
ABSTRACT
Commercially manufactured ‘jiaosu’ (fermented fruit and vegetable juices) have gained popularity in Asia recently. Like other fermented products, they have a high microbial diversity and richness. However, no published study has yet described their microbiota composition. Thus, this work aimed to obtain the full-length 16S rRNA profiles of jiaosu using the PacBio single-molecule, real-time sequencing technology. We described the bacterial microbiota of three jiaosu products purchased from Taiwan and Japan. Bacterial sequences from all three samples distributed across seven different phyla, mainly Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Forty-three genera were identified (e.g. Ochrobactrum, Lactobacillus, Mycobacterium, and Acinetobacter). Fifty-five species were identified (e.g. Ochrobactrum lupini, Mycobacterium abscessus, Acinetobacter johnsonii, Lactobacillus paracasei, Lactobacillus delbrueckii, and Petrobacter succinatimandens). No pathogen sequences were identified within the entire dataset. Moreover, only a low proportion of sequences represented common skin microflora and the food hygiene indicator Escherichia/Shigella, suggesting overall acceptable sanitary conditions during the manufacturing process.
RESUMEN
Recientemente, en Asia han ganado popularidad los jiaosu (jugos fermentados de fruta y verduras) preparados en forma comercial. Al igual que otros productos fermentados, éstos contienen una elevada diversidad y riqueza microbiana. Sin embargo, no existen estudios publicados que describan su composición microbiótica. Por ello, el presente estudio se propuso obtener los perfiles completos de 16S rRNA de jiaosu, utilizando tecnología de secuenciación a tiempo real de una sola molécula. Esta metodología permitió obtener la microbiota bacteriana de tres productos jiaosu adquiridos en Taiwán y Japón e identificar las secuencias bacterianas de las tres muestras distribuidas en siete filos diferentes, principalmente proteobacteria, firmicutes, actinobacteria y bacteroidetes. Se registraron 43 géneros (esto es, Ochrobactrum, Lactobacillus, Mycobacterium y Acinetobacter) y 55 especies (Ochrobactrum lupini, Mycobacterium abscessus, Acinetobacter johnsonii, Lactobacillus paracasei, Lactobacillus delbrueckii y Petrobacter succinatimandens). Por otra parte, en todo el conjunto de datos no se identificó ninguna secuencia patógena. Además, solo en una proporción reducida de secuencias se detectó la microflora común de la piel y el indicador de higiene alimentaria Escherichia/Shigella; ello sugiere que se trabajó en aceptables condiciones higiénicas generales durante el proceso de manufactura.
Introduction
Commercially manufactured ‘jiaosu’ (fermented fruit and vegetable juices) have gained popularity in Asia in recent years. These products have traditionally been made in small-scale and in local households. However, due to modernization of people’s lifestyle, market demand, and the advent in fermentation technologies, more and more jiaosu are commercially available and consumed. Generally, they are made by fermenting single or mixed fruits and/or vegetables together with honey, with/without addition of starter culture like lactic acid bacteria (LAB) and other value-adding ingredients. Usually, they are made in glass containers having an airlock system that allows the maintenance of anaerobic conditions meanwhile enables the release of carbon dioxide produced during the anaerobic fermentation. The fermentation process takes at least three months before the fermented juice is ready for consumption.
Commercial advertisements claim that drinking jiaosu confers numerous health-promoting functions to consumers, e.g. improving metabolism, enhancing antioxidant capacity, anti-aging, detoxification, body slimming, reducing blood lipid, liver protection, relieving alcohol hangover, and boosting immunity. However, only some of these beneficial effects have been demonstrated experimentally. For example, fermented juices of germinated seeds, sprouts of lentil and cowpea possess various in vitro desirable activities, including angiotensin-converting enzyme inhibitory activity, α-amylase and α-glucosidase inhibitory activities, bile acid-binding capacity, antioxidant activity, and hemagglutinating activity (Simsek, El, Kancabas Kilinc, & Karakaya, Citation2014). Tomato and feijoa juices have been shown to be good fermentable carriers for Lactobacillus plantarum, which improves intestinal barrier function in vitro (Valero-Cases, Roy, Frutos, & Anderson, Citation2017). Moreover, fermented orange beverage could reduce cardiovascular risk in mice (Escudero-López et al., Citation2015), while fermented beetroot juice could modulate the gut epithelium microbiota beneficially and reduce β-glucuronidase activity in rats (Klewicka, Zduńczyk, Juśkiewicz, & Klewicki, Citation2015). In human, consuming pomegranate juice for 3 weeks could significantly enhance plasma and urinary anti-oxidative activities (Gouda, Moustafa, Hussein, & Hamza, Citation2015), and daily intake of fermented citrus juice could alleviate perennial allergic rhinitis (Harima-Mizusawa et al., Citation2016).
Like other fermented products, jiaosu contain rich microbial content, which participates actively in the fermentation process and contributes significantly to the product sensory quality (Citation2017; Di Cagno et al., Citation2016; Yu et al., Citation2015) and functionality (Bansal, Mangal, Sharma, & Gupta, Citation2016). However, to our knowledge, no published work has yet focused on describing the microbial community of jiaosu. The microbiota profile of a fermented product does not only reveal its potential functions but also information regarding food safety. The application of the PacBio single-molecule, real-time (SMRT) sequencing technology in full-length 16S rRNA profiling has been applied to depict the microbiota composition in different samples, including silage (Bao et al., Citation2016), koji (Hui et al., Citation2017), infant milk powder (Zheng et al., Citation2016), and cheese (Li et al., Citation2017). This technique is advantageous over other currently available strategies in microbiota profiling because of its capacity in producing long sequence reads and thus providing high-resolution taxonomic profiles to the species level.
The current work serves as a pilot study aiming to profile the bacterial microbiota of several commercially available jiaosu products originated from Taiwan and Japan using the full-length 16S rRNA-PacBio SMRT sequencing approach. To the best of our knowledge, this is the first report comprehensively describing the bacterial microbiota of jiaosu products; thus, this report is of interest to both the scientific and industrial sectors.
Materials and methods
Fermented fruit juices
Three fermented fruit juices of different origins were collected. The samples, FY4 and TY8, were produced in Taiwan, while JY1 was originated from Japan ().
Table 1. Sample information.
Tabla 1. Información de las muestras
Extraction of DNA
The DNA was extracted with a modified CTAB method because of the high sugar content in the samples (Clarke, Citation2009). Briefly, 1 mL of sample was transferred to a 10 mL centrifuge tube, snap-frozen with liquid nitrogen, and thawed at 65ºC. The freeze and thaw process was repeated thrice. Then, 3 mL of DNA extraction buffer was added. The DNA extraction buffer was made by dissolving 100 mmol/L Tris-HCI, 100 mmol/L EDTA, 1.5 mol/L NaCl, 1% CTAB, 100 mmol/L Na2HPO4 in double deionized water. Afterward, a solution containing 0.3 mL SDS (10%), 0.7 mL NaCl (4 mol/L), and 20 μL proteinase K (10 μg/mL) was added to the tube with the sample. The mixture was incubated at 50ºC with shaking at 260 rpm/min for 2 h before centrifugation at 8944 g for 10 min. The supernatant was transferred to a fresh centrifuge tube, and DNA was extracted by gently mixing with 300 μL of absolute ethanol and chloroform/isoamyl alcohol (24:1). The supernatant was collected. The extraction step was repeated twice, and the supernatants were pooled. Two volumes of absolute ethanol and 0.1 volume of sodium acetate were added to precipitate the DNA for 30 min. The precipitated DNA was then collected by centrifugation (12,879 g) for 10 minutes, washed twice with 70% ethanol, vacuum dried, and dissolved in 10–20 μL Tris-EDTA buffer. The quality of DNA was checked with a spectrophotometer to ensure an optical density 260nm/280nm ratio of 1.8–2.0 and on an agarose gel. The DNA was stored at −20ºC before polymerase chain reaction (PCR).
Amplification of 16S rRNA and PacBio SMRT sequencing
The bacterial microbiota was profiled by amplifying the 16S rRNA by PCR before barcoded SMRT sequencing. The universal primers targeting to the full-length 16S rRNA, 27F (5ʹ-AGAGTTTGATCGTGGCTCA-3ʹ) and reverse 1492R (5ʹ-ACCTTGTTACGACTT-3ʹ), were used for PCR amplification (Lane, Citation1991). The PCR reaction mix contained a total of 50 µL, containing 5 µL of 10x EasyTaq buffer, 4 µL of 2.5 mM dNTPs, 1 µL each of the 10 µM forward and reverse primers, 0.5 µL of EasyTaq DNA polymerase (final concentration of 2.5 units), 100 ng of template DNA, and 37 µL double deionized water (reagents purchased from TransGen Biotech, Beijing, China). The PCR program conditions were as follows: a first step of initial denaturation at 95ºC for 5 min; 30 cycles of denaturation at 95ºC for 1 min, annealing at 60ºC for 45 s, and extension at 72ºC for 2 min; and a final extension step at 72ºC for 10 min. Amplicons of anticipated size were purified and quantified by the Agilent DNA1000 Kit and the Agilent 2100 Bioanalyzer (Agilent Technologies) according to the instructions of the manufacturer. The SMRT sequencing was performed with P6-C4 chemistry on a PacBio RS II instrument (Pacific Biosciences).
Sequence processing and analysis
The protocol RS_ReadsOfinsert.1 within the PacBio SMRT Portal version 2.7 was used for raw sequence processing. The raw sequence filtering parameter setup was according to a previous study (i.e. a minimum full pass and a minimum predicted accuracy of 5 and 90, respectively; a minimum and maximum read lengths of inserts of 1400 and 1800, respectively) (Hou et al., Citation2015). Mainly, the Quantitative Insights In to Microbial Ecology pipeline (QIIME, version 1.7) was used for extracting high-quality sequences (Caporaso, Kuczynski et al., Citation2010). The software, PyNAST and UCLUST, were applied to cluster the high-quality sequences under 100% similarity to obtain representative sequences (Caporaso, Bittinger et al., Citation2010; Edgar, Citation2010). Representative unique sequences were further classified into operational taxonomic units (OTUs) using UCLUST with a cut-off value of 98.6% (Lozupone & Knight, Citation2005). ChimeraSlayer was applied to remove chimeric sequences in the OTUs dataset (Haas et al., Citation2011). The Ribosomal Database Project (RDP-II) classifier with a minimum bootstrap threshold of 80% was used to assign OTUs taxonomically (Cole et al., Citation2007). Data are presented in mean ± standard error of the mean (SEM). The alpha diversity was evaluated by calculating the Shannon, Simpson’s, chao1, and number of observed OTUs. The Venn diagrams were generated by jvenn (Bardou, Mariette, Escudié, Djemiel, & Klopp, Citation2014).
Data availability
The sequence data reported in this study have been deposited in the Metagenomic Rapid Annotations using Subsystems Technology (MG-RAST) database (Project number: mgp83646).
Results
Sequencing depth and alpha-diversity
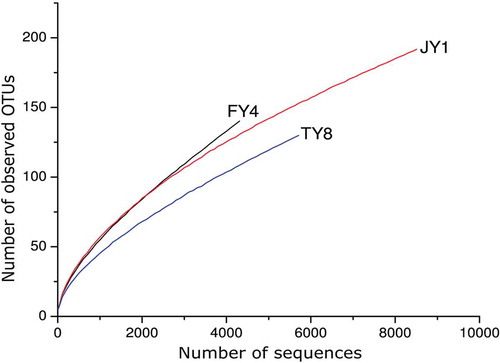
The number of high-quality sequences obtained after being processed with the QIIME pipeline was 4400, 5729, and 8624 for the samples FY4, TY8, and JY1, respectively. The Shannon diversity index curves of all three samples reached plateau (Figure S1), while rarefaction curves did not saturate (), suggesting most microbial diversity was already captured, although new phylotypes might still be identified with further sequencing. The bacterial richness in the samples was evaluated with the Shannon, Simpson’s, chao1 indexes, as well as the number of observed species at the cut-off at 4310 sequences per sample ().
Table 2. Alpha-diversity of samples.
Tabla 2. Diversidad alfa de las muestras.
Figure 1. Rarefaction curves of samples.
Figura 1. Curvas de rarefacción de las muestras
Bacterial distribution at different taxonomic levels
Bacterial sequences from all three samples distributed across seven different phyla (, Supplementary Table S1), dominated by Proteobacteria (98.57% ± 0.46%) and followed by Firmicutes, Actinobacteria, Bacteroidetes, Tenericutes, Deinococcus-Thermus, and Armatimonadetes (each contributed to less than 1% of the total sequences). A minute proportion (0.04% ± 0.01%) of sequences could not be assigned to the phylum level ().
Figure 2. Relative abundances of bacterial taxa at phylum (A), genus (B), and species (C) levels. Taxa comprising <0.05% of the total sequences are grouped as ‘others.’
Figura 2. Abundancia relativa de taxones bacterianos a nivel de filo (A), género (B) y especie (c). Los taxones que comprenden <0.05% del total de secuencias están agrupados como others
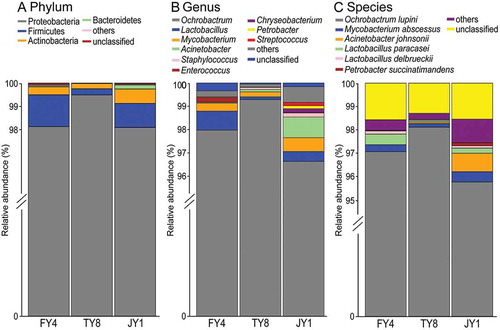
A total of 43 genera were identified across the three samples (, Supplementary Table S1). Thirty-four and nine of these genera each comprised over and below 0.05% of the total sequences, respectively. The leading genus was Ochrobactrum (97.96% ± 0.77%), followed by the much less prevalent genera of Lactobacillus (0.46% ± 0.2%), Mycobacterium (0.38% ± 0.11%), and Acinetobacter (0.34% ± 0.27%). Genera that comprised less than 0.1% of the total sequences (in descending order of relative abundance) included Staphylococcus, Enterococcus, Chryseobacterium, Petrobacter, Streptococcus, Pseudomonas, Brevundimonas, Jeotgalicoccus, Schlegelella, Clostridium, Turicibacter, Janthinobacterium, Massilia, Atopostipes, Bacteroides, Acidovorax, Belnapia, Deinococcus, Pantoea, Rhodobacter, Escherichia/Shigella, Exiguobacterium, Pediococcus, Prevotella, Rubellimicrobium, Ruminococcus, Curvibacter, Desulfovibrio, Oscillibacter, Propionibacterium, Allobaculum, Arthrobacter, Bacillus, Facklamia, Fimbriimonas, Lysobacter, Ornithinimicrobium, Serratia, and Sphingomonas. A low proportion (0.19% ± 0.08%) of sequences could not be assigned to the genus level ().
At the species level, a total of 55 species were identified (, Table S1). Six of them had a relatively high abundance of over 0.05%, including Ochrobactrum lupini (96.98% ± 0.68%), Mycobacterium abscessus (0.30% ± 0.08%), Acinetobacter johnsonii (0.29% ± 0.25%), Lactobacillus paracasei (0.25% ± 0.11%), Lactobacillus delbrueckii (0.09% ± 0.03%), and Petrobacter succinatimandens (0.05% ± 0.04%). A low proportion (1.47% ± 0.09%) of sequences could not be assigned to the species level ().
Core microbiota of the samples
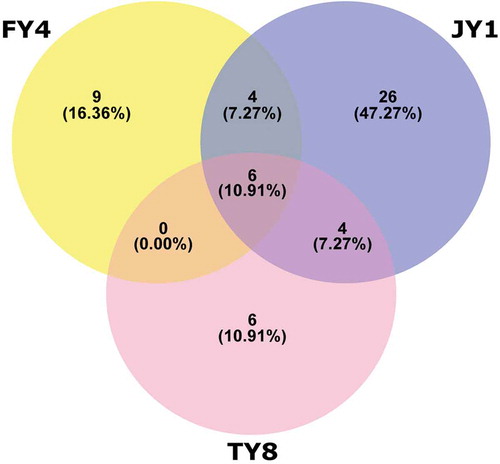
The core microbiota was defined as the bacterial population that was shared across all three samples. This common population distributed across six species, i.e. Ochrobactrum lupini, Lactobacillus paracasei, Mycobacterium abscessus, Lactobacillus delbrueckii, Staphylococcus saprophyticus, and Acinetobacter lwoffii (, ).
Table 3. Core bacterial microbiota at species level.
Tabla 3. Principales microbiotas bacterianas a nivel de especie.
Figure 3. Venn diagram illustrating bacterial distribution at species level.
Figura 3. Diagrama de Venn que muestra la distribución bacteriana a nivel de especie.
Lactobacillus subpopulation in the samples
The genus Lactobacillus plays a key role in food fermentation, and thus we also listed the Lactobacillus subpopulation composition of the samples (). Ten different Lactobacillus species were detected altogether, representing an average relative abundance of 0.45% ± 0.20% of the total sequences. Only two Lactobacillus species were shared across all three samples, which were L. paracasei and L. delbrueckii (average relative abundance of 0.25% ± 0.11% and 0.09% ± 0.03%, respectively).
Table 4. Composition of Lactobacillus subpopulation at species level.
Tabla 4. Composición de la subpoblación de Lactobacillus a nivel de especie.
Discussion
Our work collected three jiaosu samples (two from Taiwan and one from Japan) and generated their full-length 16S rRNA profiles. It is interesting to note that although the three samples were of different origins, they shared a high similarity in the overall microbiota structure at the phylum level, dominated by the phylum Proteobacteria and mainly represented by the species Ochrobactrum lupini (comprised 97.06%, 98.11%, and 95.76% of relative abundance for FY4, TY8, and JY1, respectively). Ochrobactrum lupini belongs to α-Proteobacteria, which is associated with nodulation of legumes (Trujillo et al., Citation2005). Thus, it is most likely from plant- or soil-origin in this case. The dominance of Ochrobactrum has not been commonly reported in traditionally fermented dairy foods except for koji (Hui et al., Citation2017). Weber et al. also detected Ochrobactrum lupini in raw milk by a combination of culture-dependent methods and clone libraries (Weber, Geißert, Kruse, & Lipski, Citation2014). Thus, it is not surprising to find this species occasionally in food-related samples.
The core microbes of the three samples were distributed to six different species, i.e. Ochrobactrum lupini, Lactobacillus paracasei, Mycobacterium abscessus, Lactobacillus delbrueckii, Staphylococcus saprophyticus, and Acinetobacter lwoffii. Two of them belonged to the genus Lactobacillus, which plays a major role in fermentation and is generally regarded as ‘good bacteria’. The relative abundance of Lactobacillus sequences was relatively low (comprised 0.82%, 0.12%, and 0.43% in FY4, TY8, and JY1, respectively), which could possibly be underestimated due to PCR bias. Frank et al. found that, depending on the coexisting 16S rRNA present in the samples, PCR primer sequences and conditions, the efficiency of Lactobacillus amplification varied (Frank et al., Citation2008). Nevertheless, our work did detect a rich diversity of ten different Lactobacillus species in the three samples. It is worth noting that sample FY4 was inoculated with three species of lactic acid bacteria (L. fermentum, L. acidophilus, and L. plantarum) according to the information provided by the supplier; however, none of these species were detected in the analysis. It seems that the inoculated lactobacilli species did not proliferate in the fermentation process. Alternatively, they might have proliferated at the beginning but were overtaken by other coexisting microorganisms during later stages of the ecological succession in the dynamic fermentation environment. The exact reasons remain to be further explored.
Another interesting aspect was that both FY4 and JY1 contained seven different Lactobacillus species, which were apparently more diverse compared with sample TY8 (only three different Lactobacillus species). Both FY4 and JY1 were products purchased online from well-developed companies specialized in commercial production of jiaosu, while sample TY8 was purchased from a local household. The variation in the Lactobacillus diversity might reflect intrinsic differences in the fermentation practice and production environment between well-established manufacturers and local households. Several studies suggested that regional variation exists in the microbiota composition of fermented foods (Liu et al., Citation2015; Nam, Park, & Lim, Citation2012; Sun et al., Citation2014; Zhong et al., Citation2016). The sample size of this work was rather low; nevertheless, samples were collected from two regions, Taiwan and Japan. Such geographic stratification was not observed in the Lactobacillus profile.
No common food pathogen sequences were identified in any of the samples. Moreover, a low proportion of sequences represented the common skin microflora habitants, including the genera Acinetobacter, Propionibacterium, Pseudomonas, Staphylococcus, and Streptococcus (Cogen, Nizet, & Gallo, Citation2008). The relative abundance of this subpopulation represented 0.05%, 0.23%, and 1.31% of the total sequences in samples FY4, TY8, and JY1, respectively. Among them, of particular interest is the species Acinetobacter lwoffii, which is also one of the identified core microbes. Around 25% of healthy individuals carry Acinetobacter lwoffii as normal flora. However, it is regarded as potential opportunistic pathogen in immunocompromised patients and is associated with nosocomial infections (Regalado, Martin, & Antony, Citation2009).
Food is a source of human exposure to environmental mycobacteria, which was reported in 25 of 121 food samples (20%) (Primm, Lucero, & Falkinham, Citation2004). A low proportion of sequences (0.3%) analyzed in this work represented the species Mycobacterium abscessus. Mycobacterium abscessus is a water borne environmental bacterium; however, it is emerging as one of the multidrug-resistant nontuberculous mycobacteria responsible for different diseases (Novosad, Beekmann, Polgreen, Mackey, & Winthrop, Citation2016). Moreover, a low relative abundance of Escherichia/Shigella-corresponding sequences (0.02%) was detected exclusively in the sample FY4. Escherchia/Shigella is an indicator for fecal contamination, as its natural habitat is animal gastrointestinal tract.
Although these potential pathogens and hygiene indicators comprised only a small fraction of sequences, they might represent low level of environmental contamination during the manufacturing process. These microbes mainly originate from water sources, human skin, and animal gastrointestinal tract. Thus, it would be important for the workers to comply with good manufacturing practices, as well as applying set point and controls of processing and storage areas in the facility to control bacterial growth on the raw food materials, water sources, equipment, and products.
In conclusion, this work generated the full-length 16S rRNA profiles of three jiaosu products of Taiwan and Japan. Although the limited sample size did not allow further statistical analysis, a direct comparison of the bacterial microbiota composition between samples did not suggest region-specific stratification of the microbial community. The current study is limited to sequence-based data; it will enhance the validity of this work in the aspects of food safety assurance and profiling of lactic acid bacteria (particularly potential probiotics) by complementing traditional microbiological cultivation methods and toxicity testing.
Author contribution
LYK wrote the manuscript. DM, JD, and HW performed the experiments. QH analyzed the data. LYK, QH, and HZ designed the study.
Supplemental Material
Download Zip (35.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Bansal, S., Mangal, M., Sharma, S. K., & Gupta, R. K. (2016). Non-dairy based probiotics: A healthy treat for intestine. Critical Reviews in Food Science and Nutrition, 56, 1856–1867.
- Bao, W., Mi, Z., Xu, H., Zheng, Y., Kwok, L. Y., Zhang, H., & Zhang, W. (2016). Assessing quality of Medicago sativa silage by monitoring bacterial composition with single molecule, real-time sequencing technology and various physiological parameters. Sciences Reports, 6, 28358.
- Bardou, P., Mariette, J., Escudié, F., Djemiel, C., & Klopp, C. (2014). jvenn: An interactive Venn diagram viewer. BMC Bioinformatics, 15, 293.
- Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., & Knight, R. (2010). PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics, 26(2), 266–267.
- Caporaso, J. G., Kuczynski, J, Stombaugh, J, Bittinger, K., Bushman, F. D, Costello, E. K., … Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5), 335–336.
- Clarke, J. D. (2009). Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harbor Protocols pdb.prot5177-pdb.prot5177. doi:10.1101/pdb.prot5177
- Cogen, A. L., Nizet, V., & Gallo, R. L. (2008). Skin microbiota: A source of disease or defence?: Skin microbiota. British Journal of Dermatology, 158, 442–455.
- Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., … Tiedje, J. M. (2007). The ribosomal database project (RDP-II): Introducing myRDP space and quality controlled public data. Nucleic Acids Research, 35, D169–D172.
- Di Cagno, R., Filannino, P., & Gobbetti, M. (2017). Lactic acid fermentation drives the optimal volatile flavor-aroma profile of pomegranate juice. International Journal of Food Microbiology, 248, 56–62.
- Di Cagno, R., Filannino, P., Vincentini, O., Lanera, A., Cavoski, I., & Gobbetti, M. (2016). Exploitation of Leuconostoc mesenteroides strains to improve shelf life, rheological, sensory and functional features of prickly pear (Opuntia ficus-indica L.) fruit puree. Food Microbiol, 59, 176–189.
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19), 2460–2461.
- Escudero-López, B., Berná, G., Ortega, Á., Herrero-Martín, G., Cerrillo, I., Martín, F., & Fernández-Pachón, M.-S. (2015). Consumption of orange fermented beverage reduces cardiovascular risk factors in healthy mice. Food and Chemical Toxicology, 78, 78–85.
- Frank, J. A., Reich, C. I., Sharma, S., Weisbaum, J. S., Wilson, B. A., & Olsen, G. J. (2008). Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Applied and Environmental Microbiology, 74, 2461–2470.
- Gouda, M., Moustafa, A., Hussein, L., & Hamza, M. (2015). Three week dietary intervention using apricots, pomegranate juice or/and fermented sour sobya and impact on biomarkers of antioxidative activity, oxidative stress and erythrocytic glutathione transferase activity among adults. Nutritional Journal, 15. doi:10.1186/s12937-016-0173-x
- Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., … Birren, B. W. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21, 494–504.
- Harima-Mizusawa, N., Kano, M., Nozaki, D., Nonaka, C., Miyazaki, K., & Enomoto, T. (2016). Citrus juice fermented with Lactobacillus plantarum YIT 0132 alleviates symptoms of perennial allergic rhinitis in a double-blind, placebo-controlled trial. Beneficial Microbes, 7, 649–658.
- Hou, Q., Xu, H., Zheng, Y., Xi, X., Kwok, L.-Y., Sun, Z., … Zhang, W. (2015). Evaluation of bacterial contamination in raw milk, ultra-high temperature milk and infant formula using single molecule, real-time sequencing technology. Journal of Dairy Science, 98, 8464–8472.
- Hui, W., Hou, Q., Cao, C., Xu, H., Zhen, Y., Kwok, L.-Y., … Zhang, W. (2017). Identification of microbial profile of Koji using single molecule, real-time sequencing technology. Journal of Food Science, 82, 1193–1199.
- Klewicka, E., Zduńczyk, Z., Juśkiewicz, J., & Klewicki, R. (2015). Effects of lactofermented beetroot juice alone or with N-nitroso-N-methylurea on selected metabolic parameters, composition of the microbiota adhering to the gut epithelium and antioxidant status of rats. Nutrients, 7, 5905–5915.
- Lane, D. J. (1991). 16S/23S rRNA sequencing. In E. Stackebrandt & M. Goodfellow (Eds.), Nucleic acid techniques in bacterial systematics (pp. 115–175). New York City, NY: John Wiley and Sons.
- Li, J., Zheng, Y., Xu, H., Xi, X., Hou, Q., Feng, S., … Sun Z (2017). Bacterial microbiota of Kazakhstan cheese revealed by single molecule real time (SMRT) sequencing and its comparison with Belgian, Kalmykian and Italian artisanal cheeses. BMC Microbiol, 17. doi:10.1186/s12866-016-0911-4
- Liu, W., Zheng, Y., Kwok, L.-Y., Sun, Z., Zhang, J., Guo, Z., … Zhang, H. (2015). High-throughput sequencing for the detection of the bacterial and fungal diversity in Mongolian naturally fermented cow’s milk in Russia. BMC Microbiol, 15, 45.
- Lozupone, C., & Knight, R. (2005). UniFrac: A new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology, 71, 8228–8235.
- Nam, Y.-D., Park, S., & Lim, S.-I. (2012). Microbial composition of the Korean traditional food “kochujang” analyzed by a massive sequencing technique. Journal of Food Science, 77, M250–M256.
- Novosad, S. A., Beekmann, S. E., Polgreen, P. M., Mackey, K., & Winthrop, K. L.; M. abscessus Study Team. (2016). Treatment of Mycobacterium abscessus infection. Emergency Infection Diseases, 22, 511–514.
- Primm, T. P., Lucero, C. A., & Falkinham, J. O. (2004). Health impacts of environmental mycobacteria. Clinical Microbiology Reviews, 17, 98–106.
- Regalado, N. G., Martin, G., & Antony, S. J. (2009). Acinetobacter lwoffii: Bacteremia associated with acute gastroenteritis. Travel Medica Infection Diseases, 7, 316–317.
- Simsek, S., El, S. N., Kancabas Kilinc, A., & Karakaya, S. (2014). Vegetable and fermented vegetable juices containing germinated seeds and sprouts of lentil and cowpea. Food Chemistry, 156, 289–295.
- Sun, Z., Liu, W., Bao, Q., Zhang, J., Hou, Q., Kwok, L., … Zhang, H. (2014). Investigation of bacterial and fungal diversity in tarag using high-throughput sequencing. Journal of Dairy Science, 97, 6085–6096.
- Trujillo, M. E., Willems, A., Abril, A., Planchuelo, A.-M., Rivas, R., Ludena, D., … Velazquez, E. (2005). Nodulation of Lupinus albus by strains of Ochrobactrum lupini sp. nov. Applied and Environmental Microbiology, 71, 1318–1327.
- Valero-Cases, E., Roy, N. C., Frutos, M. J., & Anderson, R. C. (2017). Influence of the fruit juice carriers on the ability of Lactobacillus plantarum DSM20205 to improve in vitro intestinal barrier integrity and its probiotic properties. Journal of Agricultural and Food Chemistry, 65, 5632–5638.
- Weber, M., Geißert, J., Kruse, M., & Lipski, A. (2014). Comparative analysis of bacterial community composition in bulk tank raw milk by culture-dependent and culture-independent methods using the viability dye propidium monoazide. Journal of Dairy Science, 97, 6761–6776.
- Yu, Y., Xiao, G., Xu, Y., Wu, J., Fu, M., & Wen, J. (2015). Slight fermentation with Lactobacillus fermentium improves the taste (Sugar: AcidRatio) of citrus (Citrus reticulata cv. chachiensis) juice. Journal of Food Science, 80, M2543–M2547.
- Zheng, Y., Xi, X., Xu, H., Hou, Q., Bian, Y., Yu, Z., … Zhang, H. (2016). Using PacBio long-read high-throughput microbial gene amplicon sequencing to evaluate infant formula safety. Journal of Agricultural and Food Chemistry, 64, 6993–7001.
- Zhong, Z., Hou, Q., Kwok, L., Yu, Z., Zheng, Y., Sun, Z., … Zhang, H. (2016). Bacterial microbiota compositions of naturally fermented milk are shaped by both geographic origin and sample type. Journal of Dairy Science, 99, 7832–7841.