
ABSTRACT
The Rho family small GTPases and their effectors, including PAKs, are extensively studied in the context of the actin cytoskeleton, excitatory synaptic function, spine morphology and memory formation. However, their roles in inhibitory synaptic function remain poorly understood. We have recently shown that PAK1 is a potent regulator of GABAergic synaptic transmission. Thus, disruption of PAK1 leads to significant impairments in inhibitory postsynaptic currents which are manifested as reduced GABA presynaptic releases. Interestingly, this effect of PAK1 is distinct from its previously known role in spines and excitatory synaptic transmission in that it is independent of postsynaptic actin, but requires retrograde messengers produced and released from the postsynaptic neurons to suppress presynaptic GABA releases. We have further identified eCBs as the retrograde messengers and shown that PAK1 regulates the eCB signaling via restricting the tissue level of AEA by promoting synaptic expression of COX-2, a key enzyme to oxidize AEA. These results have established a novel pathway whereby PAK1, and by extension Rho proteins, regulates cellular processes, synaptic function and behaviors and have important implications in understanding and treating various diseases linked to PAKs and Rho signaling.
Introduction
The Rho family GTPases are members of Ras superfamily small GTPases and act as cell signaling transducer in a wide range of cellular activities, including actin cytoskeleton organization, cell polarity, microtubule dynamics, and gene expression.Citation1 Rho proteins are guanine nucleotide binding proteins, cycling between the active GTP bound and inactive GDP bound forms, and this switch process is executed temporally and spatially by ganuine nucleotide exchange factor (GEF), and GTPases activating protein (GAP). Although diverse in function, Rho GTPases, specifically Rac1, Cdc42 and RhoA, are particularly well known as the central regulator of the actin cytoskeleton, and in doing so, they modulate a series of actin-based neural properties such as spine morphology, synaptic plasticity and neuronal migration.Citation2
P21-activated kinases (PAKs) are a family of serine/threonine kinases that are divided to 2 groups according to the structure and activation modes.Citation3,4 The group1 PAKs consisting of PAK1-3 are particularly interesting because their activity can be potently regulated by both active Rac1 and Cdc42, whereas the group II PAKs (PAK4-6) can only be regulated by Cdc42. Accordingly, extensive studies have shown that PAKs are critically involved in the regulation of the actin cytoskeleton, spine morphology, synaptic plasticity and memory formation.Citation5-9 Unfortunately, most of the studies on PAKs have been focused on excitatory synaptic function and their role in inhibitory synaptic transmission remains unknown. Interestingly, PAKs are known to be affected in a number of brain diseases and remarkably PAK1 inhibitors are capable of rescuing some of the key deficits associated with these diseases, including Neurofibromatosis model of autism, Alzheimer disease, Fragile X syndrome and schizophrenia.Citation10-14 Similarly, PAK3 is also lined to intellectual disability.Citation15 Importantly, a common change of these brain disease is altered excitatory/inhibitory (E/I) balance,Citation16 suggesting that PAKs may also be involved in inhibitory function.
Proper E/I balance depends on coordinated excitatory and inhibitory synaptic regulation, and therefore changes in either or both transmission modes would disrupt E/I balance and result in dysfunction of neural system. In the mammalian brain, excitatory synaptic transmission is mainly mediated by glutamate via activating various glutamate receptors, particularly AMPA receptors (AMPARs) and NMDA receptors (NMDARs). In contrast, inhibitory synaptic transmission is largely conducted by GABA via activating GABA receptors that causes hyperpolarization of membrane potential (). Although it is well established that both Rho proteins and PAKs are critical for glutamatergic transmission, whether they are important in GABAergic transmission is yet to be investigated.
Figure 1. PAK1 regulation of excitatory and inhibitory synaptic transmission via distinct mechanisms. At excitatory synapses, PAK1 and PAK3 regulate spine morphology and synaptic plasticity via LIMK/cofilin-mediated actin reorganization, whereas at GABAergic synapses PAK1 promotes neurotransmitter release by restricting the level of the retrograde messenger eCB, AEA, through recruiting COX-2 at the synapse.
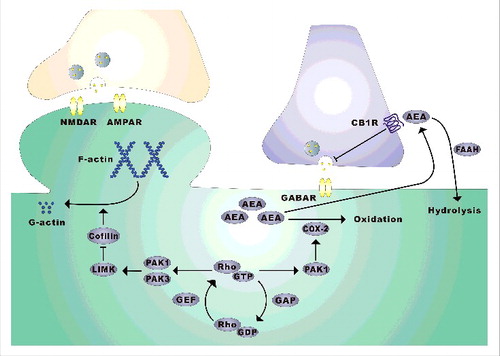
One of the key systems that is likely important to maintain E/I balance is the endocannabinoids (eCBs), the endogenous homolog of ΔTHC, which is the active ingredient in marijuana.Citation17 These small lipid molecules, which are synthesized and secreted from postsynaptic neurons to suppress presynaptic neurotransmitter release via CB1 receptors, have been demonstrated as key retrograde messengers to modulate both excitatory and inhibitor neurotransmission.Citation18 Importantly, altered eCB signaling has been found to be linked to a wide range of brain diseases, including autism and fragile X syndrome.Citation19,20
In the study reported in eLife,Citation21 we systematically investigated the role of PAK1, a key member of the PAK family proteins that is highly expressed in the brain, in the regulation of GABA transmission. We show that PAK1 is a potent, positive regulator of GABA transmission. Surprisingly, we found that this effect of PAK1 is independent of actin regulation, but instead through restricting eCB signaling. These findings established a novel signaling process by which PAK1 and Rho GTPases regulate brain function and dysfunction.
Results
We started to address the role of PAK1 in inhibitory synaptic transmission by comparing E/I balance between PAK1 KO mice and wild type (WT) using electrophysiological recordings of synaptic currents from CA1 pyramidal neurons of the hippocampus, a key brain region critical for memory formation. We found that the E/I ratio was significantly increased in PAK1 KO mice compared to WT, and this increase in E/I ratio was due to a specific reduction in GABAergic synaptic transmission. The impaired GABAergic transmission in PAK1 KO mice was not likely caused by developmental perturbations because acute application of the PAK1 inhibitors (IPA3 or peptides) directly into CA1 neurons of hippocampal slices had similar effects on inhibitory post synaptic currents (IPSCs). These inhibitor experiments also indicate that inhibition of postsynaptic PAK1 was sufficient to reduce IPSCs, suggesting that CA1 neurons are the primary source of PAK1 to exert this effect.
To probe the nature of the changes in IPSCs, we recorded spontaneous and miniature IPSCs (s/mIPSC). We found that only frequency, but not the amplitude, of s/mIPSC was reduced in PAK1 KO neurons. Importantly, postsynaptic infusion of the PAK1 inhibitor IPA3 also reduced the frequency, but not the amplitude of s/mIPSCs, again supporting that the effect of PAK1 was not related to development and that postsynaptic neurons are the primary source of PAK1. Although reduced frequency in s/mIPSCs may also result from postsynaptic changes (e.g. silencing of GABA receptors or elimination of the synapse), our data that none of these factors were altered in PAK1 KO mice strongly suggested that presynaptic changes may be responsible for the reduced s/mIPSCs. Therefore, we concluded that retrograde messengers must be involved in mediating the effect of PAK1 to reduce presynaptic GABA release.
It is known that eCBs released from postsynaptic neurons potently suppress GABA transmission and they do so by activating presynaptic CB1 receptors.Citation18,22 Therefore, it is reasonable to hypothesize that the effect of PAK1 may be mediated by eCB signaling. Consistent with hypothesis, we showed that application of the CB1 receptor antagonist AM251 increased IPSCs in WT CA1 neurons, presumably due to the blockade of the suppressing effect of eCBs on GABA release, and that this AM251 induced increase in IPSCs was dramatically elevated (by 2-folds) in PAK1 KO mice. These results suggested that the eCB signaling was enhanced by PAK1 disruption. Interestingly, we found no differences in either total or synaptic level of CB1 receptor protein, and importantly application of the CB1 receptor agonist WIN, designed to activate the presynaptic CB1 receptors, inhibited IPSCs equally well in both WT and PAK1 KO mice. These results suggested that the enhanced eCB signaling is likely caused by elevated levels of eCBs, but not by alterations in CB1 receptors and their downstream processes. Indeed, we showed that the hippocampal tissue level of anandamide (AEA), but not 2-arachidonoylglycrol (2-AG), was higher in PAK1 KO mice compared to WT controls. Therefore, we concluded that disruption of PAK1 leads to elevated AEA to suppress GABA release, resulting in reduced IPSCs. Thus, we have identified PAK1 as a potent kinase to restrict tonic AEA in the brain.
To elucidate how PAK1 regulates the AEA level in the brain, we analyzed various enzymes involved in eCB metabolic pathwayCitation18 and found that only cyclooxygenase-2 (COX-2) expression, a non-canonical but relevant pathway in the degradation of eCBs, was reduced in PAK1 KO hippocampus. In addition, the reduction in COX-2 appears to be specific to GABAergic synapse, providing a potential mechanism for the selective effect of PAK1 disruption on inhibitory transmission, but not on excitatory strength. To directly investigate whether COX-2 was responsible for the PAK1 action on IPSCs, we tested the effect of the COX-2 inhibitor Nimesulide and found that Nimesulide depressed IPSCs in WT neurons, likely due to increased eCB production, and importantly this Nimesulide induced synaptic depression was reduced in PAK KO mice. These results were consistent with the hypothesis that the effect of PAK1 is mediated by COX-2. To further confirm that the effect of COX-2 is through AEA, but not 2-AG, we used inhibitors specific to AEA ( i.e. FAAH inhibitor URB597) and 2-AG hydrolysis (i.e., MGL inhibitor JZL184), and found that ability of URB597, but not of JZL184, to reduce IPSCs was impaired in PAK1 KO mice compared to WT mice. Therefore, we conclude that the reduced GABA transmission in PAK1 KO mice is caused by reduced COX-2 expression and consequential elevation in AEA, but not 2-AG, specifically at GABAergic synapses.
Significance and discussion
Although Rho GTPases and downstream effectors have been extensively studied in the regulation of the actin cytoskeleton and excitatory synaptic function in the brain, their involvement in inhibitory transmission is poorly investigated. Our study presented in eLifeCitation21 have provided important insight on this aspect (). First, we revealed that disruption of PAK1, either genetically or pharmacologically, results in profound impairments in on GABAergic synaptic transmission. This effect occurs in the absence of any significant changes in excitatory synaptic strength, indicating that PAK1 is a potent, positive regulator of tonic inhibitory transmission. This specific effect of PAK1 on GABA transmission is surprising to us because dominant negative forms of PAK1 have been previously shown to affect spine, synaptic plasticity and memory formation.Citation6-9 One possible explanation for this apparent discrepancy is that the expression of the mutant PAK1 may also disturb other members of the PAK family. This is consistent with the observations that double KO mice lacking both PAK1 and PAK3, but not single KO lacking either PAK1 or PAK3, display severe deficits in spines and the actin cytoskeleton.Citation8 These results imply that while PAK1 and PAK3 are functionally redundant at the excitatory synapse, the role of PAK1 in inhibitory synaptic regulation is unique and/or particularly sensitive to perturbations that cannot be compensated by other members of the PAK family. The second key finding of the study is that the effect of PAK1 at the GABAergic synapse is independent of the actin cytoskeleton, but requires eCBs as retrograde messengers to suppress neurotransmitter release. To our knowledge, this is the first evidence to show that Rho signaling is involved in the regulation of the eCB system. eCBs are well known neuromodulators that can regulate a wide range of brain functions and dysfunctions, including synaptic plasticity, learning and memory, stress and addiction.Citation17,23-25 Therefore, the discovery that the eCB signaling is an effector of the Rho pathways may provide an entirely new mechanism by which Rho GTPases regulate brain function. Another exciting finding of the study is that disruption of PAK1 reduces the level of AEA, but not 2-AG, and that this effect is mediated by COX-2. This is significant because although the eCB systems, particularly their metabolic processes, have been extensively studied,Citation18 how these processes are molecularly regulated remains poorly understood. Given that Rho GTPases and PAKs can be activated by various receptors and neuronal activities,Citation1,3,4 they may act as key molecules to modulate eCB metabolism in response to brain activities and plasticity. The selective regulation of AEA by PAK1 highlights complex processes that govern the eCB signaling and indicates that additional regulatory pathways must exist to control the level of eCBs.
Clinical implications and future studies
The discovery that PAK1 regulates inhibitory synaptic strength by restricting tonic eCB signaling has a number of important implications with respect to brain disorders. It is known that PAKs are linked to a number of neurological and mental disorders and that PAK1 inhibitors are effective in ameliorating symptoms in several animal models of these diseases including autism and schizophrenia.Citation11-14 Our results that both GABA and eCB systems are target of PAK1 suggest that changes in these systems may be responsible for the pathogenic processes of PAK1-linked diseases. Indeed, alterations in GABA transmission and eCB signaling pathways are associated with a wide range of brain disorders.Citation26-29 In addition to PAKs, many other components involved in Rho signaling have also been reported to be genetically linked to neurodevelopmental and neuropsychiatric disorders. Although previous studies show that the actin cytoskeleton at the spine and excitatory synapse is a major target of Rho proteins,Citation30 our new findings suggest that GABA and eCB systems may also play an important role, and therefore the drugs that target GABA and eCB systems should be useful to treating Rho-related diseases. Finally, because PAKs, Rho proteins and eCBs are all expressed in other organs throughout the body, our findings may provide a unified mechanism for the understanding and treatment of other diseases, including inflammation, allergies and cancers. Therefore, detailed elucidation of PAK1-COX-2-AEA pathway will be of great importance in understanding the role of Rho GTPases and the eCB system in both health and diseases. Future studies include analysis of mechanisms by which PAK1 recruits COX-2 at the synapse, the role of other PAK family members in eCB regulation, and behavioral effects of the drugs that target COX-2 and the eCB system in models of brain and other diseases.
Additional information
Funding
References
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420(6916):629-35; PMID:12478284; https://doi.org/10.1038/nature01148
- Ba W, van der Raadt J, Kasri NN. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp Cell Res 2013; 319(15):2368-74; PMID:23769912; https://doi.org/10.1016/j.yexcr.2013.05.033
- Eswaran J, Soundararajan M, Kumar R, Knapp S. UnPAKing the class differences among p21-activated kinases. Trends Biochem Sci 2008; 33(8):394-403; PMID:18639460; https://doi.org/10.1016/j.tibs.2008.06.002
- Bokoch GM. Biology of the p21-activated kinases. Ann Rev Biochem 2003; 72(1):743-81; PMID:12676796; https://doi.org/10.1146/annurev.biochem.72.121801.161742
- Zhao Z-S, Manser E. PAK family kinases: Physiological roles and regulation. Cell Logist 2012; 2(2):59-68; PMID:23162738; https://doi.org/10.4161/cl.21912
- Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW, Jia Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 2009; 56(1):73-80; PMID:18644395; https://doi.org/10.1016/j.neuropharm.2008.06.055
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron 2004; 42(5):773-87; PMID:15182717; https://doi.org/10.1016/j.neuron.2004.05.003
- Huang W, Zhou Z, Asrar S, Henkelman M, Xie W, Jia Z. p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol Cell Biol 2011; 31(3):388-403; PMID:21115725; https://doi.org/10.1128/MCB.00969-10
- Meng J, Meng Y, Hanna A, Janus C, Jia Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci 2005; 25(28):6641-50; PMID:16014725; https://doi.org/10.1523/JNEUROSCI.0028-05.2005
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011; 70(5):898-907; PMID:21658583; https://doi.org/10.1016/j.neuron.2011.05.021
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A 2007; 104(27):11489-94; PMID:17592139; https://doi.org/10.1073/pnas.0705003104
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, Tonegawa S. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci U S A 2013; 110(14):5671-6; PMID:23509247; https://doi.org/10.1073/pnas.1219383110
- Molosh AI, Johnson PL, Spence JP, Arendt D, Federici LM, Bernabe C, Janasik SP, Segu ZM, Khanna R, Goswami C, et al. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat Neurosci 2014; 17(11):1583-90; PMID:25242307; https://doi.org/10.1038/nn.3822
- Hayashi-Takagi A, Araki Y, Nakamura M, Vollrath B, Duron SG, Yan Z, Kasai H, Huganir RL, Campbell DA, Sawa A. PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proc Natl Acad Sci U S A 2014; 111(17):6461-6; PMID:24706880; https://doi.org/10.1073/pnas.1321109111
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet 1998; 20(1):25-30; PMID:9731525
- Eichler SA, Meier JC. EI balance and human diseases-from molecules to networking. Front Mol Neurosci 2008; 1:2; PMID:18946535; https://doi.org/10.3389/neuro.02.002.2008
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron 2012; 76(1):70-81; PMID:23040807; https://doi.org/10.1016/j.neuron.2012.09.020
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 2003; 4(11):873-84; PMID:14595399; https://doi.org/10.1038/nrn1247
- Foldy C, Malenka RC, Sudhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013; 78(3):498-509; PMID:23583622; https://doi.org/10.1016/j.neuron.2013.02.036
- Jung K-M, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, Ginger M, Frick A, DiPatrizio NV, Mackie K, et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun 2012; 3:1080; PMID:23011134; https://doi.org/10.1038/ncomms2045
- Xia S, Zhou Z, Leung C, Zhu Y, Pan X, Qi J, Morena M, Hill MN, Xie W, Jia Z. p21-activated kinase 1 restricts tonic endocannabinoid signaling in the hippocampus. eLife 2016; 5:e14653; PMID:27296803; https://doi.org/10.7554/eLife.14653
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 2012; 35:529-58; PMID:22524785; https://doi.org/10.1146/annurev-neuro-062111-150420
- Morena M, Patel S, Bains JS, Hill MN. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 2016; 41(1):80-102; PMID:26068727; https://doi.org/10.1038/npp.2015.166
- Zanettini C, Panlilio LV, Alicki M, Goldberg SR, Haller J, Yasar S. Effects of endocannabinoid system modulation on cognitive and emotional behavior. Front Behav Neurosci 2011; 5:57; PMID:21949506
- Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci 2015; 16(10):579-94; PMID:26373473
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008; 135(3):549-60; PMID:18984165; https://doi.org/10.1016/j.cell.2008.09.060
- Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci 2010; 30(29):9929-38; PMID:20660275; https://doi.org/10.1523/JNEUROSCI.1714-10.2010
- Radhu N, Garcia Dominguez L, Farzan F, Richter MA, Semeralul MO, Chen R, Fitzgerald PB, Daskalakis ZJ. Evidence for inhibitory deficits in the prefrontal cortex in schizophrenia. Brain 2015; 138(2):483-97; PMID:25524710; https://doi.org/10.1093/brain/awu360
- Chao H-T, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010; 468(7321):263-9; PMID:21068835; https://doi.org/10.1038/nature09582
- Govek EE, Hatten ME, Van Aelst L. The role of Rho GTPase proteins in CNS neuronal migration. Dev Neurobiol 2011; 71(6):528-53; PMID:21557504; https://doi.org/10.1002/dneu.20850