
ABSTRACT
Recombinant protein therapeutics have become increasingly useful in combating human diseases, such as cancer and those of genetic origin. One quality concern for protein therapeutics is the content and the structure of the aggregated proteins in the product, due to the potential immunogenicity of these aggregates. Collective efforts have led to a better understanding of some types of protein aggregates, and have revealed the diversity in the structure and cause of protein aggregation. In this work we used a broad range of analytical techniques to characterize the quinary structure (complexes in which each composing unit maintains native quaternary structure) of the stable non-covalent dimer and oligomers of a monoclonal IgG1λ antibody. The results supported a mechanism of intermolecular domain exchange involving the Fab domains of 2 or more IgG molecules. This mechanism can account for the native-like higher order (secondary, tertiary and disulfide bonding) structure, the stability of the non-covalent multimers, and the previously observed partial loss of the antigen-binding sites without changing the antigen-binding affinity and kinetics of the remaining sites (Luo et al., 2009, mAbs 1:491). Furthermore, the previously observed increase in the apparent affinity to various Fcγ receptors (ibid), which may potentially promote immunogenicity, was also explained by the quinary structure proposed here. Several lines of evidence indicated that the formation of multimers by the mechanism of intermolecular domain exchange took place mostly during expression, not in the purified materials. The findings in this work will advance our knowledge of the mechanisms for aggregation in therapeutic monoclonal antibodies.
Abbreviations
| IgG1λ | = | immunoglobulin G subclass 1 with lambda light chains |
| HOS | = | higher order structure |
| HMW | = | high molecular weight |
| PBS | = | phosphate-buffered saline |
| SE-HPLC | = | size exclusion high performance liquid chromatography |
| CD | = | circular dichroism |
| cHA | = | ceramic hydroxyapatite |
| AUC-SV | = | analytical ultracentrifugation sedimentation velocity |
| SPR | = | surface plasmon resonance |
| SEC-MALS | = | size exclusion chromatography coupled with multi-angle light scattering detector |
| ESI-QTOF MS | = | electrospray ionization quadrupole time-of-flight mass spectrometry |
| GdmCl | = | guanidine hydrochloride |
Introduction
Recombinant proteins play increasingly important roles in medicines for human diseases. The quality of the manufactured protein products may affect the safety and efficacy of the biotherapeutics in patients. One critical quality attribute is the aggregation of the protein molecules, which is a safety concern mainly due to the potential immunogenicity of proteinaceous particles.Citation1-4 The immune responses may correlate to the sizes, numbers, structures, and properties of the aggregates, as suggested by the results of in vitro studies in cells and in vivo studies in animal and in human.Citation5-8
A substantial body of knowledge has accumulated in the past several decades on the structures and properties of protein aggregates, which exhibit enormous diversity and complexity. All protein aggregates result from intermolecular association, which can be covalently connected or non-covalently associated. The structure of the molecules in either covalent or non-covalent aggregates can be native, native-like, partially denatured, or denatured. The non-covalent aggregates can be reversible/dissociable in non-denaturing conditions, or stable in non-denaturing conditions but dissociable in denaturing conditions. The molecules dissociated from aggregates in non-denaturing conditions can be functional, partially functional, or non-functional. Even the proteins in the aggregates can be fully or partially capable of interacting with their biologic targets. In certain cases, the higher avidity resulting from aggregation can elevate the apparent binding affinity.Citation9 With regard to the higher order structure, the molecules in both covalent and non-covalent aggregates can be spatially well ordered or random to more or less degrees. The native-like aggregates, especially with repetitive structures that mimic pathogen-associated molecular patterns (PAMP), are proposed to be more immunogenic than those randomly associated and composed of fully denatured proteins.Citation1 The mechanism is thought to be via T-cell dependent pathways that involve pattern recognition receptors (PRR) on antigen presenting cells (APC).Citation8 The understanding of the interactions of protein aggregates with the innate and the adaptive immune cells, as well as the subsequent responses that lead to clinical immune reactions, has advanced in recent years, but major gaps remain to be further resolved.Citation8 One of the limiting factors is the understanding of the structures and mechanisms of all types of protein aggregates.
We previously reported some unusual properties and behaviors of high molecular weight (HMW) species of a monoclonal antibody, named mAb1.Citation9 The higher order structure (HOS) of its dimer and oligomers were characterized using biophysical methods, and the binding to the antigen and Fcγ receptors were compared with that of the monomer using surface plasmon resonance (SPR). The dimer and oligomers of mAb1 were non-covalent but very stable, with HOS similar to the native monomer, based on the spectroscopic results. The monomer and dimer had comparable antigen-binding affinity (KD ∼0.1 nM), but the binding capacity of the dimer was lower than that of monomer by nearly 50%. On the other hand, the dimer exhibited higher apparent binding affinity than that of the monomer to all the Fcγ receptors tested in that work. The apparent affinity to low-affinity Fcγ receptors, FcγRIIA and FcγRIIIB, were further increased when mAb1 molecules formed immune complexes with its divalent antigen. The significance of the elevated binding of aggregated antibodies to FcγRs in the potential immunogenicity of the antibodies, when used as biotherapeutics, were discussed,Citation10 and further recognized in recent years.Citation8,11 This result also suggested that mAb1 dimer may present to FcγRs a spatial pattern or quinary structure that bears similarity to that of immune complex.
The term quinary structure has been used in the cell biology literature to refer to the transient macromolecular interactions, primarily arising from the crowding effect in living cells,Citation12,13 but also to the specific interactions that organize the cellular interior or form supramolecular functional assemblies, such as sequential enzyme complexes and ribosome-RNAs complexes.Citation14-16 Chen et al.Citation17 applied “quinary structure” to the weak and reversible self-association of an IgG1 molecule, infliximab, in vitro at concentrations much lower than cellular protein concentrations. In this work, it is used to refer to stable self-association via specific binding of an IgG1 molecule, mAb-PFM, at an even lower concentration. Both meanings focus on the hierarchy of protein structure, rather than function, counting the assembly of well-folded heavy chains and light chains of IgG as the quaternary structure. The heavy chains and light chains are expressed and folded as individual molecules, and their associations into functional IgG do not rely on the inter-chain disulfide bonds. However, the term quinary structure should not be applied to all protein aggregates, only those in which the secondary, tertiary, and quaternary structures are known to be largely native. This generalized use of quinary structure is, therefore, consistent with that in cell biology from the structural viewpoint.
The known mechanisms for non-covalent protein aggregates with native quinary structure include charge-charge and hydrophobic interactions. However, the charge-mediated association is unlikely to be stable enough to survive the dilution required by methods such as chromatography and SPR. Therefore, charge-based explanations are insufficient to explain the reported observations. This work focuses on further characterization of the stable non-covalent dimer of the same IgG1 molecule as mAb1 in Luo et al,Citation9 renamed mAb-PFM. Substantial evidence, based on combined results of structural and functional studies, suggest that intermolecular domain exchange in the antigen-binding fragment (Fab) domains is a likely mechanism for the stable dimer and oligomer of this molecule, which to our knowledge has not been reported for proteins as large and complex as an IgG. Detailed structural knowledge of protein aggregates is important to advance the understanding of the mechanism by which the monomers associate into aggregates, which is critical to the development of strategies to control the bioprocesses and to minimize the level of aggregation in the protein drug products. It is also valuable to the understanding of the potential immunogenicity of various types of protein aggregates.
Results
Characterizations of intact mAb-PFM and high molecular weight species
Size-exclusion high performance liquid chromatography
Two chromatographic steps, protein-A (pro-A) and ceramic hydroxyapatite (cHA),Citation18 were used to purify the recombinant monoclonal antibody, mAb-PFM (IgG1 with λ light chains), from Chinese hamster ovary (CHO) cell culture medium. Analytical size exclusion high performance liquid chromatography (SE-HPLC) was used to analyze the aggregate content of each purification intermediate under native conditions. shows a SE-HPLC profile of a pro-A purified sample, which contained large amounts of HMW species. The HMW content after the pro-A step ranged from ∼18% to ∼28%.
Figure 1. SE-HPLC analysis for mAb-PFM purification from the expression medium. (A) mAb-PFM eluted from a protein A column. (B) mAb-PFM monomer eluted from the subsequent cHA column (top panel), and the mixture of mAb-PFM monomer and HMW species stripped from the cHA column with high salt solution (bottom panel). The mobile phase was PBS. The peak identifications are as indicated in the figures.
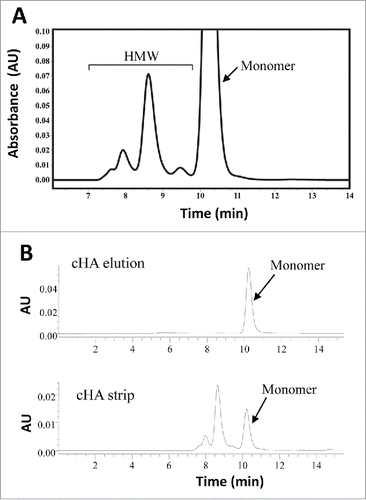
The top and the bottom panel of show the SE-HPLC profiles of the cHA-purified monomer (cHA elution) and the enriched HMW species (cHA strip), respectively. The latter was stripped from the cHA column with very high salt concentration. The purity of the monomer was >98%. The cHA strip typically contained 5–35% monomer and 65–95% HMW species, depending on the conditions.
Size distribution and HOS of mAb-PFM HMW Species
The composition of HMW species from the cHA strip was further analyzed using the analytical ultracentrifugation sedimentation velocity (AUC-SV) technique. shows a typical profile of the HMW fraction, overlaid with that of the purified monomer of mAb-PFM. The major component in the cHA HMW fraction was dimer. The trimer and tetramer, as minor components, were also resolved. shows the profiles of HMW fraction in phosphate-buffered saline (PBS), 600 mM sodium chloride or 20% ethanol (v/v). The high salt and ethanol conditions were intended to disrupt the aggregates. There are indications that these conditions may disrupt protein association by weak charge-charge or hydrophobic interactions, respectively.Citation19 The samples used in these experiments contained more monomer than the one used in the experiments shown in . However, the relative quantities of monomer, dimer and oligomers did not change significantly in the high salt or ethanol-containing solutions. These results indicated that the self-associations of mAb-PFM studied in this work were stable under these conditions, which did not support a mechanism of charge-charge or hydrophobic interaction.
Figure 2. Biophysical characterizations for the cHA column purified monomer and cHA column enriched HMW fraction. (A) Overlay of analytical ultracentrifugation sedimentation velocity (AUC-SV) profiles for the purified monomer (red) and the enriched HMW species (blue). The total protein concentrations were 0.4–0.5 mg/mL. (B) Overlay of AUC-SV profiles for the enriched HMW species in PBS (black), 600 mM NaCl (red), and in the presence of 20% ethanol (blue). The samples were obtained by ∼5-fold dilution of the HMW sample in (A) with appropriate reagents. The profiles are not normalized with respect to sample concentrations. The sedimentation coefficients in (A) and (B) were corrected for the standard state of water at 20°C. The instrumentation and experimental procedures for AUC-SV were as described previously.Citation9 (C) Overlay of far-UV CD spectra for the monomer (red) and enriched HMW species (blue) in formulation buffer (dots) and with 6 M GdmCl (dashed lines). The instrumentation and experimental procedures for far-UV CD were as described previously.Citation9 (D) Overlay of intrinsic fluorescence emission spectra of the monomer (red) and enriched HMW species (blue) in formulation buffer (solid lines) and with 6 M GdmCl (dashed lines). Each curve in (C) and (D) represents triplicate measurements averaged at each data point.
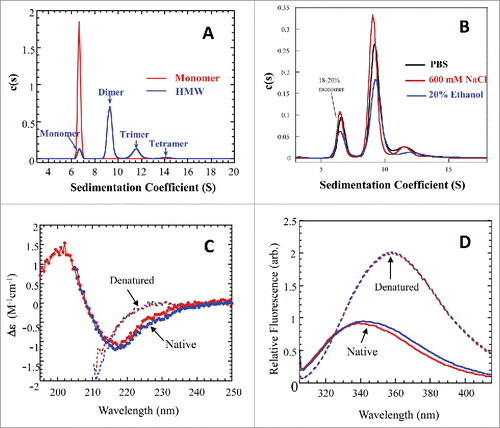
Far-UV circular dichroism (CD) and fluorescence emission spectroscopies were used to compare the apparent secondary and the tertiary structure of mAb-PFM HMW fraction (containing multiple species) and that of mAb-PFM monomer, as shown in and , respectively. The spectra of the 2 samples in 6 M guanidine hydrochloride (GdmCl) are also overlaid in the figure for comparison. Each spectrum represents an average of triplicate measurements. The far-UV CD spectra of the monomer and HMW fraction are very similar to each other in both the native and the denatured states. The spectra under the native condition suggest that the secondary structure of both monomer and HMW species of mAb-PFM consist primarily of β-sheet, as expected for a well-folded IgG antibody. The 2 samples exhibited identical fluorescence emission profiles under the denaturing condition. Under native conditions, the peak of the HMW fraction was slightly red-shifted compared with that of monomer, indicating minor differences in the tertiary structure between the 2. However, both spectra clearly indicated that the 2 materials were well folded and had largely similar tertiary structure.
Size distribution and HOS of refolded molecules of denatured mAb-PFM HMW species
shows that nearly all HMW species of mAb-PFM dissociated to monomer in 6 M GdmCl, confirming the non-covalent nature. The denatured samples were allowed to refold by diluting with PBS of 10-times volume. shows a new distribution, ∼65% monomer and ∼35% aggregates, in the refolded sample. The monomers in had lower sedimentation coefficients than that in , likely due to the differences in the shape of the molecule. When a mAb is unfolded in 6 M GdmCl, it is less globular than in native state, leading to higher frictional ratio and, therefore, lower sedimentation coefficient. The aggregates in the refolded sample consisted of significantly larger species, and were possibly formed by molecules in intermediate conformations during the refolding. They were, therefore, likely to have different structures from stable dimer and oligomers in the starting material.
Figure 3. Biophysical characterizations for the denatured and refolded mAb-PFM monomer and HMW species. (A) Overlay of AUC-SV profiles for the denatured monomer (red) and HMW species (blue). The total protein concentrations were 0.4–0.5 mg/mL. (B) AUC-SV profiles for the refolded samples of the denatured monomer and HMW species, obtained by ∼10-fold dilution with PBS, followed by overnight incubation at 20°C. The sedimentation coefficients in (A) and (B) were corrected for the standard state of water at 20°C. The instrumentation and experimental procedures for AUC-SV were as described previously.Citation9 (C) Overlay of far-UV CD spectra for the monomers isolated by SEC from the refolded samples of the denatured monomer (red) and of the denatured HMW species (blue). The instrumentation and experimental procedures for far-UV CD were as described previously.Citation9 (D) Overlay of intrinsic fluorescence emission spectra for the monomers isolated by SEC from the refolded samples of the denatured monomer (red) and of the denatured HMW species (blue). The instrumentation and conditions were as described in . Each curve in (C) and (D) represents triplicate measurements averaged at each data point.
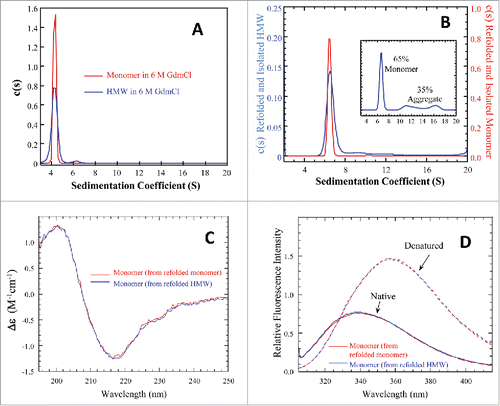
The refolded monomers of the denatured monomer and of denatured HMW fraction were isolated by SEC-HPLC. The far-UV CD and fluorescence spectra of these refolded monomers, shown in and , respectively, indicated that their structure and conformation were comparable, more so than before denaturation, and were likely to be native based on the comparison to and . Furthermore, the refolded monomers did not form dimer, indicated by the lack of a peak at ∼9 S in . This observation once again argued against a mechanism of charge-charge or hydrophobic interactions among mAb-PFM monomers in forming the dimer and HMW species in native state. More importantly, this observation demonstrated that the stable dimer/oligomers in the cell culture did not reform once the association was disrupted by denaturing the protein.
Antigen-binding of mAb-PFM monomer and HMW species
The antigen-binding kinetics and stoichiometry of HMW species of mAb-PFM (previously named mAb1), containing primarily dimer as shown in and , were compared with that of the purified monomer in the previous study using surface plasmon resonance (SPR) technique ( and in Luo et al.Citation9). The mAb-PFM samples were captured on the Biacore flow cell coated with protein A, targeting the same surface density (judged by ΔRU before and after capturing) for the monomer and the HMW samples. The binding of the infused antigen to the captured mAb-PFM on the chip was followed by a dissociation phase and the regeneration of the protein A surface for the next capturing and binding cycle. The global fitting of the curves obtained with varied antigen concentration to an appropriate kinetic model provided values of the on-rate and off-rate, on which basis the binding affinity KD were determined. The analysis also yielded Rmax values for each sample. Rmax, in general, reflects the binding capacity of the captured sample and is independent of KD. It is determined by the binding stoichiometry and the amount of active binding sites on the surface.
Based on data in of the Luo et al. article,Citation9 it was concluded that the KDs for the antigen binding of the monomer and the HMW sample, containing predominantly stable mAb-PFM dimer, were in the same order of magnitude, 60 - 110 pM. However, the Rmax values for the 2 samples, captured at equal amount by mass, were significantly different, with that of HMW species being lower than that of the monomer by ∼50%. The capture of mAb-PFM by protein A, which primarily binds to the Fc domain, ensured a configuration of mAb-PFM on the flow cell surface that the Fab domains of all molecules, including those in the stable dimer and oligomers, were equally accessible to the antigen of mAb-PFM. Therefore, the results indicating comparable KD but half of the binding capacity for the dimer vs monomer could be interpreted as showing 2 of the 4 Fabs in the stable dimer were inactive and the other 2 were as active as the Fabs in the monomer. Also, taking into account that the overall secondary and tertiary structure of the monomer and HMW species of mAb-PFM are very similar, one possible scenario for the ∼50% inactivation in mAb-PFM dimer is that dimerization occurs via a stable association of 2 Fabs, each from one of the 2 mAb-PFM monomers, and this association imposes steric interference to the antigen binding of the associated Fabs.
Table 1. Additional characterizations for comparison of mAb-PFM monomer and HMW species.
Comparisons of monomer and oligomers of mAb-PFM by other methods
Additional analytical methods were used to characterize and compare monomers and oligomers of mAb-PFM. The assays, the attributes that the assays aimed to assess, and the results are summarized in . No significant difference between the 2 samples was found by these methods. Extensive mass spectrometry studies confirmed that the molecules in the HMW fractions and in the main peak were identical in primary structure.
Characterizations of the papain-digestion fragments of mAb-PFM
Papain digestion of mAb-PFM monomer and HMW species
Papain digestion was used to test the hypothesis that the mAb-PFM HMW species, mainly dimer, were formed via stable intermolecular Fab-Fab association. The expected papain-digestion fragments of mAb-PFM monomer and dimer are illustrated in . One non-covalent Fab dimer, (Fab)2, 2 Fabs and 2 Fcs were expected for each mAb-PFM dimer, while 2 Fab and one Fc domains were expected for mAb-PFM monomer.
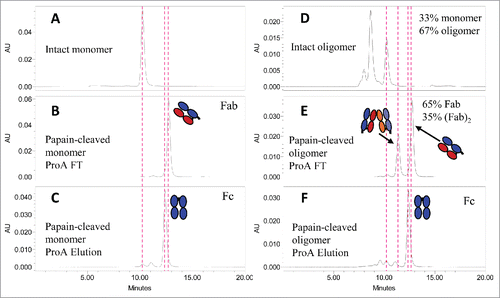
Figure 4. A: Schematic of the expected fragments resulting from papain digestion of IgG1 monomer and dimer associated via 2 Fab domains, one from each monomer. B: SDS-PAGE for the intact and the papain-digested fragments of mAb-PFM monomer and HMW species under the non-reducing (lanes 1–6) and the reducing (lanes 7–12) conditions. The fragments in the digests were fractionated by protein A column. Lanes 1 and 7: The protein A column flow-through fraction of the digested mAb-PFM monomer. Lanes 2 and 8: The protein A column eluted (Fc containing) fraction of the digested mAb-PFM monomer. Lanes 3 and 9: The protein A column flow-through fraction of the digested mAb-PFM HMW species. Lanes 4 and 10: The protein A column eluted fraction of the digested mAb-PFM HMW species. Lanes 5 and 11: Intact mAb-PFM monomer. Lanes 6 and 12: Intact mAb-PFM HMW species. The band identifications are as indicated in the figure. Invitrogen Novex Tris-Glycine 4–20% acrylamide gradient gel was used and the protein bands were stained with 0.05% Coomassie.
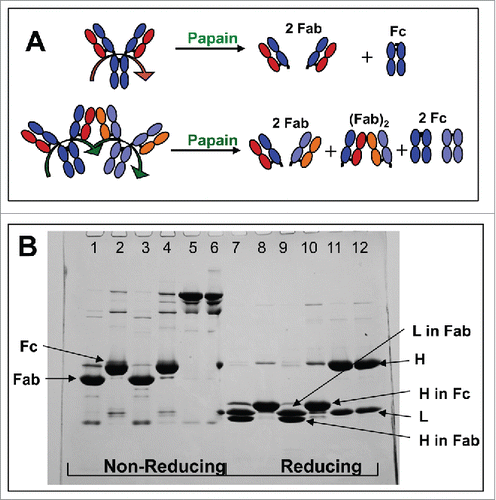
The Fcs and the Fabs in the papain digests of mAb-PFM monomer and HMW species were separated using a protein A affinity column. The components in the flow-through and the eluted fractions were analyzed using non-reducing and reducing SDS-PAGE, as shown in . The non-digested mAb-PFM monomer and HMW species were run under non-reducing (Lanes 5 and 6, respectively) and reducing (Lanes 11 and 12, respectively) conditions. The 2 samples exhibited nearly identical band patterns, with only a trace level band in Lane 6 that is larger than mAb-PFM monomer, demonstrating again the predominantly non-covalent nature of mAb-PFM HMW species. The flow-through fractions of the protein A column for papain-digested mAb-PFM monomer and HMW species were also identical under both non-reducing (Lanes 1 and 3) and reducing (Lanes 7 and 9) conditions. No dimeric Fab was observed in Lanes 3 and 9, indicating the non-covalent nature of the expected (Fab)2. The apparent size of the heavy chain (H) fragment in the papain-produced Fc (protein A eluate) was larger than the light chain (L), while the H fragment in the papain-produced Fab (protein A flow-through) was smaller than L (Lanes 9 and 10). Consequently, the apparent size of the papain-produced Fab was smaller than the Fc (protein A eluate) under non-reducing conditions (Lanes 1 and 2). These are consistent with the results of chromatography discussed below.
Mass determination of the papain-digestion fragments by SEC-MALS and mass spectrometry
The flow-through and the eluted fractions of the protein A column were also analyzed in native condition using SE-HPLC, as shown in . Consistent with the SDS-PAGE results, the hydrodynamic size of papain-produced Fab from mAb-PFM monomer () and HMW species () appeared to be smaller than that of the Fc from the same materials ( and , respectively). In contrast to the single band pattern in the SDS-PAGE for the flow through of protein A column of papain-digested mAb-PFM HMW species (, Lane 2), the SE-HPLC profile for the same fraction () revealed a larger component, which was stable and well-resolved from the Fab.
Figure 5. SE-HPLC analysis for the papain-digested fragments of mAb-PFM monomer and HMW species following the protein A column fractionation. (A and D) The intact mAb-PFM monomer and HMW species, respectively. (B and C) The protein A column flow-through and the eluted fraction, respectively, of the digested mAb-PFM monomer. (E and F) The protein A column flow-through and the eluted fraction, respectively, of the digested mAb-PFM HMW species. The mobile phase was PBS.
Multi-angle light scattering detector in-line with the size-exclusion chromatography system (SEC-MALS) provided the weight-average molar mass measurement for each chromatographic fraction, of which the concentration could be accurately determined by the absorbance. The weight-average molar masses for the peaks at ∼11.4 min and 12.8 min in were 88 kDa and 46 kDa, respectively, suggesting that the latter correspond to Fab and the former correspond to Fab dimer, (Fab)2. The weight-average molar masses of all resolved papain fragments by SEC-MALS are summarized in .
Table 2. Mass determination of the papain fragments.
According to the schemes in , all monomers produce monomeric Fab, but dimers produce Fab and (Fab)2 in 1:1 ratio in terms of mass or chromatographic peak area when monitored by UV absorbance. The starting material for papain-digested mAb-PFM HMW species consisted of ∼1/3 monomer and ∼2/3 dimer and larger (). The expected contents of papain-produced Fab and (Fab)2 should be ∼2/3 and 1/3, respectively. This ratio is consistent with the observed ratio in . The conservation of the ratios supports that the non-covalent (Fab)2 in mAb-PFM HMW species was sufficiently stable to withstand the papain digestion and the subsequent protein A and size-exclusion chromatographic conditions.
The papain-produced fragments of mAb-PFM monomer and HMW species shown in were profiled using reversed-phase liquid chromatography (RP-HPLC) coupled to UV and mass spectrometric detection. Accurate mass values were determined using electrospray ionization quadrupole time-of-flight mass spectrometry (ESI-QTOF MS). The papain digestion is known to result in heterogeneous Fab and Fcs due to ragged proteolytic cleavages in the hinge region of heavy chain near the predominant His-Thr cleavage site. The small differences in mass are detectable by mass spectrometry, but not by SEC-MALS. Multiple mass values were obtained for all Fab, (Fab)2 and Fc peaks shown in . The masses of the major component for each fragment, alongside with the calculated masses based on the corresponding sequences, as well as the weight-average molar masses by SEC-MALS, are listed in . The most noticeable discrepancy in mass values was that of (Fab)2 peak in . The average molar mass by SEC-MALS was nearly twice the mass of the Fab peak, while the mass values for Fab and (Fab)2 by mass spectrometry were identical. It is likely that the non-covalent (Fab)2 was broken apart under the denaturing conditions of reversed-phase chromatography and electrospray ionization (, Lane 3).
Antigen-binding of the papain fragments of mAb-PFM by SPR
The antigen-binding properties of the papain-produced, protein A affinity chromatography and SE-HPLC purified fragments of mAb-PFM monomer and HMW species were studied using SPR, as shown in . Due to the lack of an Fc domain in the Fabs, the experimental configuration was changed to covalently immobilizing the antigen and injecting mAb-PFM fragments as analyte at the same molar concentration. A covalent Fab dimer, F(ab’)2, was made by pepsin digestionCitation20 of mAb-PFM monomer, and used as a comparator.
Figure 6. In vitro binding capabilities of the Fcs and Fabs from papain-digested mAb-PFM monomer and HMW species, the (Fab)2 from papain-digested mAb-PFM HMW species, the (Fab’)2 from pepsin-digested mAb-PFM monomer, to the antigen of mAb-PFM by SPR (Biacore). The sensorgrams of the 2 Fc samples (isolated from the digests of monomer and HMW) and the 2 Fab samples (isolated from the digests of monomer and HMW) are respectively superimposable. All fragments were purified using protein A column followed by SE-HPLC. Each displayed sensorgram is the average of triplicate measurements, corrected for non-specific binding with a blank flow cell.
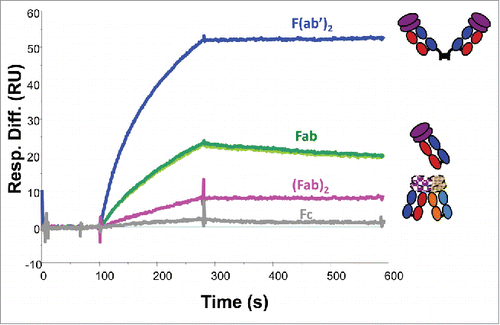
Due to the low quantities and low concentrations of the SE-HPLC purified materials, global fitting to multi-concentration biding curves was not feasible. Fitting the triplicate measurements at a single analyte concentration yielded approximate dissociation constant KD of 3 pM for F(ab’)2, and 700 pM for Fabs from both mAb-PFM monomer and mAb-PFM HMW species. The Rmax for F(ab’)2 was slightly higher than twice that for Fab, consistent with the difference in their molar masses. The apparently stronger antigen-binding of F(ab’)2 compared with Fab was mainly because of the bivalent interaction that effectively slowed down the off-rate of F(ab’)2.
The focus of this experiment was the comparison between the 2 dimer fragments, (Fab)2 vs F(ab’)2. The 2 samples at the same concentration were analyzed using the same flow cell with immobilized antigen. The triplicate binding cycles of each sample were superimposable. The fitted KD values for the 2 samples were similar, but the Rmax for (Fab)2 was less than 1/10 of that for F(ab’)2. These results indicated that the majority of (Fab)2 were incapable of binding to the antigen, which accounted for the ∼50% loss of the binding sites in intact dimer of mAb-PFM,Citation9 if the isolated (Fab)2 are the only intermolecular contacts in the intact mAb-PFM dimer.
Discussion
Evidence for self-association of mAb-PFM via intermolecular domain exchange
In-depth characterizations of mAb-PFM, an IgG1 with λ light chains, were performed in this work using a broad range of biochemical and biophysical methods, for the purpose of elucidating the association mechanism and structure for the non-covalent dimer and HMW species. First, the possibility of the self-association being induced by the differences in primary structure (amino acid sequence and posttranslational modification) was excluded by a variety of mass spectrometry methods, listed in . The mass values for the Fab and Fc domains derived from the monomer and the HMW species, shown in , also supported the conclusions in .
Three observations supported a Fab-mediated intermolecular interaction. First, papain digestion of mAb-PFM HMW species produced a second component in the flow through of the protein A column, which was ∼50% of the total Fab and had an average molar mass close to Fab dimer as determined by SEC-MALS. Second, the (Fab)2 component was largely inactive in antigen-binding as determined by SPR, corresponding to the ∼50% lower binding capacity of the intact mAb-PFM dimer. Third, ESI-QTOF mass spectrometry detected only monomeric Fab in the flow through fraction following the protein A separation and SE-HPLC purification of papain-digested mAb-PFM HMW species ((Fab)2 was non-covalent and dissociated to Fab).
Three observations argued against weak and reversible interactions on the Fab surface being the mechanism of the association. First, the intact mAb-PFM dimer and the papain-produced (Fab)2 were well resolved by AUC and SE-HPLC as stable species, with no observable concentration dependence in peak shape and peak area relative to their respective monomer. Second, mAb-PFM dimer and HMW species were not disrupted by salt at high concentration or 20% ethanol, and were only dissociated by strongly denaturing reagents (i.e., 6 M GdmCl) or under the LC/MS conditions. Third, The HMW content in purified mAb-PFM solution was very low, and did not increase over time (data not shown). The native-like secondary and tertiary structures as demonstrated by the far-UV CD and intrinsic fluorescence spectra argued against random aggregation of unfolded Fab domains being the mechanism of the association.
Two Fab domains were found to associate non-covalently and form stable dimer with nearly native secondary and tertiary structures via VH-swapped binding of the 2 Fab domains within one IgG molecule, named 2G12,Citation21 (inset in ). The discrete SEC and AUC peaks of the papain-produced Fab of 2G12 antibody exhibited larger hydrodynamic size than monomeric Fab, supporting an intramolecular Fab-Fab association. The crystal structure of 2G12 (Fab)2 showed in detail the quinary structure of the 2 Fab domains, intertwined via the swapped binding in their variable regions, i.e., the “elbow” of H in each of the 2 Fab domains was extended and twisted to allow the VH of one Fab to bind to VL of the other Fab. In this quinary structure, the quaternary structure of the constant regions of the 2 Fab domains, as well as the tertiary and the secondary structures of all 8 Ig domains of a (Fab)2, remained native. The quaternary structure of the variable regions, with swapped VH, is also close to native.
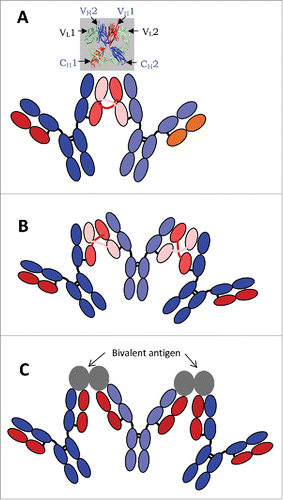
Figure 7. Schematic diagrams for IgG1 dimer (A) and trimer (B) formed via intermolecular domain exchange in the Fab domains, and for an immune complex of IgG1 with its bivalent antigen (C). The inset in (A) shows the crystal structure of the 2 Fab domains of a 2G12 antibody monomer with (intramolecular) exchanged heavy chains.Citation21
Native-like, non-covalent dimer mediated by intermolecular domain exchange had been found in a variety of proteins, due to either partial destabilization or because the protein carried certain structural determinants.Citation22 The well-studied cases include staphylococcal nuclease (1SND),Citation23 ribonuclease A (1A2W),Citation10 cyanovirin-N (3EZM),Citation24 diphtheria toxin (1DDT),Citation25 and suc 1 (1PUC)Citation26 (see Liu and EisenbergCitation27 for more examples). A diabody involving 2 scFv (single chain Fv) polypeptides from more than one scFv fusion molecule also exemplifies intermolecular domain exchange.Citation28 One commonality among these proteins is the tertiary folding of their polypeptide chains into multiple distinct domains and the association of these domains in the quaternary structure, with linker regions between the domains that are consecutive in the primary sequences. Under certain conditions one of the flexible linker regions can adopt a more extended conformation, allowing one of the domains to be dissociated from the rest of the molecule. When 2 such molecules are sufficiently close, the disengaged domain from one molecule may the disengaged domain from one molecule may replace the counterpart in the quaternary structure of the other molecule. When the reciprocal incorporation occurs, the 2 molcules form a dimer with a quinary structure exhibiting symmetrically exchanged domains between the composing monomers. The association is, therefore, non-covalent but very stable, as the quaternary structure of each composing monomer remains largely native. Oligomers can also form by this mechanism, with the exception that the replacement of domain is not mutual between 2 molcules, but rather takes place in a form of chain reaction among multiple like molecules.Citation27,29
All lines of evidence observed in this and previous work support a mechanism of intermolecular domain exchange for the non-covalent stable dimer of mAb-PFM, which involves one of the 2 Fab regions of each composing mAb molecule, as illustrated in . The exchange in the illustration corresponds to VL-VL swapping, only for convenience. It may also be VH-VH swapping, similar to the crystal structure in the inset, except that the 2 associated Fab domains in the crystal structure were from a single mAb molecule (intra-molecular domain swap). The slightly red-shifted fluorescence emission spectra of mAb-PFM HMW species may be due to the extended and twisted conformation of the “elbow” linker between the variable and the constant domains of the “cross-linking” chain, either H, as found in 2G12, or L, as illustrated in . The quinary structure of (Fab)2 from mAb-PFM stable HMW species with atomic resolution and the topology of stable HMW species of intact mAb-PFM are still pending. However, the non-covalent dimer, (“dimer 2” in Plath et al., 2016),Citation30 in the aggregates of 2 mAb molecules were shown to be more compact, using negative staining transmission electron microscopy, with 2 Fab domains side-by-side, one from each monomer. This may be a conformation of the quinary structure for a stable mAb dimer containing intermolecular domain-swapped (Fab)2. The intermolecular domain exchange was also speculated to be the mechanism for 2G12 aggregation following low pH treatment.Citation31
We note that the stable non-covalent mAb-PFM HMW species, likely resulting from intermolecular domain exchange in the Fab domain, was only found in the cell culture. The HMW content in the purified mAb-PFM material was <1%, even after being concentrated to >100 mg/mL and following long-term storage. In addition, the native-like monomer, but not the native-like non-covalent dimer, was formed in refolding of the denatured original HMW fraction of mAb-PFM. The intramolecular domain swapping similar to that found in 2G12 was not significant in purified mAb-PFM monomer, as demonstrated by the negligible (Fab)2 in the papain digest (). All these findings indicate that the “elbow” region of the swapped chain (either H or L) in the (Fab)2 of mAb-PFM HMW species is not incessantly flexible to allow the domain exchange continuously take place. Further studies are required to thoroughly understand when and how the intermolecular domain exchange takes place during mAb-PFM expression. It is also unknown why an intramolecular domain exchange, similar to that in 2G12 antibody, does not occur, despite the fact that the 2 Fab domains in one IgG molecule are close to each other at all times.
Effect on the antigen-binding
In addition to the fact that the (Fab)2 of 2G12 is formed in 2G12 monomer via intramolecular domain exchange between the 2 Fab domains of the same molecule, while (Fab)2 of mAb-PFM dimer characterized in this work is likely to be formed in mAb-PFM dimer and oligomer via intermolecular domain exchange between 2 Fab domains of 2 different mAb-PFM molecules, another significant difference between the (Fab)2 of 2G12 and (Fab)2 of mAb-PFM dimer is the activity in binding to their respective antigen. Human antibody 2G12 is a novel antibody capable of neutralizing a broad range of human immunodeficiency virus type 1 (HIV-1) by binding to the carbohydrate moieties on envelope glycoprotein.Citation32 It is challenging to obtain anti-carbohydrate antibodies with high affinity and specificity.Citation33 In a 2G12 IgG antibody, the 2 antigen-binding Fab domains are brought very close to each other by the swapping of the VH region. The close vicinity of the 2 Fab domains not only allows their binding to 2 carbohydrate of the same type in the same gp120, but also gives rise to a new binding surface that binds to the third carbohydrate of a different type on the same gp120 molecule.Citation21 Both effects contribute toward enhancing the affinity of 2G12 antibody to gp120 via the mechanism of increasing avidity. Since 2G12 naturally occurs in human, this intramolecular domain swap was recognized as an immunological solution for high-affinity carbohydrate cluster recognition. In contrast, the dimer and HMW species in mAb-PFM are aberrant products of the mAb-PFM expression process. The Fab of mAb-PFM targets a specific epitope on a large antigen protein. It is conceivable that if 2 Fab domains are brought spatially very close to each other by, e.g., domain exchange, the 2 Fab domains can interfere with each other's binding to the same epitope. The close vicinity of the 2 Fab domains in a rigid conformation prevents them from reaching 2 separate antigens of mAb-PFM. These steric effects may account for the reduced antigen-binding capability of (Fab)2 from mAb-PFM HMW species, even though the tertiary structure of the 2 Fab domains are native and the quaternary structure is native-like.
Effect on the structure and functions of the Fc domains
In the quinary structure of a mAb dimer linked by swapped chains in the Fab domains, as illustrated in , the 2 Fc domains are intact and apart from each other, and each is accessible to Fc-binding proteins, including protein A and various Fcγ receptors. The evidence for binding to protein A is the capturing of mAb-PFM dimer and HMW species on the protein A column.
The Fab-Fab association via domain exchange can propagate to involve 3 or more IgG molecules, forming oligomers as illustrated in . The Fc domains of all subunits in the oligomer should be regularly spaced and equally accessible. This quinary structure bears resemblance, from the viewpoint of IgGs, to immune complexes formed by bivalent or multivalent antigens and the IgGs, as illustrated in . One important commonality is the regular spacing and free access of the Fc domains. This structure predicts that each of the Fc domains in such dimer or oligomer is capable of binding to Fcγ receptors, which may lead to enhanced affinity via the increased avidity. The mAb-PFM HMW species exhibited such capability in the SPR studies where the receptors were immobilized,Citation9 which mimics the receptors on cell surface. The immune complexes of mAb-PFM and its antigen were shown in the same work to be larger than dimer and have higher apparent binding affinity to the immobilized FcγRs, consistent with the prediction from the structure. The elevated FcγR binding may lead to elevated Fc effector functions and immunogenicity. A cHA chromatography step was used in the purification process, which separated the dimer and oligomers of mAb-PFM from the monomer,Citation18 although a correlation between the in vitro binding results and the in vivo immunogenicity is yet to be established.
In summary, we demonstrated, using a broad range of techniques, multiple lines of evidence that support a mechanism of domain exchange in the Fab domain of mAb-PFM for the formation of stable non-covalent mAb-PFM dimer and higher order HMW species. The proposed quinary structures provide rationale for the stability of mAb-PFM dimer and other HMW species under non-denaturing conditions, the native-like higher order structure, and the partial loss of the antigen-binding sites in mAb-PFM dimer with normal affinity of the remaining sites to antigen. The proposed structure also provided a structural basis for the observed elevation in binding to FcγRs, which may be relevant to immunogenicity. In contrast, randomly associated aggregates of unfolded proteins are not expected to elicit a similar effect. This work, to our best knowledge, is the first to present supportive evidence for intermolecular domain exchange being an aggregation mechanism for molecules as large and complex as an IgG. This finding will advance the understanding of the diverse mechanisms for aggregation in therapeutic monoclonal antibodies, which may aid in the control and improvement of biopharmaceutical manufacturing processes.
Materials and methods
Materials
The Pfizer proprietary monoclonal antibody, mAb-PFM (human IgG1 with λ light chains, targeting a 25 kDa protein Ag1), was expressed in CHO cells and purified with a protein A column, followed by ceramic hydroxyapatite (cHA) column.Citation18 The HMW species were captured on the cHA column and were eluted using 1 M salt solution. Dimer was isolated from oligomers by size-exclusion chromatography. Ag1, the antigen of mAb-PFM, is a transforming growth factor superfamily member, and naturally a homodimer covalently linked by inter-chain disulfide bonds. The expression and purification of recombinant Ag1 were described previously.Citation9 All reagents from commercial sources were of the highest purity.
Methods
Protein A affinity high-performance liquid chromatography
PA ImmunoDetection® Sensor Cartridge (Applied Biosystems), containing protein A ligand covalently bound to the surface of the stationary phase, was used with Alliance HPLC system (Waters) to fractionate the Fc-containing species following the papain digestion of mAb-PFM monomer and HMW samples. Mobile phase A, containing 10 mM sodium phosphate, pH 7.0, 140 mM NaCl, was used to capture the Fc-containing species, which were eluted during a 10 min gradient ending with 100% mobile phase B (10 mM sodium phosphate, pH ∼2, 400 mM NaCl) at a flow rate of 1 mL/min. The non-Fc species were in the flow-through fraction during the wash with mobile phase A.
Size exclusion high-performance liquid chromatography and the in-line coupled multi-angle light scattering
The intact mAb-PFM species, i.e., monomer, dimer and oligomers, as well as the papain-digested mAb-PFM fragments, were resolved and analyzed using a TSKgel G3000SWXL or G4000SWXL column (7.8 mm × 30 cm, 8 µm, Tosoh Biosciences) and an Alliance HPLC system (Waters) under isocratic flow. The mobile phase contained 10 mM sodium phosphate, pH 7.4, 140 mM NaCl.
The average molar mass of each resolved peak was assessed using a multi-angle light scattering detector (DAWN HELEOS II, Wyatt Technology) in-line with SE-HPLC and an UV absorbance detector for concentration determination. The system was normalized with bovine serum albumin. The data were collected and analyzed using the manufacturer-provided software ASTRA v5.3.4.20.
Reversed-phase high-performance liquid chromatography / electrospray ionization quadruple time-of-flight mass spectrometry
Protein A elution and flow-through fractions were separated via an Agilent Zorbax Poroshell C3–300SB column (1.0 × 75 mm, 5 μm) and analyzed by a Waters Acquity Ultra Performance Liquid Chromatography (UPLC) system (Milford, MA) with a tunable, dual-wavelength UV/Vis detector monitoring at 214 nm and 280 nm. In all experiments, the flow rate was 100 µL/min, column temperature was 65°C, and a linear gradient (20 to 50% B in 17 min) with 0.05% (v/v) TFA in water (mobile phase A) and 0.05% TFA (v/v) in acetonitrile (mobile phase B) was used with a fast 0 to 20% B ramp in the first 5 min.
A high-resolution Waters Q-Tof-2 mass spectrometer (Milford, MA) was used in the positive ion mode for all online LC/UV/MS analyses, where cone voltage was set to 40 V, collision energy was set to 10 eV; desolvation and ion source block temperatures were set to 275°C and 115°C, respectively; ESI capillary voltage was set to 3000 V; and the pusher cycle time was set to 88 µs. For all analyses, ions were detected over a m/z range of 800–4300 and a total scan time of 2 s (signal accumulated for 1.9 s with an inter-scan delay of 0.1 s) was used. Prior to LC/UV/MS, the Q-Tof-2 instrument was mass calibrated with NaI.
Chromatographic data was acquired and processed using Waters Empower Pro software (Milford, MA). MassLynx 3.5 for NT (Milford, MA) was the Q-Tof-2 software for instrument control, data acquisition, calibration, and mass spectrum processing. Probabilistic maximum entropy analysis (MaxEnt-1 module, Milford, MA) was used to deconvolute the multiply-charged mass data into a zero-charge mass spectrum.
Fluorescence emission spectroscopy
Fluorescence emission spectra were collected using a Varian Cary Eclipse fluorescence spectrophotometer with a semi-micro quartz cuvette equilibrated at 20°C. The excitation wavelength was 300 nm; emission was monitored from 305 to 420 nm with 1-nm resolution. Protein samples were in either mAb-PFM formulation buffer, or PBS, or 6 M GdmCl, as indicated in the figure legends. Appropriate buffer spectra were subtracted from sample spectra.
Surface plasmon resonance
Biacore 3000 and a streptavidin (SA) chip (GE Healthcare Life Sciences) were used to test the antigen-binding affinity and capacity of mAb-PFM fragments. The antigen molecules were biotinylated and immobilized by streptavidin in the sample flow cells at low density (∼40 RU). One of the 4 flow cells was not exposed to antigen and was used as the blank control to correct for non-specific binding of the analyte. An analyte, e.g., one of the mAb-PFM fragments to be studied, was injected into one or more sample flow cell(s) and the blank flow cell to initiate the association phase of the binding, followed by the injection of the running buffer to begin the dissociation phase. A pulse injection of a pH 2.5 buffer (10 mM Na3PO4, 500 mM NaCl) removed the remaining analyte in the flow cell(s) before the next binding cycle. The immobilized antigen molecules were not removed from the SA chip by the low pH buffer. Their binding affinity to mAb-PFM fragments were not affected by the short exposure to the low pH, evidenced by the excellent reproducibility of multiple cycles of the same fragment, and excellent match between the 2 Fc and the 2 Fabs from 2 different sources (i.e., mAb-PFM monomer and HMW). All measurements were performed at 25°C, with a flow rate of 30 μL/min. The running buffer for all measurements was HBS-P (GE Healthcare) containing 0.01 M HEPES, pH 7.4, 0.15 M NaCl, 0.005% polysorbate 20. Data analyses were performed using the instrument manufacturer supplied software BiaEvaluation. Each binding sensorgram from a sample flow cell was corrected with the sensorgram from the blank flow cell of the same cycle. Each displayed sensorgram is the average of triplicate measurements.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The following current and former Pfizer employees are thanked for their assistance and helpful discussion during the study and preparation of the manuscript: Joseph McClellan, Shujun Sun, Marta Czupryn, John Steckert, Paul Thoday, Paulena Lieske, and Lucas Wafer.
References
- Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J 2006; 8:E501-E7; PMID:17025268; https://doi.org/10.1208/aapsj080359
- Bachmann MF, Fehr T, Freer G, Hengartner H, Zinkernagel RM. Correlation of tolerogenicity of a viral antigen with its immunogenicity. J Immunol 1997; 158:5106-11; PMID:9164925
- Dintzis RZ, Middleton MH, Dintzis HM. Studies on the immunogenicity and tolerogenicity of T-independent antigens. J Immunol 1983; 131:2196-203; PMID:6631009
- Henney CS, Ellis EF. Antibody production to aggregated human gamma-G-globulin in acquired hypogammaglobulinemia. N Engl J Med 1968; 278:1144-6; PMID:4171617; https://doi.org/10.1056/NEJM196805232782104
- Joubert M, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, Goletz TJ, Kerwin BA, Chirmule N, Narhi LO, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem 2012; 287:25266-79; PMID:22584577; https://doi.org/10.1074/jbc.M111.330902
- Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO. Classification and characterization of therapeutic antibody aggregates. J Biol Chem 2011; 286:25118-33; PMID:21454532; https://doi.org/10.1074/jbc.M110.160457
- Luo Q, Joubert M, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem 2011; 286:25134-44; PMID:21518762; https://doi.org/10.1074/jbc.M110.160440
- Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM. Immunogenicity of therapeutic protein aggregates. J Pharm Sci 2016; 105:417-30; PMID:26869409; https://doi.org/10.1016/j.xphs.2015.11.002
- Luo Y, Lu Z, Raso SW, Entrican E, Tangarone B. Dimers and multimers of monoclonal IgG1 exhibit higher in vitro binding affinities to Fcγ receptors. mAbs 2009; 1:491-504; PMID:20065648; https://doi.org/10.4161/mabs.1.5.9631
- Liu Y, Hart PJ, Schlunegger MP, Eisenberg D. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-A resolution. Proc Natl Acad Sci USA 1998; 95:3437-42; PMID:9520384; https://doi.org/10.1073/pnas.95.7.3437
- Wang T, Joshi SB, Kumru OS, Telikepalli S, Middaugh CR, Volkin DB. Case studies applying biophysical techniques to better characterize protein aggregates and particulates of varying size, in Biophysics for Therapeutic Protein Development, Narhi LO, Editor. 2013, Springer New York. p. 205-43; https://doi.org/10.1007/978-1-4614-4316-2_9
- McConkey EH. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci USA 1982; 79:3236-40; PMID:6954476; https://doi.org/10.1073/pnas.79.10.3236
- Wirth AJ, Gruebele M. Quinary protein structure and the consequences of crowding in living cells: leaving the test-tube behind. Bioessays 2013; 35:984-93; PMID:23943406; https://doi.org/10.1002/bies.201300080
- Edelstein SJ. Patterns in the quinary structures of proteins. Plasticity and inequivalence of individual molecules in helical arrays of sickle cell hemoglobin and tubulin. Biophys J 1980; 32:347-60; PMID:7248453; https://doi.org/10.1016/S0006-3495(80)84961-7
- Srere PA. The metabolon. Trends Biochem Sci 1985; 10:109-10; https://doi.org/10.1016/0968-0004(85)90266-X
- Monteith WB, Cohen RD, Smith AE, Guzman-Cisneros E. Quinary structure modulates protein stability in cells. Proc Natl Acad Sci USA 2015; 112:1739-42; PMID:25624496; https://doi.org/10.1073/pnas.1417415112
- Chen K, Long DS, Lute SC, Levy MJ, Brorson KA, Keire DA. Simple NMR methods for evaluating higher order structures of monoclonal antibody therapeutics with quinary structure. J Pharm Biomed Anal 2016; 128:398-407; PMID:27344629; https://doi.org/10.1016/j.jpba.2016.06.007
- Sun S, Luo Y, Jennings P. Purification of acidic proteins using ceramic hydroxyapatite chromatography, U.S. Patent number US008058407B2, 2011, Wyeth LLC: USA.
- Brandis JF, Hunt L. The thermodynamics of protein denaturation. III. The denaturation of ribonuclease in water and in aqueous urea and aqueous ethonal mixtures. J Am Chem Soc 1967; 89:4826-38; PMID:6074801; https://doi.org/10.1021/ja00995a002
- Lamoyi E. Preparation of F(ab')2 fragments from mouse IgG of various subclasses. Methods Enzymol 1986; 121:652-63; PMID:3088385; https://doi.org/10.1016/0022-1759(83)90415-5
- Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 2003; 300:2065-71; PMID:12829775; https://doi.org/10.1126/science.1083182
- Ding F, Prutzman KC, Campbell SL, Dokholyan NV. Topological Determinants of Protein Domain Swapping. Structure 2006; 14:5-14; https://doi.org/10.1016/j.str.2005.09.008
- Green SM, Gittis AG, Meeker AK, Lattman EE. One-step evolution of a dimer from a monomeric protein. Nat Struct Biol 1995; 2:746-15; PMID:7552745; https://doi.org/10.1038/nsb0995-746
- Yang F, Bewley CA, Louis JM, Gustafson KR, Boyd MR, Gronenborn AM, Clore GM, Wlodawer A. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. J Mol Biol 1999; 288:403-12; PMID:10329150; https://doi.org/10.1006/jmbi.1999.2693
- Kundu S, Jernigan RL. Molecular mechanism of domain swapping in proteins: An analysis of slower motions. Biophys J 2004; 86:3846-54; PMID:15189881; https://doi.org/10.1529/biophysj.103.034736
- Khazanovich N, Bateman K, Chernaia M, Michalak M, James M. Crystal structure of the yeast cell-cycle control protein, p13suc1, in a strand-exchanged dimer. Structure 1996; 4:299-309; PMID:8805536; https://doi.org/10.1016/S0969-2126(96)00034-2
- Liu Y, Eisenberg D. 3D domain swapping: As domains continue to swap. Protein Sci 2002; 11:1285-99; PMID:12021428; https://doi.org/10.1110/ps.0201402
- Wu AM, Tan GJ, Sherman MA, Clarke P, Olafsen T, Forman SJ, Raubitschek AA. Multimerization of a chimeric anti-CD20 single-chain Fv-Fc fusion protein is mediated through variable domain exchange. Protein Engineering 2001; 14(12):1025-33; https://doi.org/10.1093/protein/14.12.1025
- Zegers I, Deswarte J, Wyns L. Trimeric domain-swapped barnase. Proc Natl Acad Sci USA 1999; 96:818-22; PMID:9927651; https://doi.org/10.1073/pnas.96.3.818
- Plath F, Ringler P, Graff-Meyer A, Stahlberg H, Lauer ME, Rufer AC, Graewert MA, Svergun D, Gellermann G, Finkler C, et al. Characterization of mAb dimers reveals predominant dimer forms common in therapeutic mAbs. mAbs 2016; 8:928-40; PMID:27031922; https://doi.org/10.1080/19420862.2016.1168960
- Roux KH, Zhu P, Seavy M, Katinger H, Kunert R, Seamon V. Electron microscopic and immunochemical analysis of the broadly neutralizing HIV-1-specific, anti-carbohydrate antibody, 2G12. J Mol Immunol 2004; 41:1001-11; https://doi.org/10.1016/j.molimm.2004.05.008
- Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 1996; 70:1100-8; PMID:8551569
- Sterner E, Flanagan N, Gildersleeve JC. Perspectives on anti-glycan antibodies gleaned from development of a community resource database. ACS Chem Biol 2016; 11:1773-83; PMID:27220698; https://doi.org/10.1021/acschembio.6b00244