position on the concentration-response curve [Citation95].
7.2.3. Integrated PK/PD-metabolomics analysis
Longitudinal pharmacometabolomics data in conjunction with drug concentration data need to be described using PK/PD modeling in order to identify a fingerprint of pharmacologically relevant parameters such as the in vivo potencies, the maximal drug effects or the turnover rates [Citation90,Citation95] ( and ).
7.2.4. Multi-tissue biomarker evaluation
Drug concentrations and endogenous metabolites must be analyzed in multiple biofluids, such as plasma, brainECF, and CSF to understand how the plasma metabolic phenotype relates to the target site effect ().
7.2.5. Generate proof of principle for an integrated PK/PD-metabolomics approach in translational CNS drug development
A primary challenge will be the generation of proof of principle for the integrated PK/PD-metabolomics approach.
First of all, multiple same-in-class drugs are to be compared biochemically using a pharmacometabolomics approach. Haloperidol and clozapine showed different efficacy on the basis of a multivariate analysis with 58 different components of movement, as well as a multivariate evaluation with monoamines [Citation66]. Although both analyses marked the fact that haloperidol and clozapine showed different efficacy, the pattern was not similar for the behavioral and the monoamine analysis. This indicates two things: (1) a multivariate biochemical is promising with regard to understanding differences between same-in-class drugs. (2) The abovementioned analysis showed that the monoamine-based evaluation, although recognizing the pharmacological complexity, still is oversimplified to explain the behavioral outcome.
A second aspect that needs to be included to provide proof of principle is the application of longitudinal metabolomics in multiple species, including humans. Taking into account the known species differences, the interspecies metabolomes should be compared to understand and map species differences and evaluate applicability of pharmacometabolomics in translational CNS drug development [Citation73]. In particular, it will be important to validate the scaled PK/PD models in humans.
A third aspect is to relate the metabolic fingerprint to relevant clinical (side) effects. Kaddurah-Daouk et al. [Citation70] nicely showed this for risperidone, olanzapine, and aripiprazole, comparing their lipidomic profiles. Interestingly, aripiprazole showed less impact on lipids, which was associated with the absence of weight gain as a side effect. Further studies will indicate whether such approach is generally applicable in drug development.
7.3. The future of translational CNS drug development with an integrated PK/PD metabolomics approach
It is envisioned that the integration of PK/PD and pharmacometabolomics will increase the understanding of system-wide pharmacology and improve the interspecies translation of CNS drugs. Specifically, it is envisioned to enable the extraction of system-wide pharmacologically relevant parameters that can be scaled to humans. Additionally, information on biomarkers and pathways is obtained. This advancement must be seen together with the developments in the field of QSP [Citation91,Citation96,Citation97]. The integrated PK/PD-metabolomics approach reveals a PK/PD fingerprint biomarker representing the dynamics of known and unknown pathways. QSP aims to connect the cellular pathway response with the organ- or system-level response. On one hand, the integrated PK/PD-metabolomics approach can thus inform QSP models on relevant pharmacological pathways. On the other hand, QSP models can identify the mechanistic relationship between the single metabolites described by an integrated PK/PD metabolomics model.
Altogether, an integrated PK/PD metabolomics approach is envisioned to have a promising role in translational CNS drug development by providing a method to scale system-wide effects from animal to men ().
Article highlights
Translational CNS drug development is shifting from an empirical to a mechanistic approach
PK/PD modeling in conjunction with scaling principles enables the interspecies translation of pharmacological CNS effects
Pharmacometabolomics provides a mean to compare the system-wide pharmacological CNS effects in multiple species
An integrated PK/PD-metabolomics is envisioned to increase understanding of CNS drug effects and improve translational CNS drug development
To achieve an integrated PK/PD-metabolomics approach, we need multi-level biomarker evaluations, to study a large dose range, and longitudinal sampling from the brain, plasma, and CSF.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
Related Research Data
References
- World Health Organization. Neurological disorders: a public health approach. Geneva: WHO Press; 2006.
- Dumas ME, Davidovic L. Metabolic profiling and phenotyping of central nervous system diseases: metabolites bring insights into brain dysfunctions. J Neuroimmune Pharmacol. 2015;10:402–424.
- Lei S, Powers R. NMR metabolomics analysis of Parkinson’s disease Shulei. Curr Metabol. 2013;1:191–209.
- Qi Z, Yu GP, Tretter F, et al. A heuristic model for working memory de fi cit in schizophrenia ☆. BBA Gen Subj [Internet]. 2016;1860:2696–2705.
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715.
- Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov [Internet]. 2007;6:521–532. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17599084
- Soares HD. The use of mechanistic biomarkers for evaluating investigational CNS compounds in early drug development. Curr Opin Investig Drugs. 2010;11:795–801.
- Ringel M, Tollman P, Hersch G, et al. Does size matter in R&D productivity? If not, what does? Nat Rev Drug Discov [Internet]. 2013;12:901–902. Available from: http://www.nature.com/doifinder/10.1038/nrd4164
- Geerts H. Of mice and men bridging the translational disconnect in CNS drug discovery. CNS Drugs. 2009;23:915–926.
- Kaddurah-Daouk R, Krishnan KRR. Metabolomics: a global biochemical approach to the study of central nervous system diseases. Neuropsychopharmacology. 2009;34:173–186.
- de Lange ECM, Ravenstijn PGM, Groenendaal D, et al. Toward the prediction of CNS drug-effect profiles in physiological and pathological conditions using microdialysis and mechanism-based pharmacokinetic-pharmacodynamic modeling. Aaps J. 2005;7:E532–E543.
- Morgan P, van der Graaf PH, Arrowsmith J, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today [Internet]. 2012;17:419–424.
- Breimer DD, Danhof M. Relevance of the application of pharmacokinetic-pharmacodynamic modelling concepts in drug development. The “wooden shoe’ paradigm. Clin Pharmacokinet. 1997;32:259–267.
- Kaddurah-Daouk R, Weinshilboum RM. Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin Pharmacol Ther [Internet]. 2014;95:154–167. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24193171
- Pratt J, Winchester C, Dawson N, et al. Advancing schizophrenia drug discovery: optimizing rodent models to bridge the translational gap. Nat Rev Drug Discov [Internet]. 2012;11:560–579. Available from: http://www.nature.com/doifinder/10.1038/nrd3649
- Hall AM, Roberson ED. Mouse models of Alzheimer ’ s disease. Brain Res Bull [Internet]. 2012;88:3–12. Available from: http://rd.springer.com/chapter/10.1007/978-1-84628-440-3_16
- Danhof M, de Lange ECM, Della Pasqua OE, et al. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29:186–191.
- Hurko O. The uses of biomarkers in drug development. Ann N Y Acad Sci. 2009;1180:1–10.
- Wong DF, Potter WZ, Brasic JR. Proof of concept: functional models for drug development in humans. Neuropsychopharmacol Fith Gener Prog [Internet]. 2002;457–473. Available from: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.95.4307&rep=rep1&type=pdf.
- Pich EM, Vargas G, Domenici E. Biomarkers for antipsychotic therapies. Handb Exp Pharmacol Curr Antipsychotics. 2012;212:339–360.
- Lesko L, Atkinson A. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol. 2001;41:347–366.
- Atkinson AJJ, Colburn WA, DeGruttola VG, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95.
- Johnson M, Kozielska M, Reddy VP, et al. Translational modeling in schizophrenia : predicting human dopamine D 2 receptor occupancy. Pharm Res. 2016;33:1003–1017.
- Garner R, Gopalakrishnan S, McCauley JA, et al. Preclinical pharmacology and pharmacokinetics of CERC‐301, a GluN2B‐selective N‐methyl‐D‐aspartate receptor antagonist. Pharmacol Res Perspect [Internet]. 2015;3:e00198. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4777252/
- Wong YC, Ilkova T, van Wijk RC, et al. Development of a population pharmacokinetic model to predict brain distribution and dopamine D2 receptor occupancy of raclopride in non-anesthetized rat. Eur J Pharm Sci. 2017;111:514–525.
- Borsook D, Becerra L, Fava M. Use of functional imaging across clinical phases in CNS drug development. Transl Psychiat [Internet]. 2013;3:e282.
- Borsook D, Hargreaves R, Becerra L. Can functional magnetic resonance imaging improve success rates in CNS drug discovery? Expert Opin Drug Discov. 2011;6:597–617.
- Groenendaal D, Freijer J, de Mik D, et al. Influence of biophase distribution and P-glycoprotein interaction on pharmacokinetic-pharmacodynamic modelling of the effects of morphine on the EEG. Br J Pharmacol. 2007;151:713–720.
- Cox EH, Langemeijer MW, Gubbens-Stibbe JM, et al. The comparative pharmacodynamics of remifentanil and its metabolite GR90291, in a rat electroencephalogrphic model. Anesthesiology. 1999;90:535–544.
- Stevens J, Ploeger BA, Hammarlund-Udenaes M, et al. Mechanism-based PK-PD model for the prolactin biological system response following an acute dopamine inhibition challenge: quantitative extrapolation to humans. J Pharmacokinet Pharmacodyn. 2012;39:463–477.
- Danhof M, Alvan G, Dahl SG, et al. Mechanism-based pharmacokinetic-pharmacodynamic modeling-a new classification of biomarkers. Pharm Res. 2005;22:1432–1437.
- Mager DE, Woo S, Jusko WJ. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab Pharmacokinet. 2009;24:16–24.
- de Lange ECM, Danhof M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet [Internet]. 2002;41:691–703. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12162757
- Shen DD, Artru AA, Adkison KK. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev. 2004;56:1825–1857.
- Lin J. CSF as a surrogate for assessing CNS exposure: an industrial perspective. Curr Drug Metab [Internet]. 2008;9:46–59. Available from: http://www.scopus.com/inward/record.url?eid=2-s2.0-38849136513&partnerID=tZOtx3y1
- de Lange EC. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS [Internet]. 2013;10:12. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3602026&tool=pmcentrez&rendertype=abstract
- de Lange ECM, Hammarlund-Udenaes M. Translational aspects of blood-brain barrier transport and central nervous system effects of drugs: from discovery to patients. Clin Pharmacol Ther. 2015;97:380–394.
- Lange ECMD, Brink WJVD, Yamamoto Y, et al. Novel CNS drug discovery and development approach: model-based integration to predict neuro-pharmacokinetics and pharmacodynamics. Expert Opin Drug Discov [Internet]. 2017;12:1–12. Available from: https://www.tandfonline.com/doi/full/10.1080/17460441.2017.1380623
- Yamamoto Y, Välitalo PA, Berg D-JVD, et al. A generic multi-compartmental CNS distribution model structure for 9 drugs allows prediction of human brain target site concentrations. Pharm Res [Internet]. 2016. DOI:10.1007/s11095-016-2065-3.
- Loryan I, Sinha V, Mackie C, et al. Molecular properties determining unbound intracellular and extracellular brain exposure of CNS drug candidates. Mol Pharm. 2015;12:520–532.
- Yamamoto Y, Välitalo PA, Huntjens DR, et al. Predicting drug concentration-time profiles in multiple CNS compartments using a comprehensive physiologically-based pharmacokinetic model. CPT Pharmacomet Syst Pharmacol. 2017;6:765–777.
- Visser SAG, Smulders CJGM, Reijers BPR, et al. Mechanism-based pharmacokinetic-pharmacodynamic modeling of concentration-dependent hysteresis and biphasic electroencephalogram effects of alphaxalone in rats. J Pharmacol Exp Ther [Internet]. 2002;302:1158–1167. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12183676
- Danhof M, de Jongh J, De Lange ECM, et al. Mechanism-based pharmacokinetic-pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol. 2007;47:357–400.
- Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31:510–518.
- de Witte WEA, Vauquelin G, van der Graaf PH, et al. The influence of drug distribution and drug-target binding on target occupancy: the rate-limiting step approximation. Eur J Pharm Sci [Internet]. 2017;1–6. DOI:10.1016/j.ejps.2017.05.024.
- Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–478.
- Chien JY, Friedrich S, Heathman MA, et al. Pharmacokinetics/pharmacodynamics and the stages of drug development : role of modeling and simulation. 2005;7:E544–E559.
- Adolph E. Quantitative relations in the physiological constitutions of mammals. Science (80-.) 1949;109:579–585.
- Bertalanffy LV. Quantitative laws in metabolism and growth. Q Rev Biol [Internet]. 1957;32:217–231. Available from: http://linksource.ebsco.com/FullText.aspx?linkout=http://search.ebscohost.com/login.aspx?direct=true&scope=site&db=mnh&AN=21800635&ErrorURL=http://linksource.ebsco.com/error.aspx
- West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276:122–126.
- Zuideveld KP, van der Graaf PH, Peletier LA, et al. Allometric scaling of pharmacodynamic responses: application to 5-HT 1A receptor mediated responses from rat to man. Pharm Res. 2007;24:2031–2039.
- Kantae V, Krekels EHJ, Ordas A, et al. Pharmacokinetic modeling of paracetamol uptake and clearance in zebrafish larvae: expanding the allometric scale in vertebrates with five orders of magnitude. Zebrafish [Internet]. 2016;13:504–510. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27632065
- Westerhout J, Danhof M, Lange ECMD. Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J Pharm Sci. 2011;100:3577–3593.
- Ball K, Bouzom F, Scherrmann J-M, et al. Physiologically based pharmacokinetic modelling of drug penetration across the blood–brain barrier—towards a mechanistic IVIVE-based approach. AAPS J [Internet]. 2013;15:913–932. Available from: http://link.springer.com/10.1208/s12248-013-9496-0
- Westerhout J, Ploeger B, Smeets J, et al. Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J. 2012;14:543–553.
- Kalvass JC, Olson ER, Cassidy MP, et al. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50,u and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther. 2007;323:346–355.
- Gosset JR, Beaumont K, Matsuura T, et al. A cross-species translational pharmacokinetic-pharmacodynamic evaluation of core body temperature reduction by the TRPM8 blocker PF-05105679. Eur J Pharm Sci [Internet]. 2017;0–1. DOI:10.1016/j.ejps.2017.06.009.
- Gotta V, Yu Z, Cools F, et al. Application of a systems pharmacology model for translational prediction of hERG-mediated QTc prolongation. Pharmacol Res Perspect. 2016;4:1–17.
- van den Brink W, Palic S, Köhler I, et al. Access to the CNS: biomarker strategies for dopaminergic treatments. Pharm Res. 2018;35:64.
- Bianchi MT, Pathmanathan J, Cash SS. From ion channels to complex networks: magic bullet versus magic shotgun approaches to anticonvulsant pharmacotherapy. Med Hypotheses [Internet]. 2009;72:297–305.
- Xuan J, Pan G, Qiu Y, et al. Metabolomic profiling to identify potential serum biomarkers for schizophrenia and risperidone action. J Proteome Res. 2011;10:5433–5443.
- López-Muñoz F, Baumeister AA, Hawkins MF, et al. The role of serendipity in the discovery of the clinical effects of psychotropic drugs: beyond of the myth. Actas Esp Psiquiatr. 2012;40:34–42.
- Sun J, Xu H, Zhao Z. Network-assisted investigation of antipsychotic drugs and their targets. Chem Biodivers. 2012;9:900–910.
- Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov [Internet]. 2004;3:353–359. Available from: http://www.nature.com/doifinder/10.1038/nrd1346
- Margineanu DG. Neuropharmacology beyond reductionism - A likely prospect. BioSystems [Internet]. 2016;141:1–9.
- Carlsson A, Waters N, Holm-Waters S, et al. Interactions between monoamines, glutamate, and GABA in schizophrenai: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260.
- Greef JVD, Mcburney RN. Rescuing drug discovery: in vivo systems pathology and systems pharmacology. Nat Rev Drug Discov. 2005;4:961–968.
- Kumar B, Prakash A, Ruhela RK, et al. Potential of metabolomics in preclinical and clinical drug development. Pharmacol Rep [Internet]. 2014;66:956–963.
- Kaddurah-Daouk R, Weinshilboum R. Pharmacometabolomics research network. metabolomic signatures for drug response phenotypes: pharmacometabolomics enables precision medicine. Clin Pharmacol Ther [Internet]. 2015;98:71–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25871646
- Kaddurah-Daouk R, McEvoy J, Baillie RA, et al. Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol Psychiatry. 2007;12:934–945.
- Lamers RJAN, Van Nesselrooij JHJ, Kraus VB, et al. Identification of an urinary metabolite profile associated with osteoarthritis. Osteoarthr Cartil. 2005;13:762–768.
- van der Greef J, Adourian A, Muntendam P, et al. Lost in translation? Role of metabolomics in solving translational problems in drug discovery and development. Drug Discov Today Technol. 2006;3:205–211.
- Blais EM, Rawls KD, Dougherty BV, et al. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat Commun [Internet]. 2017;8:1–15.
- Wishart DS, Jewison T, Guo AC, et al. HMDB 3.0-the human metabolome database in 2013. Nucl Acids Res. 2013;41:801–807.
- Quinones MP, Kaddurah-Daouk R. Metabolomics tools for identifying biomarkers for neuropsychiatric diseases. Neurobiol Dis [Internet]. 2009;35:165–176.
- Kaddurah-Daouk R, Bogdanov MB, Wikoff WR, et al. Pharmacometabolomic mapping of early biochemical changes induced by sertraline and placebo. Transl Psychiat [Internet]. 2013;3:e223. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3566722&tool=pmcentrez&rendertype=abstract
- Rozen S, Cudkowicz ME, Bogdanov M, et al. Metabolomic analysis and signatures in motor neuron disease. Metabolomics. 2005;1:101–108.
- Jimhez-Jimcnez FJ, Molina A, Vargas C, et al. Neurotransmitter amino acids in cerebrospinal fluid of patients with Parkinson ’ s disease. J Neurol Sci. 1996;141:39–44.
- Brown M, Dunn WB, Ellis DI, et al. A metabolome pipeline : from concept to data to knowledge. Metabolomics. 2005;1.
- Bartel J, Krumsiek J, Theis FJ. Statistical methods for the analysis of high-throughput metabolomics data. Comput Struct Biotechnol J [Internet]. 2013;4:e201301009. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3962125&tool=pmcentrez&rendertype=abstract
- Krumsiek J, Suhre K, Illig T, et al. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst Biol. 2011;5:21.
- Xia J, Wishart DS. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucl Acids Res. 2010;38:71–77.
- Danhof M. Systems pharmacology - Towards the modeling of network interactions. Eur J Pharm Sci [Internet]. 2016. DOI:10.1016/j.ejps.2016.04.027.
- Jin JY, Almon RR, DuBois DC, et al. Modeling of corticosteroid pharmacogenomics in rat liver using gene microarrays. J Pharmacol Exp Ther. 2003;307:93–109.
- Wang J, Reijmers ÆT, Chen ÆL. Systems toxicology study of doxorubicin on rats using ultra performance liquid chromatography coupled with mass spectrometry based metabolomics. Metabolomics. 2009;5:407–418.
- Lamers RJ, DeGroot J, Spies-Faber EJ, et al. Identification of disease- and nutrient-related metabolic fingerprints in osteoarthritic Guinea pigs. J Nutr. 2003;133:1776–1780.
- ’T Hart BA, Vogels JTWE, Spijksma G, et al. 1H-NMR spectroscopy combined with pattern recognition analysis reveals characteristic chemical patterns in urines of MS patients and non-human primates with MS-like disease. J Neurol Sci. 2003;212:21–30.
- Yao D, Shi X, Wang L, et al. Characterization of differential cocaine metabolism in mouse and rat through metabolomics-guided metabolite profiling. Drug Metab Dispos. 2013;41:79–88.
- National Research Council (US). Committee on applications of toxicogenomics to cross-species extrapolation. Application of Toxicogenomics to Cross-Species Extrapolation: A Report of a Workshop; 2005.
- Kohler I, Hankemeier T, Graaf PHVD, et al. European Journal of Pharmaceutical Sciences Integrating clinical metabolomics-based biomarker discovery and clinical pharmacology to enable precision medicine. Eur J Pharm Sci [Internet]. 2017;0–1. DOI: 10.1016/j.ejps.2017.05.018
- Hasselt JGCV, Graaf PHVD. Towards integrative systems pharmacology models in oncology drug development. Drug Discov Today Technol [Internet]. 2015;15:1–8.
- Kantae V, Krekels EHJ, Esdonk MJV, et al. Integration of pharmacometabolomics with pharmacokinetics and pharmacodynamics: towards personalized drug therapy. Metabolomics [Internet]. 2017;13:9. Available from: http://link.springer.com/10.1007/s11306-016-1143-1
- Ellero-Simatos S, Lewis JP, Georgiades A, et al. Pharmacometabolomics reveals that serotonin is implicated in aspirin response variability. CPT Pharmacomet Syst Pharmacol [Internet]. 2014;3:e125. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25029353
- Rasmussen MA, Colding-Jørgensen M, Hansen LT, et al. Multivariate evaluation of pharmacological responses in early clinical trials - a study of rIL-21 in the treatment of patients with metastatic melanoma. Br J Clin Pharmacol. 2010;69:379–390.
- van den Brink WJ, Elassaiss-Schaap J, Gonzalez-Amoros B, et al. Multivariate pharmacokinetic/pharmacodynamic (PKPD) analysis with metabolomics shows multiple effects of remoxipride in rats. Eur J Pharm Sci [Internet]. 2017;109:431–440. [cited 2017 Sep 13]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0928098717304888
- Sorger PK, Allerheiligen SRB, Altmann RB, et al. Emergence of quantitative and systems pharmacology: a white paper. NIH QSP Work [Internet]. 2011;1–48. Available from: papers2://publication/uuid/135d6564-7ee8-4a97-ba4b-be47e3369b3b
- Iyengar R, Zhao S, Chung S-W, et al. Merging Systems Biology with Pharmacodynamics. Sci Transl Med. 2012;4:126ps7–126ps7.
- Knight-Schrijver VR, Chelliah V, Cucurull-Sanchez L, et al. The promises of quantitative systems pharmacology modelling for drug development. Comput Struct Biotechnol J [Internet]. 2016;14:363–370.
- Geerts H, Spiros A, Roberts P, et al. Has the time come for predictive computer modeling in CNS drug discovery and development? CPT Pharmacometrics Syst Pharmacol [Internet]. 2012;1:e16. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3600733&tool=pmcentrez&rendertype=abstract
- de Lange ECM, Danhof M, de Boer AG, et al. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood–brain barrier. Brain Res Rev [Internet]. 1997;25:27–49. Available from: http://www.sciencedirect.com/science/article/pii/S0165017397000143
- de Lange ECM. Microdialysis in drug development. Microdialysis Drug Dev [Internet]. 2013;13–33. Available from: http://link.springer.com/10.1007/978-1-4614-4815-0
- Qu Y, Olson L, Jiang X, et al. Evaluating PK/PD relationship of CNS drug by using liquid chromtography/tandem mass spectrometry coupled to in vivo microdialysis. In: J Prasain, editor. Tandem mass spectrometry - Applications and principles. Rijeka: InTech; 2012. p. 421–440.
- Ravenstijn PG, Drenth H-J, O’Neill MJ, et al. Evaluation of blood-brain barrier transport and CNS drug metabolism in diseased and control brain after intravenous L-DOPA in a unilateral rat model of Parkinson’s disease. Fluids Barriers CNS [Internet]. 2012;9:4. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3298802&tool=pmcentrez&rendertype=abstract
- Huang M, Panos JJ, Kwon S, et al. Comparative effect of lurasidone and blonanserin on cortical glutamate, dopamine, and acetylcholine efflux: role of relative serotonin (5-HT) 2A and da D2 antagonism and 5-HT1A partial agonism. J Neurochem. 2014;128:938–949.
- Kao C-Y, Anderzhanova E, Asara JM, et al. NextGen brain microdialysis: applying modern metabolomics technology to the analysis of extracellular fluid in the central nervous system. Mol Neuropsychiatry. 2015;1:60–67.
- Goetghebeur PJ, Swartz JE. True alignment of preclinical and clinical research to enhance success in CNS drug development: a review of the current evidence. J Psychopharmacol [Internet]. 2016;30:586–594. Available from: http://jop.sagepub.com/cgi/doi/10.1177/0269881116645269
- Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73.
- Greef JVD, Martin S, Juhasz P, et al. The art and practice of systems biology in medicine : mapping patterns of relationships. J Proteome Res. 2007;6:1540–1559.
- Chokkathukalam A, Kim D, Barrett MP, et al. Stable isotope-labeling studies in metabolomics : new insights into structure and dynamics of metabolic networks. Bioanalysis. 2014;6:511–524.
- Bueschl C, Krska R, Kluger B, et al. Isotopic labeling-assisted metabolomics using LC-MS. Anal Bioanal Chem. 2013;405:27–33.