
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Arginase 2 (ARG2) is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine. The dysregulated expression of ARG2 within specific tumor microenvironments generates an immunosuppressive niche that effectively renders the tumor ‘invisible’ to the host’s immune system. Increased ARG2 expression leads to a concomitant depletion of local L-arginine levels, which in turn leads to suppression of anti-tumor T-cell-mediated immune responses. Here we describe the isolation and characterization of a high affinity antibody (C0021158) that inhibits ARG2 enzymatic function completely, effectively restoring T-cell proliferation in vitro. Enzyme kinetic studies confirmed that C0021158 exhibits a noncompetitive mechanism of action, inhibiting ARG2 independently of L-arginine concentrations. To elucidate C0021158’s inhibitory mechanism at a structural level, the co-crystal structure of the Fab in complex with trimeric ARG2 was solved. C0021158’s epitope was consequently mapped to an area some distance from the enzyme’s substrate binding cleft, indicating an allosteric mechanism was being employed. Following C0021158 binding, distinct regions of ARG2 undergo major conformational changes. Notably, the backbone structure of a surface-exposed loop is completely rearranged, leading to the formation of a new short helix structure at the Fab-ARG2 interface. Moreover, this large-scale structural remodeling at ARG2’s epitope translates into more subtle changes within the enzyme’s active site. An arginine residue at position 39 is reoriented inwards, sterically impeding the binding of L-arginine. Arg39 is also predicted to alter the pKA of a key catalytic histidine residue at position 160, further attenuating ARG2’s enzymatic function. In silico molecular docking simulations predict that L-arginine is unable to bind effectively when antibody is bound, a prediction supported by isothermal calorimetry experiments using an L-arginine mimetic. Specifically, targeting ARG2 in the tumor microenvironment through the application of C0021158, potentially in combination with standard chemotherapy regimens or alternate immunotherapies, represents a potential new strategy to target immune cold tumors.
Introduction
Tumors are exquisitely adept at circumventing a host’s adaptive and innate immune defenses, allowing neoplastic cells to establish and cancers to grow unchecked,Citation1 effectively rendering themselves ‘invisible’ to the host’s immune system. Understanding the different immunosuppressive mechanisms used by tumors will allow development of effective anti-cancer drugs and treatments, particularly when combined with standard chemotherapeutic regimens. The inception and maintenance of an immune-privileged niche by a tumor can occur in many ways.Citation2 Indeed, the sheer number and variety of immunosuppressive mechanisms adopted by tumors reflect the success and importance of this generalized immune escape strategy for their growth and survival.Citation3 Examples of immune escape strategies used by tumors include reducing their inherent immunogenicity , evolving novel survival mechanisms or resistance to host anti-tumor immune responses, and actively nurturing an immunosuppressive tumor microenvironment. An example of the latter is the tumor-driven increase in the expression of amino-acid degrading enzymes such as indoleamine 2,3-dioxygenase (IDO), tryptophan 2,3-dioxygenase (TDO), arginase 1 (ARG1) or arginase 2 (ARG2) to promote immunological tolerance.Citation4 Increasing the local concentration of amino acid metabolizing enzymes can be driven by the tumor itself, or through the recruitment of specific tolerogenic myeloid populations to the tumor microenvironment.Citation5
Arginine, a semi-essential amino acid in mammals used for the biosynthesis of proteins, creatine and agmatine, is critical for several cellular processes that include tissue repair, cell viability and immune function preservation.Citation6 T cells are auxotrophic for L-arginine, and their ability to proliferate is directly linked to the availability of sufficiently high levels of extracellular arginine within the tissue microenvironment. A reduction in extracellular arginine levels can lead to a loss of CD3ζ chain expression and T-cell anergy, with a concomitant dampening of T-cell-mediated anti-tumor immune responses.Citation7 Mammals express two arginase isoenzymes, each differing in their respective subcellular locations. ARG1 is a cytosolic protein, predominantly expressed in the liver, that plays a key role in the urea cycle, although the protein can also be found extra-hepatically. ARG2 is a mitochondrial protein whose expression is normally limited to the brain, small intestine, kidney, and certain monocytic lineages.Citation8 The upregulated expression of ARG2, and not ARG1, has been described in multiple tumor types,Citation9,Citation10 cancer-associated fibroblasts,Citation11 and T cells themselves.Citation7 The specific upregulation and extracellular release of ARG2 has recently been reported in acute myeloid leukemia (AML) patients, leading to a reduction in circulating arginine levels,Citation12,Citation13 a concomitant impairment of T-cell proliferation and a skewing of monocytic populations to a more M2-like immunosuppressive phenotype.Citation12 In support of these general observations, an in vitro study using multiple murine renal cell carcinoma (mRCC) cell lines described the increased intracellular expression and extracellular release of ARG2, but not ARG1.Citation14 This specific increase in ARG2 expression led to depletion of extracellular arginine and a concomitant decrease in T-cell CD3ζ expression. Inhibition of this extracellular ARG2 activity using the small molecule arginase inhibitor, Nω-hydroxy-nor-L-arginine (nor-NOHACitation15) significantly suppressed cell growth in the high expressing ARG2 mRCC CL-19 cell line. Moreover, the targeted depletion of intracellular ARG2 through ARG2 gene silencing, deletionCitation10, or microRNA expressionCitation16 has shown clear anti-tumor efficacy.
Small molecule inhibitors of arginases, such as nor-NOHA, have shown anti-tumor effects in in vivo mouse models.Citation17 For example, CB-1158, a small-molecule inhibitor of ARG1, reduces tumor growth in mouse cancer models. CB-1158 is currently being tested in clinical trials for the treatment of solid tumors both as a standalone monotherapy and in conjunction with anti-PD-1 treatment.Citation18 Although targeting arginases with small-molecule inhibitors has merit, unwanted liver toxicity might occur through the disruption of the urea cycle. We hypothesized that a neutralizing monoclonal antibody targeting tumor-expressed ARG2 found specifically within the extracellular microenvironment would circumvent any potential adverse effects caused by the nonspecific targeting of intracellular arginase. The high sequence homology between ARG1 and ARG2 at their respective active sites makes finding conventional, competitive substrate mimetics with specificity for one isoform over the other hard to achieve. In contrast, raising an antibody to epitopes found within areas of sequence divergence outside of the common substrate binding domain would enable specificity to be achieved, mitigating any off-target, ARG1-mediated effects. Moreover, an ARG2-specific therapeutic antibody, with its relatively longer half-life and better bioavailability, would be expected to have a better pharmacokinetic profile than a small-molecule inhibitor.Citation19
Here, we describe the discovery and characterization of a human antibody, C0021158, that binds specifically to human ARG2 and not to its paralogue, ARG1. C0021158 is a high affinity, potent ARG2 inhibitor that can fully reverse ARG2-mediated inhibition of T-cell proliferation in vitro. The co-crystal structure of C0021158 antigen-binding fragment (Fab) in complex with homo-trimeric human ARG2 is also presented. Binding of C0021158 leads to significant conformational changes at the contact site on ARG2 (epitope), which in turn results in subtle changes within the enzyme’s active site that account for C0021158’s novel noncompetitive allosteric mode of inhibition. A second ARG2-specific inhibitory antibody, C0021181, was identified that also uses the same structural mechanism of inhibition as C0021158. The isolation of two unique but sequence-related antibodies with a shared allosteric mechanism of antibody-mediated inhibition raises the possibility of a currently unknown in vivo regulatory system for ARG2, involving a functional partner protein binding at the regulatory site targeted by C0021158.
Results
Discovery and affinity optimization of ARG2 inhibitory antibodies
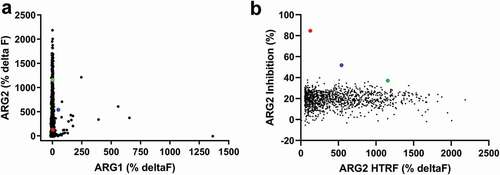
ARG2 binding antibodies were isolated from naïve phage libraries displaying human antibody single-chain variable fragment (scFv)Citation20-Citation22 by performing soluble selections on biotinylated recombinant human ARG2 trimer. Selection outputs were screened in parallel for ARG2 binding specificity and their ability to inhibit ARG2’s in vitro enzymatic activity. Initially, a biochemical homogeneous time-resolved fluorescence (HTRF®) assay was used to screen selection outputs for selective binding to ARG2 (vs. human ARG1; )). The same selection outputs were also screened for their ability to inhibit the conversion of L-arginine to urea and ornithine in an in vitro biochemical ARG2 functional assay ()). Despite the majority of HTRF® binders being specific for ARG2, remarkably few scFvs showed significant inhibition of ARG2 in the enzyme activity assay. The level of inhibition displayed by the majority of the scFvs selected was similar to the background level exhibited by an irrelevant (non-ARG2-binding) scFv, CEA6 (21.0% ± 3.9%). Notably, only two clones were able to inhibit ARG2 by greater than 50%, namely C0020185 (84.7%) and C0020187 (51.9%). The clear disparity between the high number of ARG2-specific binders obtained and the very low number of potent neutralizing scFvs suggests that true function-blocking epitopes are rare.
Figure 1. High throughput screening of ARG2 phage display round two outputs identified large numbers of human ARG2-specific binders, but few ARG2 inhibitors. Representative results illustrating that (a) clones sampled from ARG2 phage display selection outputs are highly specific for binding to ARG2 and not its paralogue, ARG1. (n = 1593/4400; ARG2 deltaF >100%, ARG1 deltaF <100%). In contrast, selection outputs contained (b) significantly fewer clones with an ability to inhibit recombinant human ARG2 (n = 1235). Lead candidate clones are highlighted: C0020185 (![]()
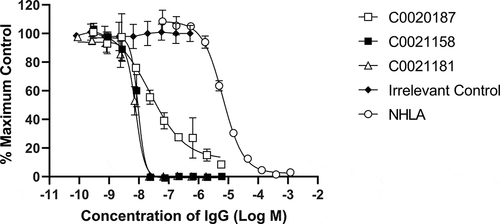
Three ARG2 inhibitory clones (C0020185, C0020186, and C0020187) were subsequently chosen for expression as purified scFv and their binding specificity for recombinant ARG2 over ARG1 confirmed by HTRF® (,)). These three clones were all able to inhibit recombinant human ARG2 in a scFv-dependent manner, yielding IC50 values of 145.8 nM ± 9.1 nM (C0020185), 807.4 nM ± 312 nM (C0020186) and 446.8 nM ± 84.5 nM (C0020187). All three clones were more potent in the ARG2 inhibition assay than the small molecule arginase inhibitor NG-hydroxy-L-arginine (NHLA; IC50 20640.0 nM ± 3933.8 nM) ()).
Figure 2. A lead panel of purified scFv specifically bind to and inhibit the enzymatic function of recombinant human trimeric ARG2. Representative data further characterizing clones C0020185 (Δ), C0020186 (▲) and C0020187 (□) originally identified in the parallel in vitro ARG2 biochemical and inhibition high throughput screens. Each clone binds (a) recombinant biotinylated trimeric human ARG2, but not (b) recombinant biotinylated trimeric human ARG1 and (c) effectively neutralizes recombinant trimeric human ARG2 in a scFv concentration-dependent manner. Direct binding was measured using HTRF®, titrating in purified scFv and using recombinant biotinylated human ARG2 or ARG1 trimer at a fixed concentration of 12 nM or 24 nM, respectively. A hook effect was observed at highest concentrations of scFv used.Citation23 ARG2 enzyme inhibition assay plotted as percentage. IC50 values ± standard deviation were determined for C0020185 (145.8 nM ± 9.1 nM), C0020186 (807.4 nM ± 312 nM) and C0020187 (446.8 nM ± 84.5 nM). The small molecule Arginase inhibitor NG-hydroxy-L-arginine (NHLA; ○) was used as a positive control (IC50 20640.0 nM ± 3933.8 nM). HTRF® and enzyme inhibition assay data points represent the mean of duplicate wells ± standard deviation across independent experiments (n = 2 and n = 4, respectively).
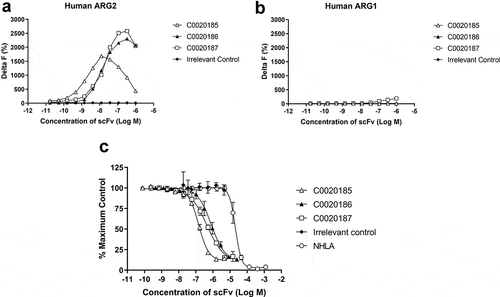
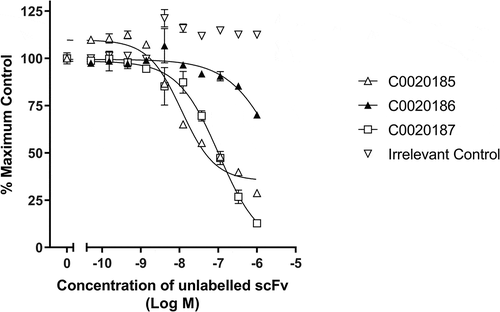
HTRF® competitive binding assays suggested that the three ARG2-inhibitory antibodies selected for further characterization share a similar or at least over-lapping epitope. This observation is consistent with the paucity of inhibitory ARG2 antibodies identified and suggests that epitope space capable of inhibiting the enzyme is restricted (, Figure S1). The three inhibitory scFvs were converted to a human immunoglobulin G1 (IgG1) format, and their ability to bind and inhibit ARG2 was re-examined (Figure S2). Notably, C0020185’s ability to inhibit in the ARG2 enzyme inhibition assay was significantly impaired, relative to when screened as a scFv. The two remaining clones (C0020186 and C0020187) also showed a drop-off in potency upon conversion to IgG, but to a lesser extent. This behavior contrasts with potency gains typically observed when scFvs are reformatted to IgG and is indicative of an altered epitope or angle of binding between these two antibody formats.Citation24 The loss of potency observed with C0020185 IgG meant that this specific clone was not progressed further. In contrast, because C0020187 consistently performed well in the enzyme inhibition assay as either scFv or IgG and had the highest affinity for recombinant human ARG2, this clone was chosen for affinity optimization and functional characterization.
Figure 3. Lead ARG2 function-blocking clones appear to bind a common or overlapping epitope. Representative data showing that unlabeled C0020185 (Δ), C0020186 (▲) and C0020187 (□) scFv can compete away DyLight®650 labeled C0020187 scFv bound to human ARG2. Direct binding was measured using HTRF®. Labeled C0020187 scFv and biotinylated recombinant human ARG2 trimer were used at a fixed assay concentration of 100 nM and 12 nM, respectively. Assay data points represent the mean of duplicate wells ± standard deviation. Reciprocal competition experiments using DyLight®650-labeled C0020185 or C0020186 were also performed yielding similar results.
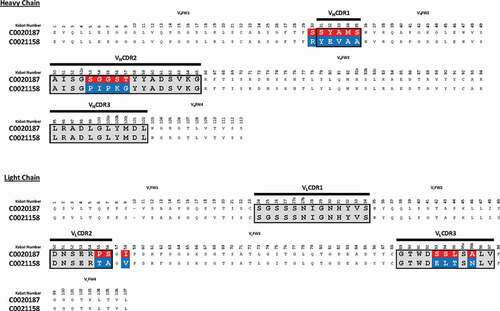
C0020187 was affinity optimized using a comprehensive affinity maturation campaign that targeted, in a nonbiased manner, the parental scFv’s entire sequence, as described in detail by Chan et al.Citation25 The main thrust of this work sought to exploit and recombine advantageous mutations across all six of C0020187’s hypervariable complementarity-determining regions (CDRs). Importantly, lead optimized scFvs continued to be screened for ARG2 specificity and inhibition at each stage of the affinity optimization process, helping to ensure that C0020187’s inhibitory epitope was retained. This extensive campaign of affinity optimization and screening delivered a panel of unique, but sequence-related (Figure S3a), ARG2 inhibitory clones that retained specificity for ARG2 over ARG1. ScFv clones were reformatted to human IgG1 and the corresponding Fab fragment to enable functional screening in a T-cell proliferation assay and to determine antibody affinity using bio-layer interferometry, respectively. Using this approach, several significantly more tightly binding antibodies were identified, including C0021158, C0021181, C0021177, and C0021144 (KD values of 173 pM, 288 pM, 173 pM and 338 pM, respectivelyCitation25). The lead antibody C0021158 differs from the parent antibody C0020187 by 1 residue in VHFW1 (Kabat residues: S30R), 5 residues in VHCDR1 (Kabat residues: S31Y; Y32E; A33V; M34A; and S35A), 5 in VHCDR2 (Kabat residues: S53P; G54I; G55P; S56K; T57G), 2 in VLCDR2 (Kabat residues: P55T, S56A), 1 residue in VLFW3 (Kabat residue: I58V) and 4 in VLCDR3 (Kabat residues: S93E, S94L, L95T, A95bN) (). It is interesting to note that all the amino acid substitutions selected for in the CDRs are non-conservative in terms of biophysical properties and/or structure, which implied a significant change in the mode of binding to ARG2. This was later confirmed by comparison of crystal structures solved for the parent (C0020187) and selected affinity matured (C001158 and C001181) inhibitory antibodies bound to ARG2.Citation25 In addition, no amino acid changes were seen in either VLCDR1 or VHCDR3, which suggests that interactions made by these CDR loops with ARG2 are critically important and intolerant to changes that might alter specific paratope/epitope interactions. The structural and epitope interaction rationale behind the antibody sequence evolution seen during the affinity maturation process is described and discussed in detail in a related paper.Citation25
Figure 4. Amino acid sequence alignment of C0020187 and C0021158. Complementary-determining regions (CDRs) are delineated by black lines. Amino acid differences between the parent clone C0020187 and the affinity matured lead C0021158 are highlighted in white text on a red and blue background, respectively. All residues numbered according to Kabat.Citation26 All the CDR residue substitutions are non-conservative.
The ARG2 binding affinity of 173 pM for C0021158 equates to an approximately 50-fold improvement in affinity relative to the parent antibody, C0020187. No binding of C0021158 to human ARG1 was detected using bio-layer interferometry.Citation25 Commensurate with an increase in affinity, C0021158 displayed significant improvements in in vitro potency, fully inhibiting recombinant human ARG2, with an IC50 of 18.5 ± 5.1 nM as an IgG (). There is also a significant change in the steepness of the inhibitory profile seen for C0021158 compared to its parent C0020187 (), with C0020187 appearing to show negative cooperativity in inhibition, implying three non-equivalent binding sites on the ARG2 trimer. This again suggests a substantial change in the binding mode of the affinity-matured antibody.
Figure 5. Recombinant human ARG2 is fully inhibited by C0021158 and C0021181 IgG1 in vitro. Representative results showing that C0021158 and C0021181 inhibit recombinant human ARG2 activity in vitro, with a IC50 (value ± standard error mean) of 18.5 ± 5.1 nM and 10.7 ± 2.3 nM, respectively. C0020187 (□), C0021158 (■), C0021181 (Δ) and irrelevant isotype control (♦). Human IgG1s were titrated into the assay, while maintaining a fixed concentration of recombinant ARG2. The arginase inhibitor NG-hydroxy-L-arginine (NHLA; ○) has an IC50 of 5046.0 ± 801 nM. Incomplete inhibition with C0020187 precludes assigning a definitive IC50 value for this IgG. Data points represent the mean of duplicate wells ± standard deviation across at least three independent experiments.
C0021158 fully restores ARG2-mediated T-cell proliferation in vitro
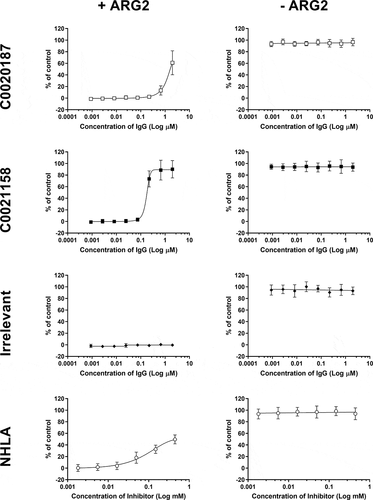
The release of ARG2 by AML blasts into the plasma of AML patients is known to inhibit T-cell proliferation in vitro.Citation12 We therefore investigated whether our affinity-matured lead, C0021158, could relieve ARG2-dependent inhibition of T-cell proliferation in vitro (). The presence of 15 µg/ml recombinant human ARG2 is sufficient to fully inhibit the proliferation of CD3/CD28 co-stimulated T-cells. The addition of the parental antibody, C0020187, partially restored the ARG2-dependent inhibition of T-cell proliferation (EC50 > 2 µM). In contrast, C0021158 was able to fully restore T-cell proliferation with an EC50 value of 157 nM. No restoration of T-cell proliferation was observed with the irrelevant isotype control (human IgG1).
Figure 6. C0021158 IgG1 fully reverses ARG2-mediated inhibition of T-cell proliferation in vitro. Representative data showing that C0021158 (■) can relieve ARG2-mediated suppression of T-cell proliferation in vitro, with a mean EC50 value of 157 nM. C0020187 (□) IgG1 and the small molecule arginase inhibitor NG-hydroxy-L-arginine (NHLA; ○) partially restore the T-cell proliferation response. An irrelevant IgG1 (♦) did not relieve ARG2-mediated T-cell suppression. Cells incubated in the presence/absence of recombinant human trimeric ARG2 (15 µg/ml final concentration) plus a titration of antibodies or NHLA. BrdU incorporation was used as a direct measure of cell proliferation 78 hours after addition of ARG2 and antibodies or NHLA. The data pertaining to C0021158, C0020187 and irrelevant IgG1 represents the average percent proliferation of control (± standard deviation) from two independent experiments, with each experiment using a different donor. NHLA data represents the average percent proliferation of control (± standard deviation) of three independent experiments, using one, two and two different donors, respectively. T-cell proliferation data collected for NHLA was gathered using different donors to those used for C0021158, C0020187 and irrelevant IgGs. Two or more replicate data points were taken for each test concentration.
C0021158 inhibits ARG2 with a noncompetitive mode of action
The specific mechanism of inhibition adopted by C0021158 was investigated. The activity of trimeric ARG2 was measured at defined concentrations of C0021158 IgG1 while titrating different concentrations of arginine into the assay (). Using a Michaelis-Menten least squares fit model Vmax and KM values were determined. (). Changing the concentration of C0021158 substantially reduced the enzyme’s Vmax without significantly affecting the KM value. This observation strongly suggests that C0021158 has a noncompetitive inhibitory mechanism of action and that the antibody binds equally well to both the substrate-free form and substrate-bound form of ARG2.Citation27
Figure 7. C0021158 inhibits recombinant ARG2 with a noncompetitive mechanism of action. The activity of trimeric ARG2 was measured at defined concentrations of C0021158 IgG1 while titrating different concentrations of L-arginine into the assay. Changing the concentration of C0021158 reduces the enzyme’s Vmax without significantly affecting its KM. Michaelis-Menten least squares fit model used to determine Vmax and KM values for a given concentration of C0021158 ().
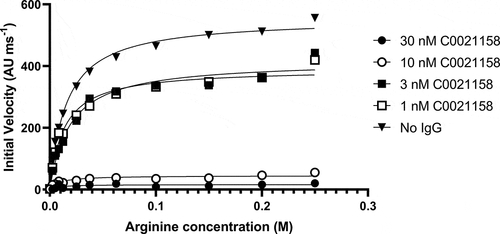
Table 1. Vmax and KM values for the inhibition of recombinant human ARG2 trimer by C0021158 IgG1.
Structures of inhibitory affinity matured antibodies bound to ARG2
To determine the molecular basis of the observed noncompetitive mechanism of ARG2 inhibition, representative antibodies from the different affinity-matured sequence families were selected for X-ray crystallography studies and produced as Fabs. After extensive screening and optimization of crystallization conditions, atomic resolution diffraction data sets were acquired for two distinct inhibitory Fabs (C0021158 and C0021181) bound to full-length, trimeric ARG2 (332 residues per subunit excluding the N-terminal mitochondrial transit peptide) and for four Fabs alone (see Table S1), which were all solved by molecular replacement to reveal the structures for both the isolated Fabs and bound complexes. The ARG2-Fab complexes share many features, which are described in detail below, with both containing the expected 3 Fab molecules bound per ARG2 trimer to form a highly symmetric complex.
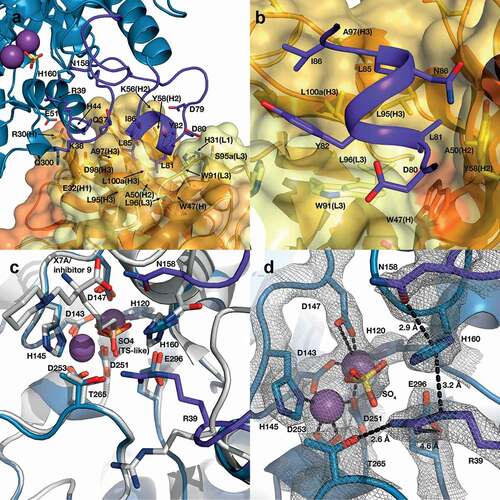
The crystal structure of the ARG2-C0021158 complex was refined to 2.4 Å in space group H 3, with a final R/Rfree of 0.25/0.28. The asymmetric unit contains a single ARG2 subunit and bound Fab molecule, with the 3 Fabs bound per enzyme trimer revealed to bind to the base of the ARG2 trefoil and each interact with a single subunit (,)). The antigen-binding domains (VH and VL) of the Fab are well defined in the electron density, with a clearly defined interface and orientation with respect to ARG2 ()). In contrast, the constant domains of both heavy (CH) and light chain (CL) of the bound Fab are less defined within the electron density, which suggests some degree of disorder and/or mobility for these regions, with the structure reported here representing the most common orientation. The three individual Fabs associated with the ARG2 trimer show no evidence of significant interaction, with the closest contact (~5.1 Å) seen between Lys75 (heavy chain) and Gln79 (light chain). This interaction is bridged by a SO4 ion and appears to be a weak crystal contact.
Figure 8. ARG2 trimer (blue) bound to C0021158 Fab (VH/CH in orange and VL/CL in light orange). (a) ‘Top down’ and (b) ‘side’ view. In the active sites, manganese atoms are shown as purple spheres and a sulfate ion as sticks. (c) Close-up of the boxed region in (b). Free ARG2 (PDB-ID 4HZE, light grayCitation28) was superimposed on C0021158 Fab-bound ARG2 (excluding the regions of conformational change, shown in dark purple). Key active site residues associated with antibody-induced inhibition of ARG2 are shown as sticks. The CDRs on the Fab are shown in yellow (VH) and light yellow (VL).
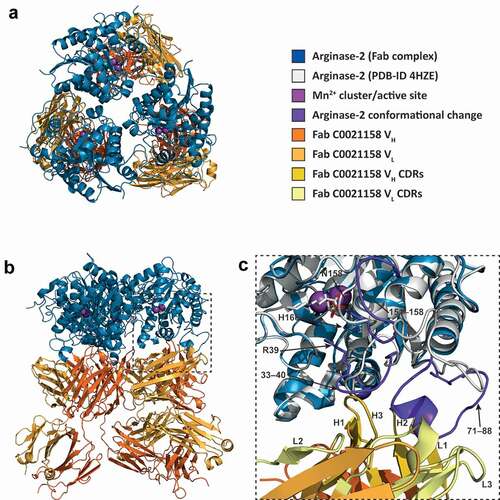
C0021158 interacts with ARG2 mainly via a hydrophobic cleft formed by several of the CDRs (VHCDR2, VHCDR3 and VLCDR3) (,)), which opens slightly upon binding via a rotation of the outer β-sheet of the VH domain by approximately 10° with respect to VL (Figure S4a). Several regions of ARG2 show major conformational changes induced by C0021158 binding, but particularly the surface-exposed loops formed by residues 71–88. There the backbone structure is completely rearranged, including the formation of a short helix (residues 81–85) at the antibody–enzyme interface () and ,)). This large conformational change is accompanied by smaller structural changes within two adjacent surface loops of ARG2 (residues 33–40 and 151–158), which also contribute to the interface with the Fab ()).
Figure 9. Structure of the human ARG2/C0021158 Fab binding interface and Fab-induced changes within ARG2’s active site. (a) Side view of the binding interface between C0021158 Fab and ARG2, with interacting side chains shown as sticks. Key active site residues involved in antibody-induced inhibition (Arg39, Asn158 and His160) are also shown as sticks for clarity. The Fab (VH/CH in orange and VL/CL in light orange, CDRs in yellow and light yellow for VH and VL, respectively) is shown as a cartoon inside a semi-opaque surface representation. ARG2 is shown as blue cartoon, with the region undergoing conformational change upon binding shown in dark purple, manganese ions shown as purple spheres and sulfate shown as sticks. (b) Close-up top-down view of the hydrophobic cleft between VH and VL and the central single-turn helix of the epitope on ARG2. (c) A close-up of the ARG2 active site when bound to C0021158 Fab (blue, with conformational changes upon binding in dark purple) superimposed with free ARG2 bound to an inhibitor (light gray, PDB-ID 4HZECitation28). Critical catalytic and coordinating side chains are shown as sticks. (d) Electron density map (2Fo – Fc, contoured at 2.0 σ (gray mesh) for regions of ARG2 within 2.0 Å of highlighted residues and ligands at the active site of ARG2 when bound to C0021158. Complexation of the dimanganese center and other interactions of interest are shown with black dashes.
The structure of a second affinity-matured inhibitory antibody (C0021181) bound to full-length trimeric ARG2 was determined to a slightly lower resolution of 2.9 Å (space group P 3 2 1), with a final R/Rfree of 0.27/0.32. C0021181 was revealed to bind in essentially the same position and orientation with respect to ARG2 as seen for the C0021158-containing complex ( and Figure S5a and S5b). There is again no evidence of a significant interaction between the 3 Fabs associated with the ARG2 trimer, with the closest contact between the variable domains (~ 5.7 Å) involving Lys75 (heavy chain) and Arg61 (light chain). A few additional close contacts (~3.0 Å) are made by the side chain of Lys206 (heavy chain) with residues 158–161 (heavy chain); however, these appear to represent weak crystal contacts. Interestingly, despite significant sequence and structural differences in VHCDR1 and VHCDR2 (Figure S3a and S3b), binding of C0021181 induces nearly identical conformational changes in ARG2 to those observed for C0021158 (), S5c and S6a). The induced conformational changes also translate into nearly identical, well-defined rearrangements in the active site (Figure S6b and S6c). This clearly points to a shared allosteric mechanism of ARG2 inhibition for both antibodies.
The closely related structures obtained for the two inhibitory Fabs bound to ARG2 clearly reveal that three distinct regions of ARG2 are involved in interactions with the antibodies (residues 37–51, 79–86 and 299–308, as shown schematically in Figure S7a), which partially overlap with the regions found to show major conformational changes induced by antibody binding (residues 33–40, 71–88 and 151–158), as illustrated in ). A major feature at the heart of the ARG2 contact surface for both inhibitory Fabs is the formation of a hydrophobic single-turn α-helix (residues 81–85 on ARG2), which sits in a hydrophobic cleft formed by VHCDR2, VHCDR3 and VLCDR3 (,) and Figure S5c). In both inhibitory complexes, this central hydrophobic contact interface is surrounded by a network of polar interactions (,)). As predicted by the enzyme kinetics studies described above, the inhibitory antibodies do not directly block access to the ARG2 active site, indicating an allosteric mechanism of inhibition, which is mediated via the conformational changes in ARG2 induced by antibody binding and is discussed in detail below.
Allosteric mechanism of ARG2 inhibition by affinity-matured antibodies
The affinity-matured ARG2-specific antibodies that we have identified and characterized clearly inhibit via an allosteric mechanism involving antibody-induced conformational changes in ARG2. Superimposition of the backbone structure of ARG2 (residues 25–32, 41–70, 89–150, 159–329) bound to C0021158 with the previously reported structure of the enzyme containing a transition-state-like inhibitor in the active site (PDB-ID: 4HZECitation28) reveals only a single major change in the active site region, which involves a reorientation of the loop containing Arg39 ()), together with associated more distant changes in residues 151–158. The large antibody-induced change in the loop composed of residues 33–40 results in the side chain of Arg39 pointing toward the active site, where it forms a strong hydrogen bond with Thr265 (2.6 Å between heavy atoms) and a weak ionic interaction with Glu296 (4.6 Å between heavy atoms), as highlighted in ). The guanidinium head group of Arg39 also becomes positioned approximately 3 Å from the side chain of His160, which allows for a favorable cation-π stacking interaction ()). It should be noted that the side chain of Arg39 is very well defined by the electron density observed for both Fab-bound Arginase-2 structures reported here and shows no evidence of significant mobility () and Figure S6c). Although less dramatic, the side chain of Asn158 is also rotated toward the substrate binding pocket by both inhibitory antibodies binding, which places it close to where the amino acid moiety of the substrate would be positioned and potentially restricting substrate binding.
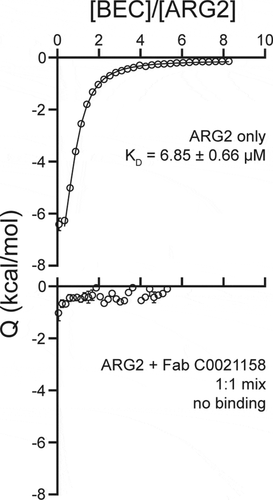
The structural comparisons between Fab-bound and free ARG2 clearly imply a key role for the induced movement of the Arg39 side chain to the active site on antibody binding in mediating the inhibition of activity. His160 has previously been suggested to play an essential role in arginase catalysis, with the histidine side chain proposed to act as a proton donor responsible for transferring an H+ to the newly formed ornithine side chain, thereby facilitating product release.Citation29 This is supported by a reported study of the effect of an H141N substitution in Arginase 1 (homologous to H160N in Arginase 2), which resulted in a variant with only approximately 10% of the wild-type protein’s catalytic activity.Citation30 In the case of binding by the inhibitory antibodies described here, induced interaction of the positively charged Arg39 side chain with the imidazole ring of His160 would be expected to lead to a dramatic reduction in the pKA of the His160 side chain, precluding its involvement as a proton donor in catalysis. We have calculated theoretical pKA values for His160 within the active site environment seen in both free and C0021158-bound ARG2. These calculations predict a dramatic decrease in the pKA for His160, changing from approximately 7 to 2, due to the movement of the Arg39 side chain. This change in the pKA may not be sufficient to explain the ability of the antibodies to completely inhibit catalysis, as one might observe some residual activity due to proton donation from the bulk solvent.Citation31 Consequently, we probed the catalytic consequences of the movement of the Arg39 side chain by examining in silico docking of the substrate to both free and C0021158-bound forms of ARG2, which unexpectedly suggested that the substrate is unable to bind in a catalytically competent position due to the antibody-induced interaction of Arg39 with His160 (Figure S8b). In contrast, the substrate readily docked in a catalytically competent state for the free enzyme (Figure S8a). The tentative conclusions from the in silico docking studies were further supported by isothermal calorimetry (ITC) measurements with the small molecule active site inhibitor S-(2-Boronoethyl)-L-cysteine hydrochloride (BEC), which was found to bind to the free enzyme with an expected KD of approximately 7 µM, but showed no interaction with the C0021158-bound enzyme (). It should be noted that BEC is a transition state mimic, which adopts a similar binding mode to ARG2 as expected for the substrate (also see PDB ID 1PQ3Citation32).
Figure 10. Isothermal titration calorimetry of the ARG2 small-molecule inhibitor S-(2-boronoethyl)-L-cysteine hydrochloride (BEC) with free ARG2 or ARG2-C0021158 Fab complex. Upper panel: Free ARG2. BEC (0.75 mM) was titrated to a solution containing 20 µM of human ARG2 (monomer concentration). Data was fitted to a one-to-one binding model using a 68.3% confidence interval as error. Lower panel: ARG2 bound with C0021158 Fab in a 1:1 mix. BEC at a concentration of 0.50 mM was titrated into a solution containing 20 µM of human ARG2 (monomer concentration) and 20 µM of C0021158 Fab.
Discussion
In this study, we report the targeted inhibition of ARG2 via a novel antibody-mediated noncompetitive mechanism of action, exemplified by the affinity-matured lead C0021158. Solving the structure of the ARG2/Fab C0021158 complex identified subtle changes within the active site that occurs upon antibody binding, providing a clear mechanistic rationale for the antibody’s potent inhibitory effect. These changes, most notably the inward reorientation of Arg39, sterically impede the binding of the L-arginine substrate and prevent its turnover.
The parallel screening of naïve phage display selection outputs in both ARG2-specificity and ARG2 inhibition screens yielded significant numbers of clones with the desired specificity, but very few inhibitory clones. Our report mirrors another similar phage display antibody discovery campaign against the serine protease fibroblast activation protein where large numbers of specific binders were identified, but only a few, low potency, inhibitory clones.Citation33 In our lead isolation campaign, the high local sequence and structural homology shared between the ARG1 and ARG2 active site regions (Figure S7b and S7c) coupled with our decision to screen out ARG1/ARG2 cross-reactive clones made the likelihood of identifying epitopes specifically binding to the conserved active site region unlikely. There are examples of therapeutically relevant enzymes being efficiently inhibited with selective, targeted monoclonal antibodies,Citation34,Citation35 but these antibodies typically exert their inhibitory effect through a competitive mechanism of action, barring access of a large macromolecular protein substrate to the enzyme’s active site, often mimicking naturally evolved inhibitory ligands.Citation36 From a therapeutic standpoint, an antibody exerting a truly allosteric, noncompetitive mechanism of inhibition is often preferable, being both independent of substrate concentration and less prone to acquired resistance through gain of function mutations within the enzyme’s active site. In addition, the relatively small size of the L-arginine substrate coupled with the concave topology of the enzyme’s active site makes the identification of non-allosteric inhibitors less likely. For a competitive antibody to bind to ARG2 and effectively block ingress of L-arginine into the active site, the antibody’s paratope must be sufficiently large and complementary in shape to this concave epitope. Unlike camelid and shark antibodies, the antigen binding surface (i.e., paratope) of human scFv or Fab antibody libraries typically form flat, grooved or concave surfaces.Citation37 The identification of naturally occurring antibodies with paratopes containing extended CDR loops capable of protruding into the active site cleft is therefore correspondingly rare, but not unprecedented.Citation38-Citation40
Overall, our analysis of the major conformational changes induced in ARG2 by inhibitory antibody binding support an innovative allosteric mechanism of inhibition. The combined effects of the Arg39 and Asn158 movement on removing the ability of His160 to act as a proton donor in catalysis, together with preventing the binding of substrate in a catalytically competent position, provide a detailed molecular understanding of the complete inhibition of ARG2 activity achieved by the antibodies. It is especially striking that both the C0021158 and C0021181 inhibitory antibodies induced essentially identical conformational changes and active site side-chain rearrangements in ARG2, even though the interactions at the ARG2-antibody interface are only partially conserved (Figure S7a). This allosteric mechanism of action, whereby substantial conformational changes induced at the antibody binding surface on ARG2 are propagated through the protein to the active site, raises the intriguing question of whether the inhibitory antibodies identified are mimicking a currently unknown in vivo regulatory system for ARG2, involving a functional partner protein binding to the site recognized by the inhibitory antibodies. This ability to tightly regulate the activity of extracellular ARG2 would be consistent with the level of control expected for a key modulator of T-cell activation.
Materials and methods
Phage display isolation of anti-ARG2 antibodies
Five naïve human scFv phage display librariesCitation20-Citation22 were used in parallel for antibody isolation (selections). Three consecutive rounds of selections were performed using biotinylated recombinant human ARG2 trimer at decreasing concentrations (100 nM, 50 nM and 25 nM, respectively), followed by sequence analysis and screening. ScFv were amplified by polymerase chain reaction (PCR) from pCantab6 using vector-specific primers.Citation22,Citation41 Selections were completed in the presence or absence of a 10-fold Molar excess of recombinant human ARG1 trimer (deselection), relative to the concentration of human ARG2 antigen used at any given round. Phage were incubated with ARG2 in Dulbecco’s phosphate-buffered saline (dPBS) and 3% (w/v) Marvel milk powder at room temperature for 2 hours. ARG2-bound scFv-phage were captured on streptavidin-coated paramagnetic beads (Dynabeads®), eluted, infected into E. coli TG1 and rescued for the next round of selection.Citation42
ARG2 and ARG1 biochemical HTRF® FRET binding assay
Representative scFv from round two phage display selection outputs were screened in a biochemical homogenous time-resolved fluorescence resonance energy transfer assay (HTRF® FRET, Cisbio) for specific binding to recombinant human ARG2. FRET signal between bound c-myc-tagged scFv and biotinylated recombinant ARG2 was detected using anti-c-mycXL665, and streptavidin cryptate, respectively (CisBio). In brief, selection outputs were screened as bacterial periplasmic extracts prepared in assay buffer (200 mM Tris buffer pH 7.4, 0.5 mM ethylenediaminetetraacetic acid (EDTA), 0.5 M sucrose). Five-microliter volumes of periplasmic extract, 12 nM biotinylated recombinant human ARG2 trimer, 6.67 nM streptavidin cryptate and 40 nM anti-c-mycXL665 were combined and added to a single well of a Greiner® 384 well assay plate (Greiner Bio-one). All assay component dilutions were performed in dPBS containing 0.4 M potassium fluoride (KF) and 0.1% bovine serum albumin. Assay plates were incubated overnight at 4°C prior to reading time resolved fluorescence on a Pherastar plate reader (PerkinElmer) using an excitation wavelength of 320 nm and measuring the emission at 620 nm and 665 nm (100 flashes).
Data were analyzed by calculating % Delta F values for each sample according to the following equation.
HTRF® screens were internally controlled for scFv expression by the inclusion of a stripy assay plate consisting of alternating columns of positive (ARG2-binding) and negative (CEA6; non-ARG2-binding) scFv clones (n = 8 replicate wells per column or ‘stripe’). Bacterial periplasmic extracts for the stripy plate were prepared in an identical manner to that used to generate each test plate. Cross-reactive scFv clones were identified via a parallel assay using recombinant human trimeric ARG1 enzyme in place of ARG2.
In vitro ARG2 enzyme inhibition assay
Round two phage display selection outputs were screened as bacterial periplasmic extracts prepared in assay buffer (200 mM Tris buffer pH 7.4, 0.5 mM EDTA and 0.5 M sucrose). 10 µl of scFv extract was added to a Greiner bio-one® 384 well assay plate (Greiner Bio-one), followed by 5 µl of 1.6 µg/ml recombinant trimeric human ARG2 diluted in 4x enzyme buffer (40 mM MnCl2, 100 mM Tris-HCl, pH7.4), before incubating overnight at 4°C. 5 µl 100 mM L-arginine substrate diluted in water, pH 7.4 was then added before incubating the assay plate at room temperature for 1 hour. Urea was detected by the simultaneous addition of equal volumes of O-phthaldialdehyde (1.5 mM O-phthaldialdehyde, 7.5% H2SO4, 0.03% Brij L23) and N-(1-napthyl)ethylenediamine (1.66 mM N-(1-napthyl)ethylenediamine, 81 mM boric acid, 22.5% H2SO4, 0.03% Brij L23) followed by an 18 minute incubation at room temperature before quantifying the colorimetric product on a Pherastar plate reader, reading at 505 nm (PerkinElmer).
Background control wells were defined for each plate by the omission of substrate. Maximum signal control wells were defined for each plate by using a negative control unpurified scFv (CEA6) in place of the test scFv sample. Data were analyzed by calculating % inhibition for each sample according to the following equation.
Like the ARG2 HTRF® screens, each ARG2 enzyme inhibition assay was internally controlled for scFv expression by the inclusion of a stripy assay plate.
ARG1 and ARG2 biochemical HTRF® binding assay using purified scFv fragments
ScFv were expressed in bacterial periplasm as described in Dobson, C. L. et al.Citation43 and purified as described in Osbourn et al.Citation44 An 11-point dilution series of purified scFv were screened in the same biochemical HTRF® binding assay previously described for unpurified scFv. Concurrent screens testing binding to ARG1 and ARG2 were undertaken.
In vitro ARG2 enzyme inhibition assay using purified scFv fragments
Serial dilutions of purified scFvCitation43,Citation44 were screened for inhibition of recombinant ARG2 as previously described for unpurified scFv. ScFv inhibition was compared to the small molecule Arginase inhibitor NHLACitation45 (Millipore).
Lead isolation scFv HTRF® epitope competition assay
A panel of purified lead isolation scFvs were directly labeled with DyLight650, (Thermo). All assay component dilutions were performed in dPBS containing 0.4 M KF and 0.1% bovine serum albumin. In brief, 2.5 µl volumes of streptavidin cryptate, DyLight650 labeled scFv, recombinant biotinylated ARG2 and unlabeled competitor scFv and were added to a well of a of a Greiner® 384 well assay plate (Greiner Bio-one). A final assay concentration of 100 nM labeled scFv, 3 nM biotinylated recombinant ARG2 and 1.69 nM streptavidin cryptate was used. Plates were incubated overnight at 4°C before reading time resolved fluorescence on a Pherastar plate reader (PerkinElmer) using an excitation wavelength of 320 nm and measuring the emission at 620 nm and 665 nm (100 flashes). Resolved fluorescence was calculated as delta F (%).
Conversion of scFv to IgG and Fab
The reformatting of scFv into human IgG1 format was performed according to Persic et al.Citation46 The reformatting of scFv to human Fab was performed using modified versions of the same IgG1 vectors described originally in Persic et al.Citation46
In vitro T-cell proliferation assay
Peripheral blood mononuclear cells (PBMCs) were isolated from leukocyte cones following Ficoll plaque gradient separation (GE Healthcare). T-cell populations were isolated from PBMCs by negative selection using a Human T Cell Enrichment Kit (Stemcell Technologies). 40000 cells/well were seeded in complete growth media (RPMI 1640, 5% human serum albumin) into 96-well clear TC treated microplates (Greiner-Bio One). T cells were activated by the addition of ImmunoCult CD3/CD28 (Stemcell Technologies) used at a dilution of 1:40 and the plate incubated 37°C, 5% CO2 for 45 minutes. All antibodies were diluted in sterile Tris-buffered saline (TBS)/NaCl Buffer before 20 µl was added to the assay plate. The plate was incubated for 30 minutes at 37°C, 5% CO2. Recombinant human ARG2 was added to a final assay concentration of 15 µg/ml. The total assay volume per well was 200 µl. Cells were treated with test antibodies for a total of 96 hours at 37°C, 5% CO2. Assessment of T-cell proliferation was performed using the Bromodeoxyuridine (BrdU) Cell Proliferation Enzyme-Linked Immunosorbent Assay Kit (Roche). BrdU labeling reagent was added 78 hours after antibody treatment and the plate incubated for a further 18 hours at 37°C, 5% CO2. Proliferation data were expressed as percentage of control and graphical plots were constructed using a four-parameter fit and EC50 values determined using model 205 in XLFit (IDBS).
Anti-ARG2 antibody inhibitory mechanism of action
The inhibitory mechanism of action for anti-ARG2 antibodies was investigated using the in vitro ARG2 enzyme inhibition assay described. In brief, four concentrations of test IgG were prepared (10, 3, 1 and 0.3 µM). Test IgGs were then added to an 11-point dilution of L-arginine substrate, with a top concentration of 500 mM and bottom concentration of 5 mM. The reaction was allowed to proceed for 80, 60, 45, 30, 15 or 5 minutes before the addition of the urea detection reagents O-phthaldialdehyde and N-(1-napthyl)ethylenediamine. Raw data derived from each concentration of antibody was plotted as a function of time for each concentration of L-arginine and fitted using a linear fit model (GraphPad Prism) to derive initial velocities (slope values). Initial velocity data was plotted against L-arginine concentration for each concentration of antibody. The maximum velocity (Vmax) and enzyme/substrate affinity (KM) values at each inhibitor concentration was solved by fitting each trace to the following Michaelis-Menten model:
Isothermal titration calorimetry
Isothermal titration calorimetry experiments were undertaken at 25°C using a MicroCal VP ITC system (Malvern Panalytical Limited). A 750 µM solution of BEC was titrated into a 20 µM solution of recombinant human ARG2 (monomer concentration). Thermograms were integrated using NITPIC software and binding isotherms were fitted using SEDPHAT software, using a one-binding site model. Binding of BEC to ARG2 in the presence of bound C0021158 Fab was undertaken by pre-incubating a 1:1 20 µM mixture of C0021158 Fab and recombinant human ARG2, for 1 h at 20°C, before the addition of BEC.
ARG2 expression and purification
Human ARG2 (residues 23–354, C-terminal His6-tag) was cloned into vector pLEICS-50 (C-terminal His6-tag) and used to transform E. coli BL21 (DE3) cells and to create frozen glycerol stocks (30% glycerol) from a single colony. A small scrape of the frozen glycerol stock was used to inoculate a starter culture of 50 ml Luria-Bertani-Ampicillin (LB-Amp), which was grown overnight (37°C, 200 rpm). 2 × 500 ml LB-Amp main culture were inoculated with the starter culture to an OD600 nm of 0.2 and incubated at 37°C and 150 rpm to reach an OD600 nm of 1.0 after 3 h, when the culture was cooled to 20°C and induced with 100 µM isopropyl β-d-1-thiogalactopyranoside. After overnight expression the culture was centrifuged for 10 min at 4°C and 4500 × g, and the cell pellet was collected and frozen. Subsequently, the thawed pellet was resuspended in 50 ml lysis/binding buffer (50 mM Tris pH 8.0, 300 mM NaCl, 20 mM imidazole, 6% glycerol, 5 mM β-mercaptoethanol, 200 µM MnCl2, 20 µg/l DNase I, 100 µM phenylmethylsulfonyl fluoride (PMSF), 1 tablet cOmplete inhibitor (Roche)). The cells were cracked using sonication on ice (8 × 30 s pulse, 30 s cool-down) and the lysate was cleared by centrifugation for 20 minutes at 4°C and 48000 × g. The lysate was loaded onto a Ni-NTA column and eluted with a gradient of elution buffer (50 mM Tris pH 8.0, 300 mM NaCl, 500 mM imidazole, 5 mM β-mercaptoethanol, 200 µM MnCl2). The fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and the ARG2-containing fractions (12 ml) were pooled, concentrated to 4 ml (centrifugal filter unit with 10 kDa molecular weight cutoff) and gel filtrated on a Superdex 200 16/600 column into 20 mM Tris pH 8.0, 150 mM NaCl and 200 µM MnCl2. The fractions of the peak corresponding to the ARG2 trimer were analyzed, pooled and concentrated to 1.3 mg/ml. The concentrated ARG2 was aliquoted and flash frozen for storage.
Fab C0021144 crystallization, data collection and structure determination
The Fab was concentrated in TBS pH 7.4 to 22 mg/ml and pipetted as sitting drops (0.13 µl Fab + 0.13 µl reservoir solution using an Oryx8 robot) into JSCG+ (Molecular Dimensions) and AmSO4 Suite (Qiagen) screens. JCSG+ condition A9 (200 mM NH4Cl, 20% PEG3350) yielded several rhombic crystals of up to 100 µm in length after 2 days at 18°C. The crystals were cryo-protected by adding 50% PEG400 to the crystallization drop, through which the crystals were pulled, and cryo-cooled in liquid N2. Data of three crystals were collected at DLS beamline i04 and merged, which extended the resolution to 2.4 Å and indexed in space group H 3 2. The data were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The structure was then solved by molecular replacement in PHASERCitation49 using a homology model calculated by the PIGSPro web server,Citation50 and was subsequently refined using REFMAC5Citation51 and COOT,Citation52 with TLS refinement turned on during the final cycles (final R/Rfree = 0.21/0.24).
Fab C0021158 crystallization, data collection and structure determination
The Fab was concentrated in TBS pH 7.4 to 20 mg/ml and pipetted as sitting drops (0.15 µl Fab + 0.15 µl reservoir solution using an Oryx8 robot) into JCSG+ and PACT screens. PACT condition A9 (100 mM sodium acetate buffer pH 5.0, 20% PEG6000, 200 mM LiCl) yielded layered, irregular crystals of approx. 130 × 130 x 150 µm after 2 days at 18°C. The crystal was cryo-cooled in liquid N2 and data collection was performed at ESRF beamline ID23-1.Citation53 One crystal diffracted to approx. 1.9 Å and indexed in space group P 21. The data were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The structure was then solved by molecular replacement in PHASERCitation49 using a high-resolution structure of Fab C0021144 (PDB-ID 6SRV, this study), and was subsequently refined using REFMAC5Citation51 and COOTCitation52 (final R/Rfree = 0.24/0.27).
Fab C0021177 crystallization, data collection and structure determination
The Fab was concentrated in TBS pH 7.4 to 17 mg/ml and pipetted as sitting drops (0.16 µl Fab + 0.16 µl reservoir solution using an Oryx8 robot) into JCSG+ and PACT screens. PACT condition D4 (100 mM MMT (malic acid, MES, Tris) pH 7.0, 25% PEG1500) yielded a single rhombic crystal of approx. 80 × 35 x 35 µm after more than 6 months at 18°C. The crystal was cryo-cooled in liquid N2 and data collection was performed at the BESSY II macromolecular beamline MX-14-1.Citation54 The crystal diffracted to approx. 2.2 Å and indexed in space group P 212121. The data were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The structure was then solved by molecular replacement in PHASERCitation49 using a high-resolution structure of Fab C0021144 (PDB-ID 6SRV, this study), and was subsequently refined using REFMAC5Citation51 and COOTCitation52 (final R/Rfree = 0.25/0.32).
Fab C0021181 crystallization, data collection and structure determination
The Fab was concentrated in TBS pH 7.4 to 18 mg/ml and pipetted as sitting drops (0.13 µl Fab + 0.13 µl reservoir solution using an Oryx8 robot) into JCSG+ and PACT screens (Molecular Dimensions). PACT condition A4 (100 mM SPG (succinic acid, phosphate, glycine) buffer pH 7.0, 25% PEG1500) yielded a crystal of approx. 100 µm after 2 days at 18°C. The crystal was cryo-cooled in liquid N2 and data collection was performed at ESRF beamline ID23-1.Citation53 The crystal diffracted to approx. 1.7 Å and indexed in space group P 21. The data were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The structure was then solved by molecular replacement in PHASERCitation49 using a high-resolution structure of Fab C0021144 (PDB-ID 6SRV, this study), and was subsequently refined using REFMAC5Citation51 and COOTCitation52 (final R/Rfree = 0.19/0.22).
ARG2/Fab C0021158 complex formation, crystallization, data collection and structure determination
ARG2 and Fab C0021158 were mixed in a molar ratio 1:1.04 at 1.2 mg/ml and incubated at room temperature for 1 h. The complex was then concentrated to 8.7 mg/ml and 20% (v/v) of diluted microseeds for cross-seeding were added to a final complex concentration of 6.9 mg/ml. Microseeds were produced from ARG2 + Fab C0021181 crystals grown in 100 mM MIB (malonate:imidazole:boric acid = 2:3:3) pH 10.0,Citation55 20% glycerol, 10% PEG4000, 45 mM NaNO3, 45 mM Na2HPO4, 45 mM (NH4)2SO4, by crushing the crystals in the well with a thin glass rod, vortexing the debris for 2 min with a glass bead in 50 µl reservoir solution, pelleting large pieces by centrifugation at 16000 × g and finally diluting them 500 times with more reservoir solution. The crystallization was set up as sitting drops (0.25 µl complex/microseeds + 0.25 µl reservoir solution using an Oryx8 robot) at 18°C in JCSG+ and SG-1 screens (Molecular Dimensions). The condition yielding the best-diffracting crystal contained 2 M (NH4)2SO4. The crystals grew within two days to an irregular shape, with dimensions of approx. 60 µm × 60 µm × 60 µm. After adding 3 M (NH4)2SO4 and 5% glycerol for cryo-protection, the crystal was cryo-cooled in liquid N2 and data collection was performed at the BESSY II macromolecular beamline MX-14-1,Citation54 where a dataset diffracting to 2.4 Å was obtained. The data were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The data indexed in space group H 3 and was twinned. The structure was then solved by molecular replacement in PHASERCitation49 using a high-resolution ARG2 structure (PDB-ID: 4HZECitation28) and a high-resolution structure of the free Fab C0021158 (PDB-ID 6SRX, this study) as search models. The asymmetric unit contained one copy of ARG2 and one copy of the Fab. Automated and manual refinement was then performed by alternatingly using REFMAC5Citation51 (with twin- and TLS refinement turned on after most of the model was built) and COOT,Citation52 respectively. Refined R/Rfree of the completed model were 0.25/0.28, and the twin fractions were refined to H K L = 0.80/K H-L 0.20.
ARG2/Fab C0021181 complex formation, crystallization, data collection and structure determination
ARG2 and Fab C0021181 were mixed in a molar ratio 1:1.05 at 0.7 mg/ml and incubated at room temperature for 1 h. The complex was then concentrated to 6.5 mg/ml and crystallization was set up as sitting drops (0.5 µl complex + 0.5 µl reservoir solution using an Oryx8 robot (Douglas Instruments)) at 18°C. The condition yielding the best-diffracting crystals contained 100 mM MMT (malic acid:MES:Tris = 1:2:2) pH 5.0,Citation55 20% glycerol, 10% PEG4000, 15 mM NaNO3, 15 mM Na2HPO4, 15 mM (NH4)2SO4 and was derived from a Morpheus (Molecular Dimensions) screen condition. Buffers with pH values higher than 8.0 also yielded crystals, which were larger half-hexagon prisms, but showed worse diffraction characteristics. Crystals grew typically within a day to a thin, hexagonal prism shape, with a diameter of approx. 100 µm and a thickness of approx. 20 µm. Crystals were cryo-cooled in liquid N2 and data collection was performed at the BESSY II macromolecular beamline MX-14-1.Citation54 Most crystals diffracted to approx. 3.5 Å or worse, but a few specimens diffracted up to 2.9 Å. Two datasets of the best crystal were processed and scaled with XDSCitation47 and merged using AIMLESS.Citation48 The crystals indexed in space group P 6 2 2 and appeared to be twinned, indicating that the space group was wrong. The data were reindexed in P 6, P 3 2 1, P 3 1 2 and P 3 and subjected to molecular replacement trials searching for all possible solutions in the individual point groups using PHASER.Citation49 A high-resolution ARG2 structure (PDB-ID: 4HZECitation28) and a high-resolution structure of the free Fab C0021181 (PDB-ID 6SS0, this study) were used as search models. By comparing the electron density quality of the solutions, Rfree values and validation using ZANUDA,Citation56 the real space group was found to be P 3 2 1, with twinning still present. The asymmetric unit contained one copy of ARG2 and one copy of the Fab, but only the VL and VH domains of the Fab could be placed easily. The CH and CL domains are most likely present in several orientations, which is reflected by the increased B factor and worse quality of the electron density compared to the rest of the structure. Automated and manual refinement was then performed by alternatingly using REFMAC5Citation51 (with twin- and TLS refinement turned on after most of the model was built) and COOT,Citation52 respectively. Refined R/Rfree of the completed model were 0.27/0.32, and the twin fractions were refined to H K L = 0.50/-H -K L 0.50.
Structural modeling and docking
Structural modeling and docking were performed using Schrödinger Maestro release 2017–2.Citation57 Protein structures were prepared for modeling using the default settings of the Protein Preparation Wizard. The apo structures were generated using the structure of ARG2 bound to BEC (PDB ID: 1PQ3Citation32) and antibody complex structures, and sulfate and BEC ligand were converted to water (i.e., everything except the oxygen atom near the manganese cluster was deleted and hydrogen atoms were added). These structures were then also prepared for modeling using the Protein Preparation Wizard. pKa values were calculated with PROPKACitation58 implemented in the Protein Preparation Wizard. Ligands were docked using the default parameters of the induced fit docking procedure,Citation57,Citation59-Citation61 except that re-docking was performed with XP settings. The transition state was docked into ligand and sulfate bound structures, whereas the substrate was docked into the prepared apo structures. The product-bound state was manually built from the transition state model and refined as described above.
Abbreviations
| AML | = | Acute myeloid leukaemia |
| ARG1 | = | Arginase 1 |
| ARG2 | = | Arginase 2 |
| BEC | = | S-(2-boronoethyl)-L-cysteine hydrochloride |
| BrdU | = | Bromodeoxyuridine |
| CD | = | Cluster of differentiation |
| CDR | = | Complementarity-determining region |
| CH | = | Constant heavy chain domain |
| CL | = | Constant light chain domain |
| dPBS | = | Dulbecco’s phosphate-buffered saline |
| EDTA | = | Ethylenediaminetetraacetic acid |
| ELISA | = | Enzyme-linked immunosorbent assay |
| Fab | = | Fragment, antigen binding |
| FRET | = | Fluorescence resonance energy transfer |
| FW | = | Framework domain |
| HTRF® | = | Homogenous time resolved fluorescence |
| IgG | = | Immunoglobulin G |
| ITC | = | Isothermal calorimetry |
| KF | = | Potassium fluoride |
| LB | = | Luria-Bertani |
| n | = | Number of separate experiments |
| NHLA | = | NG-hydroxy-L-arginine |
| nor-NOHA | = | Nω-hydroxy-nor-L-arginine |
| PBMC | = | Peripheral blood mononuclear cell |
| PCR | = | Polymerase chain reaction |
| PD-1 | = | Programmed cell death protein 1 |
| PMSF | = | Phenylmethylsulfonyl fluoride |
| scFv | = | Single-chain variable fragment |
| TBS | = | Tris-buffered saline |
| VH | = | Variable heavy chain domain |
| VL | = | Variable light chain domain |
Disclosure of Potential Conflicts of Interest
The authors report no conflict of interest.
Supplemental Material
Download Zip (109.8 MB)Acknowledgments
The authors thank Diamond Light Source (proposal mx19880), the European Synchrotron Radiation Facility (proposal mx1984) and the Helmholtz-Zentrum Berlin, BESSY II (proposal 182-07165-ST) for beam time to collect diffraction data. The authors would particularly thank the staff of the beamlines I04 and I04-1 (DLS), ID23-1 (ESRF) and MX-14-1 (HZB, BESSY II) for support with crystal testing and data collection.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Muenst S, Laubli H, Soysal SD, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: therapeutic concepts. J Intern Med. 2016;279:541–17. doi:https://doi.org/10.1111/joim.12470.
- Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2016;39:1–6. doi:https://doi.org/10.1016/j.coi.2015.10.009.
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi:https://doi.org/10.1146/annurev.immunol.25.022106.141609.
- Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy. 2017;9:83–97. doi:https://doi.org/10.2217/imt-2016-0118.
- Umansky V, Adema GJ, Baran J, Brandau S, Van Ginderachter JA, Hu X, Jablonska J, Mojsilovic S, Papadaki HA, De Coaña YP, et al. Interactions among myeloid regulatory cells in cancer. Cancer Immunol Immunother. 2019;68:645–60. doi:https://doi.org/10.1007/s00262-018-2200-6.
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54. doi:https://doi.org/10.1038/nri1668.
- Martí I Líndez -A-A, Dunand-Sauthier I, Conti M, Gobet F, Núñez N, Hannich JT, Riezman H, Geiger R, Piersigilli A, Hahn K, et al. Mitochondrial arginase-2 is a cell‑autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight. 2019;4:e132975. doi:https://doi.org/10.1172/jci.insight.132975.
- Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336(Pt 1):1–17. doi:https://doi.org/10.1042/bj3360001.
- Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, McConville C, Sbirkov Y, Wheeler K, Bendle G, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015;75:3043–53. doi:https://doi.org/10.1158/0008-5472.CAN-14-3443.
- Zaytouni T, Tsai PY, Hitchcock DS, DuBois CD, Freinkman E, Lin L, Morales-Oyarvide V, Lenehan PJ, Wolpin BM, Mino-Kenudson M, et al. Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat Commun. 2017;8:242. doi:https://doi.org/10.1038/s41467-017-00331-y.
- Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, Kanai Y, Hiraoka N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One. 2013;8:e55146–e. doi:https://doi.org/10.1371/journal.pone.0055146.
- Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P, Cerundolo V, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013;122:749–58. doi:https://doi.org/10.1182/blood-2013-01-480129.
- Miraki-Moud F, Ghazaly E, Ariza-McNaughton L, Hodby KA, Clear A, Anjos-Afonso F, Liapis K, Grantham M, Sohrabi F, Cavenagh J, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–68. doi:https://doi.org/10.1182/blood-2014-10-608133.
- Tate DJ Jr., Vonderhaar DJ, Caldas YA, Metoyer T, Patterson J, Aviles DH, Zea AH. Effect of arginase II on L-arginine depletion and cell growth in murine cell lines of renal cell carcinoma. J Hematol Oncol. 2008;1:14. doi:https://doi.org/10.1186/1756-8722-1-14.
- Pudlo M, Demougeot C, Girard-Thernier C. Arginase inhibitors: a rational approach over one century. Med Res Rev. 2017;37:475–513. doi:https://doi.org/10.1002/med.21419.
- Wang Y, Zhao P, Qian D, Hu M, Zhang L, Shi H, Wang B. MicroRNA-613 is downregulated in HCMV-positive glioblastoma and inhibits tumour progression by targeting arginase-2. Tumour Biol. 2017;39:1010428317712512. doi:https://doi.org/10.1177/1010428317712512.
- Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi:https://doi.org/10.1158/0008-5472.CAN-04-0465.
- Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101. doi:https://doi.org/10.1186/s40425-017-0308-4.
- Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–27. doi:https://doi.org/10.1038/nrc1913.
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS, et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–14. doi:https://doi.org/10.1038/nbt0396-309.
- Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods. 2006;313:129–39. doi:https://doi.org/10.1016/j.jim.2006.04.002.
- Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel. 2009;22:159–68. doi:https://doi.org/10.1093/protein/gzn058.
- Einhorn L, Krapfenbauer K. HTRF: a technology tailored for biomarker determination-novel analytical detection system suitable for detection of specific autoimmune antibodies as biomarkers in nanogram level in different body fluids. Epma J. 2015;6:23. doi:https://doi.org/10.1186/s13167-015-0046-y.
- Zhou Y, Goenaga AL, Harms BD, Zou H, Lou J, Conrad F, Adams GP, Schoeberl B, Nielsen UB, Marks JD, et al. Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Mol Cancer Ther. 2012;11:1467–76. doi:https://doi.org/10.1158/1535-7163.MCT-11-1038.
- Chan DTY, Jenkinson L, Haynes SW, Austin M, Diamandakis A, Burschowsky D, Seewooruthun C, Addyman A, Fiedler S, Ryman S, et al. Extensive sequence and structural evolution of Arginase 2 inhibitory antibodies enabled by an unbiased approach to affinity maturation. Proc Natl Acad Sci USA. 2020;117:16949–60. doi:https://doi.org/10.1073/pnas.1919565117.
- Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol. 1991;147:1709–19.
- Strelow J, Dewe W, Iversen PW, Brooks HB, Radding JA, McGee J, Weidner J. Mechanism of action assays for enzymes. In: Sittampalam GS, Grossman A, Brimacombe K, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, et al. editors. Assay guidance manual. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004;65–86.
- Van Zandt MC, Whitehouse DL, Golebiowski A, Ji MK, Zhang M, Beckett RP, Jagdmann GE, Ryder TR, Sheeler R, Andreoli M, et al. Discovery of (R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J Med Chem. 2013;56:2568–80. doi:https://doi.org/10.1021/jm400014c.
- Cox JD, Kim NN, Traish AM, Christianson DW. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat Struct Biol. 1999;6:1043–47. doi:https://doi.org/10.1038/14929.
- Cavalli RC, Burke CJ, Kawamoto S, Soprano DR, Ash DE. Mutagenesis of rat liver arginase expressed in Escherichia coli: role of conserved histidines. Biochemistry. 1994;33:10652–57. doi:https://doi.org/10.1021/bi00201a012.
- Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–57. doi:https://doi.org/10.1038/383554a0.
- Cama E, Colleluori DM, Emig FA, Shin H, Kim SW, Kim NN, Traish AM, Ash DE, Christianson DW. Human Arginase II: crystal structure and physiological role in male and female sexual arousal†,‡. Biochemistry. 2003;42:8445–51. doi:https://doi.org/10.1021/bi034340j.
- Zhang J, Valianou M, Simmons H, Robinson MK, Lee HO, Mullins SR, Marasco WA, Adams GP, Weiner LM, Cheng JD, et al. Identification of inhibitory scFv antibodies targeting fibroblast activation protein utilizing phage display functional screens. FASEB J. 2013;27:581–89. doi:https://doi.org/10.1096/fj.12-210377.
- Fischer T, Riedl R. Inhibitory antibodies designed for matrix metalloproteinase modulation. Molecules. 2019;24:2265. doi:https://doi.org/10.3390/molecules24122265.
- Santamaria S, de Groot R. Monoclonal antibodies against metzincin targets. Br J Pharmacol. 2019;176:52–66. doi:https://doi.org/10.1111/bph.14186.
- Sela-Passwell N, Kikkeri R, Dym O, Rozenberg H, Margalit R, Arad-Yellin R, Eisenstein M, Brenner O, Shoham T, Danon T, et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat Med. 2011;18:143–47. doi:https://doi.org/10.1038/nm.2582.
- Lauwereys M, Ghahroudi MA, Desmyter A, Kinne J, Holzer W, De Genst E, Wyns L, Muyldermans S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. Embo J. 1998;17:3512–20. doi:https://doi.org/10.1093/emboj/17.13.3512.
- Matz H, Dooley H. Shark IgNAR-derived binding domains as potential diagnostic and therapeutic agents. Dev Comp Immunol. 2019;90:100–07. doi:https://doi.org/10.1016/j.dci.2018.09.007.
- Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305:1770–73. doi:https://doi.org/10.1126/science.1101148.
- Nam DH, Rodriguez C, Remacle AG, Strongin AY, Ge X. Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc Natl Acad Sci USA. 2016;113:14970–75. doi:https://doi.org/10.1073/pnas.1609375114.
- McCafferty J, Fitzgerald KJ, Earnshaw J, Chiswell DJ, Link J, Smith R, Kenten J. Selection and rapid purification of murine antibody fragments that bind a transition-state analog by phage display. Appl Biochem Biotechnol. 1994;47:157–71. discussion 71-3. doi:https://doi.org/10.1007/BF02787932.
- Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–96. doi:https://doi.org/10.1016/0022-2836(92)90639-2.
- Dobson CL, Main S, Newton P, Chodorge M, Cadwallader K, Humphreys R, Albert V, Vaughan TJ, Minter RR, Edwards BM, et al. Human monomeric antibody fragments to TRAIL-R1 and TRAIL-R2 that display potent in vitro agonism. mAbs. 2009;1:552–62. doi:https://doi.org/10.4161/mabs.1.6.10057.
- Osbourn JK, McCafferty J, Derbyshire EJ, Waibel R, Chester KA, Boxer G, Allen D. Isolation of a panel of human anti-CEA single chain Fv from a large phage display library. Tumor Target. 1999;4:150–57.
- Daghigh F, Fukuto JM, Ash DE. Inhibition of rat liver arginase by an intermediate in NO biosynthesis, NG-hydroxy-L-arginine: implications for the regulation of nitric oxide biosynthesis by arginase. Biochem Biophys Res Commun. 1994;202:174–80. doi:https://doi.org/10.1006/bbrc.1994.1909.
- Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi:https://doi.org/10.1016/S0378-1119(96)00628-2.
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi:https://doi.org/10.1107/S0907444909047337.
- Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D. 2013;69:1204–14. doi:https://doi.org/10.1107/S0907444913000061.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi:https://doi.org/10.1107/S0021889807021206.
- Marcatili P, Rosi A, Tramontano A. PIGS: automatic prediction of antibody structures. Bioinformatics. 2008;24:1953–54. doi:https://doi.org/10.1093/bioinformatics/btn341.
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–67. doi:https://doi.org/10.1107/S0907444911001314.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi:https://doi.org/10.1107/S0907444910007493.
- Nurizzo D, Mairs T, Guijarro M, Rey V, Meyer J, Fajardo P, Chavanne J, Biasci J-C, McSweeney S, Mitchell E, et al. The ID23-1 structural biology beamline at the ESRF. J Synchrotron Radiat. 2006;13:227–38. doi:https://doi.org/10.1107/S0909049506004341.
- Mueller U, Förster R, Hellmig M, Huschmann FU, Kastner A, Malecki P, Pühringer S, Röwer M, Sparta K, Steffien M, et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: current status and perspectives. Eur Phys J Plus. 2015;130:141. doi:https://doi.org/10.1140/epjp/i2015-15141-2.
- Newman J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr D Biol Crystallogr. 2004;60:610–12. doi:https://doi.org/10.1107/S0907444903029640.
- Lebedev AA, Isupov MN. Space-group and origin ambiguity in macromolecular structures with pseudo-symmetry and its treatment with the program Zanuda. Acta Crystallogr D Biol Crystallogr. 2014;70:2430–43. doi:https://doi.org/10.1107/S1399004714014795.
- Schrödinger release 2017–2. New York (NY): Schrödinger, LLC; 2019. p. 2017. https://www.schrodinger.com/platform.
- Sondergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7:2284–95. doi:https://doi.org/10.1021/ct200133y.
- Farid R, Day T, Friesner RA, Pearlstein RA. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem. 2006;14:3160–73. doi:https://doi.org/10.1016/j.bmc.2005.12.032.
- Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des. 2006;67:83–84. doi:https://doi.org/10.1111/j.1747-0285.2005.00327.x.
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–53. doi:https://doi.org/10.1021/jm050540c.