
Abstract
Cellular quiescence (G0) is a sleep-like cellular state that allows cells to maintain the ability to re-enter and exit the proliferative cycle. Quiescent cancer cells are considered a therapeutic challenge since they are often resistant to conventional chemoradiotherapy resulting in disease progression and relapse. To date, the majority of published studies have focused on identifying molecules that target proliferating tumor cells while limited information is available on methods to target quiescent cancer cells. This review provides a summary of the relevant molecular mechanisms underlying quiescence maintenance and pharmacological interventions aimed at either eliminating this cancer cell subpopulation or indefinitely maintaining the quiescence state. Furthermore, the irradiation responses of quiescent cancer cells, in particular, the influence of manipulating intratumor hypoxia and radiation properties on radiosensitivity, are discussed. The main aim of this article is to provide a theoretical basis for the effective targeted killing of dormant tumor cells.
Quiescent tumor cells represent a clinical problem
Quiescence is defined as a reversible cellular state from which the cells are able to re-enter the proliferative cycle in response to certain promitogenic factors, including cell-extrinsic environmental signals (e.g. injury or tumor acidification) and cell-intrinsic regulatory mechanisms [Citation1,Citation2]. Scientific literature to date supports the existence of quiescent cancer cells in many tumor types, including breast, liver and pancreatic cancer, acute myeloid leukaemia, melanoma, and glioblastoma [Citation3,Citation4]. In earlier studies, cells were induced to quiescence via serum starvation (whereby cells at low density were grown in serum-free medium) or contact inhibition (cells were grown in 10% serum until complete confluence) [Citation5–7]. Quiescent cells were confirmed based on the lack of expression of Ki67 (a cell proliferation marker), negative EDU incorporation [Citation7] or low rate of BrdU incorporation [Citation3], and low Erk 1/2: p38 MAPK ratio [Citation6–8]. Moreover, mVenus-p27K−, a fusion protein composed of the fluorescent protein mVenus and a defective mutant of p27 combined with Fucci probe could effectively recognize and distinguish cells at G0 from those at G1 during the G0-G1 transition state [Citation9,Citation10].
Quiescent cells initiate a series of programs that protect against cellular stress and toxicity. Although the protected state is favorable for long-term maintenance of the quiescent state, activation of these pathways in tumor cells may play a crucial role in higher resistance to conventional cancer therapies that largely target proliferating cells [Citation3,Citation5,Citation11,Citation12]. Quiescent tumor cells are considered significantly less radio/chemosensitive with a greater repair capacity than cycling cells [Citation11,Citation13,Citation14]. Resistance of G0 cells from breast, pancreatic, ovarian tumors and squamous cell carcinomas to traditional therapeutic patterns has been reported [Citation3,Citation15]. In addition, quiescent mesenchymal stem cells are more refractory to heat stress (HS) than growing cells. Quiescent cells exposed to HS show no alterations in reactive oxygen species (ROS) production but the genes involved in antioxidant defense are mostly silenced [Citation16]. Therefore, the development of new and effective therapeutic strategies that target both quiescent and proliferating tumor cells remains an urgent medical requirement.
Relevant molecular mechanisms (gene signatures) of tumor cell quiescence
The majority of cells from both unicellular to multicellular organisms spend part of their lifetime in quiescence, a temporary nonproliferating state [Citation17]. As a single-cell eukaryote, yeast presents an excellent model organism to investigate cell division and quiescence [Citation18,Citation19]. The advantage of studying quiescence in free-living, unicellular organisms is the lack of intervention or genetic manipulation required to track transition in or out of the quiescent state [Citation20]. S-adenosylmethionine synthetase (Sam1), a family of SHK gene products, is implicated in cell growth and proliferation as well as maintenance and termination of the G0 phase in fission yeast [Citation19]. Meanwhile, ssd1 and the cell wall integrity pathway promote entry, maintenance, and recovery from the quiescent state in budding yeast [Citation20]. To date, studies on Saccharomyces cerevisiae are broadly responsible for demonstrating the functional importance of nuclear organization. Previous research has shown that following carbon source exhaustion, the silencing factor Sir3 drives the telomeres of G0 cells to form a discrete, large cluster (hyper-cluster) at the center of the nucleus, resulting in spatial reorganization of the genome that favors long-term survival during quiescence. Notably, ROS produced by mitochondrial activity commit cells to form a telomere hypercluster upon starvation and sustain longevity of G0 cells [Citation21].
The fission yeast Schizosaccharomyces pombe (S. pombe) is an ideal model species for studying the quiescence phase of mammalian cancer cells owing to easy control of the switch between proliferative and G0 phases based on the availability of nitrogen in the media, simple gene editing, and proteins with high homology to their counterparts in mammalian species [Citation22,Citation23]. An accepted finding is that quiescent cells must sustain mitotic competence (MC) to re-enter the cell cycle. Researchers have identified 85 genes essential for fission yeast to maintain MC during the quiescent phase induced by nitrogen deprivation. Notably, nearly half of these genes are associated with cancer, suggesting a close link between the maintenance of MC in G0 cells and malignant division of cancer cells. In addition, protein phosphatases and related regulators are closely associated with maintenance of MC. In particular, Nem1-Spo7 and Ned1 function as a regulator and target, respectively. Nem1-Spo7 plays a critical role in protecting the nucleus from autophagy through manipulation of Ned1 [Citation23,Citation24]. These genes constitute the landscape of genetic regulation of MC, offering a possible therapeutic approach against cancer progression. Data from the collective shuttle studies between yeast and mammalian cells should facilitate clarification of the molecular mechanisms underlying tumorigenesis and improve screening for antitumor targets and drugs.
Similar to the induction of the G0 phase in proliferative cells of fission yeast, cultured mammalian cells can be induced to enter the G0 phase in response to contact inhibition or serum starvation [Citation24]. Importantly, the endless diversity of cells and habitats excludes the existence of a universal quiescence program [Citation17]. Previous studies have identified embryonic stem cell-expressed RAS (ERAS) specifically expressed in quiescence hepatic stellate cells (HSCs) and significantly downregulated during HSC activation owing to an increase in promotor DNA methylation. ERAS targets AKT through two different pathways driven by PI3K and mTORC2 in G0 cells whereas in activated HSCs, RAS signaling is transferred to RAF-MEK-ERK [Citation25]. ERAS is thus responsible for the maintenance of quiescence in HSCs. Another earlier study reported that quiescence is regulated by TGF-β-dependent signature genes, which drives the chemoresistance of tumor-propagating cells (TPC) in squamous cell carcinoma. TGF-β inhibition enhances the chemosensitivity of TPCs through preventing entry into the G0 phase [Citation15]. Dynamic changes in expression of the chemokine receptor, CXCR4, in tumor cells are associated with entry and exit of dormancy. Downregulation of CXCR4 in metastasized breast cancer cells is related to their dormancy [Citation26]. In the human mammary epithelial cell line, MCF10A, unresolved DNA replication errors in mother cells are transmitted to daughter cells, causing p21-dependent entry of daughter cells into quiescence after mitosis with a duration related to the degree of inherited damage [Citation27]. The quiescence state is further associated with a signature of Wnt pathway genes in myogenic cells. Induction of Rgs2 and Dkk3, the quiescence-induced regulators of canonical Wnt signaling, in G0 is necessary for clonogenic self-renewal [Citation28]. Emerging evidence suggests that cancer cells under duress enter dormancy after cannibalism of mesenchymal stem/stromal cells [Citation29]. The quiescence-related molecules and their functional mechanisms are listed in . Clarification of the molecular mechanisms that control the quiescent cell state may open up a new paradigm for specifically targeting and eliminating quiescent cancer cells, potentially leading to significantly improved survival prospects.
Table 1. Molecular mechanisms (gene signatures) of quiescence maintenance.
Treatment strategies targeting quiescent cancer cells
In the face of numerous drug attacks, some cancer stem cells (CSCs) enter a dormant state, thereby escaping the drug killing effect. Three distinct approaches to therapeutic interventions are suggested: (i) to allow cells to remain dormant indefinitely in a “harmless” state, (ii) to reactivate dormant cells and increase their sensitivity to anti-proliferative drugs, and (iii) to eradicate dormant cells [Citation12,Citation31].
Strategies to keep quiescence tumor cells “asleep”
Antibodies against NET-remodeled laminin
Therapies designed to prevent the awakening of dormant cells have the potential to improve the survival prospects of cancer patients. Researchers have identified a new pathway that causes dormant cancer cells to reawaken and metastasize. Albrengues et al. [Citation39] proposed that persistent lung inflammation and concomitant formation of neutrophil extracellular traps (NET) stimulate dormant cells to exit quiescence. NETs concentrate neutrophil proteases (neutrophil elastase and matrix metalloproteinase 9) at their substrate protein laminin, inducing sequential proteolytic laminin remodeling and activation of integrin α3β1 signaling in cancer cells. Antibodies against NET-remodeled laminin could effectively block the awakening of dormant cancer cells (). However, this study focused on animal experiments. Further experimental data are required for validation of these findings, along with identification of the corresponding drug carrier.
Figure 1. NET produced during inflammation awakens dormant tumor cells.
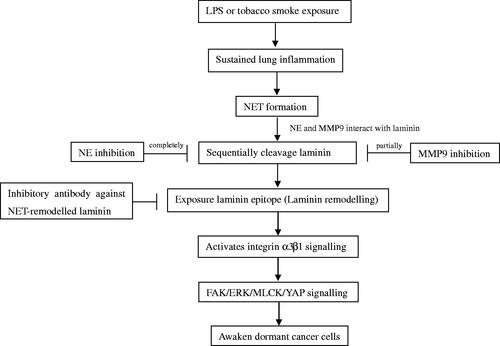
Figure 2. A regulatory network affecting the depth of quiescence.
Inhibitors of histone acetyltransferases KAT6A/B
Lysine acetyltransferase (KAT)-mediated histone acetylation plays a crucial role in chromatin organization and function. KAT6A and KAT6B, the targets of chromosomal translocation, have been identified in various cancer types, supporting the feasibility of suppressing the expression and activity of these molecules as a therapeutic strategy for cancer. Inhibitors of KAT6A/B are reported to induce senescence and cell cycle exit [Citation40,Citation41]. For instance, WM-8014 and WM-1119 suppress tumor gene transcription by blocking histone acetylation so that cancer cells fall into a state of permanent senescence without damaging normal cells, thus minimizing the toxic side effects of treatment [Citation40].
Efipladib
Cytosolic phospholipase A2 alpha (cPLA2α) is critical for regulating cell cycle re-entry in quiescent cancer cells. In quiescent endothelial cells, cPLA2 is inactivated via sequestration at the Golgi apparatus and subsequently released and activated in proliferating cells [Citation42]. cPLA2α inhibition is indispensable for impeding quiescent prostate cell re-entry into the cell cycle. Earlier studies have indicated that Efipladib, a selective inhibitor of cPLA2α, suppresses accumulation of cyclin/CDK and phosphor-pRb during cell cycle entry. Simultaneously, inhibition of cPLA2α upon cell cycle entry prevents recovery of the components involved in DNA synthesis and Skp2 while retaining p27 [Citation6]. Other studies suggest that diminished cPLA2α activity leads to lower cyclin D1, higher p27Kip1, and a remarkable decrease in kinase activity related to Cdk4 [Citation43]. Meanwhile, ablation of cPLA2α markedly improves chemosensitivity in cervical cancer via inhibiting β-catenin signaling [Citation44]. Collectively, our data suggest that targeting of cPLA2α provides an effective treatment modality for preventing the progression and recurrence of cancer.
Other pathways
Dormant cells are characterized by increased p38 MAPK and decreased ERK1/2 activities, and the p38 MAPKhigh/ERKlow phenotype is widely used as a marker of the dormant state [Citation31]. Therefore, strategies for inducing and/or maintaining the p38 signaling pathway in dormant disseminated tumor cells (DTC) should theoretically lead to sustenance of tumor dormancy [Citation45]. Conversely, suppression of MAPK signaling and urokinase receptor- and integrin-mediated ERK signaling upregulated in DTC steer cells into a dormant state [Citation31,Citation45]. The ERK inhibitor, U0126, can effectively maintain the dormancy of disseminated breast cancer [Citation31]. Meanwhile, substantial evidence has implicated Src kinase in multiple cellular processes and revealed a central role in the regulation of diverse signal transduction pathways related to tumorigenesis and development [Citation46]. Src kinase activity is responsible for the maintenance of dormancy in breast cancer cells. Src inhibitors, such as AZD0530 and PP1, or Src knockdown have been shown to cause nuclear accumulation of p27 and block the proliferative outbreak of dormant breast cancer cells and metastatic lesion formation [Citation47].
Moreover, dormant colorectal cancer cells are reported to respond to itraconazole, which suppresses the Wnt pathway through non-canonical Hedgehog signaling. Itraconazole treatment initially caused a proliferative burst, forcing dormant cells to cycle briefly and subsequently enter irreversible G1 cell cycle arrest and senescence [Citation31,Citation48]. Furthermore, tubeimoside-1 potently suppressed the growth of human prostate cancer cells via inducing oxidative stress-mediated apoptosis and G0/G1 phase arrest [Citation49]. Arctigenin, the active component of burdock root, enhanced p27Kip1 protein levels through inhibition of Akt and stimulation of FOXO3a activity, in turn, suppressing CDK2 kinase activity and finally inducing overall inhibition of HSC proliferation and G0/G1 phase arrest [Citation50]. Altogether, our findings suggest that dormancy can be effectively sustained through inhibition of proliferative signaling, activation of dormant pathways or delivering the components of dormant niches.
Strategies to promote cell cycle re-entry in quiescence cells
Reduction of the depth of cellular quiescence
Chronological age significantly influences quiescent cell survival and exit efficiency. Additionally, quiescent cells show an age-dependent loss of ability to proliferate [Citation51]. Long-term cultured quiescent fibroblasts cannot re-enter the proliferation cycle [Citation38]. Consequently, the quiescent state is a continuum that evolves over time, with early and deep quiescence being distinguishable by the capability of cells to re-enter the proliferative cycle [Citation51].
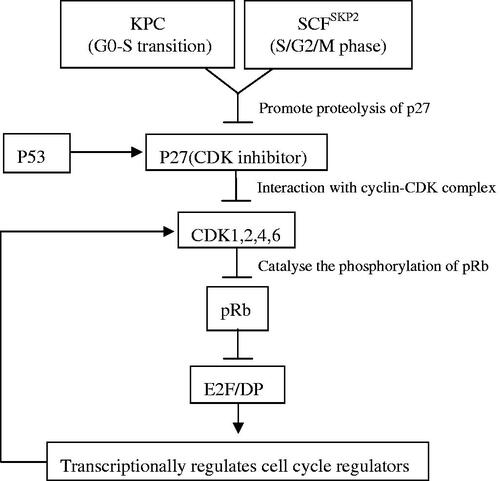
Reducing quiescence depth may awaken dormant CSCs, thus increasing their susceptibility to existing anticancer agents [Citation7,Citation52]. The Rb-E2F bistable switch plays critical roles in quiescence control. Destruction of the Rb-E2F gene network often leads to uncontrolled cell division and cancer formation [Citation52]. pRb is indispensable for suppressing cell cycle entry and acts through binding and inhibition of the transcriptional activator E2F, which regulates numerous target genes involved in the cell cycle, DNA replication, growth and differentiation [Citation10]. Consistently, emerging evidence has shown that quiescence depth can be tuned to different degrees by altering the expression of various proteins in the Rb-E2f network () [Citation52]. Natural compounds purified from Ganoderma lucidum, ganodermanondiol, and ergosterol peroxide, play inhibitory roles via reducing the quiescence depth of cancer cells. Previous research has confirmed that these two compounds not only effectively inhibit rapidly proliferating cells but also activate the Rb-E2F bistable switch in quiescent slow-cycling cells, thereby reducing the quiescence depth of dormant cancer cells and enhancing their sensitivity to chemotherapeutic drugs [Citation7]. Further studies on the effects of natural Ganoderma lucidum compounds on target quiescent cancer cells may present a new avenue for the development of novel chemotherapeutic agents.
DYRK1A/B inhibitors
As mentioned above, DYRK1A and DYRK1B kinases promote dormancy in a variety of ways. Inhibition of DYRK1A via administration of harmine enhanced the efficacy of imatinib in gastrointestinal stromal tumors through activating the cell cycle, supporting the potential of targeting DYRK1A for more efficient imatinib responses in future therapeutic interventions [Citation53]. The protein kinase, DYRK1B, is regarded as a druggable protein that regulates the transition of the G0/G1-S phase. Similarly, accumulating evidence suggests that depletion or inhibition of DYRK1B forces quiescent cancer cells to re-enter the cell cycle, resulting in cancer cell sensitization to the cytotoxic effects of drugs targeting proliferating cells [Citation31,Citation34].
Other strategies
TβR1 kinase inhibitors and TGF-β neutralizing antibodies are believed to play a major role in activating dormant cancer cells and consequently rendering them susceptible to chemotherapeutic drugs in vitro and in vivo [Citation12]. Furthermore, supplementation with insulin-like growth factor 1 receptor (IGF1-R) that targets quiescent cancer cells effectively promotes tumor responsiveness to conventional chemotherapy [Citation54]. JARID1B, an H3K4me3 demethylase, is overexpressed in a variety of cancers. In oral squamous cell cancer, JARID1B silencing weakens sphere formation and CSC marker expression. G0-like cells are primed to enter a JARID1Bhigh state and acquire CSC-like functions via upregulation of PI3K-dependent JARID1B. In summary, blocking entry into the G0-like state coupled with PI3K inhibition may present an effective therapeutic approach [Citation55]. These encouraging findings justify continued research efforts to develop effective approaches to disrupt the quiescent state and reverse the chemoresistance of non-cycling cancer cells.
Strategies to kill quiescent cells
Proteasome inhibition
The ubiquitin-proteasome system (UPS) is essential for degradation of aberrantly folded proteins and short-lived proteins involved in cell cycle progression, apoptosis, and signal transduction [Citation5,Citation56,Citation57]. Tumor cells utilize UPS to achieve aberrant proliferation and survival [Citation58]. Earlier findings support the utility of proteasomes as potential drug targets for some cancer types. Proteasome storage granules (PSG) enhance resistance to genotoxic stress and confer cell fitness during aging [Citation59]. Upon entry of Saccharomyces cerevisiae into the stationary phase, massive re-localization of proteasome subunits occurs from the nucleus into motile cytosolic granules, thereby triggering PSG formation. This phenomenon is rapidly and completely reversed upon exit from quiescence [Citation59,Citation60]. Notably, ubiquitin is involved in regulating proteasome dynamics between proliferation and quiescence in yeast [Citation59]. In addition, Spg5, a quiescence-specific positive regulator of the proteasome, directly binds to components of the proteasome and governs its function in quiescence [Citation56].
Proteasome inhibitors are capable of blocking the proteolytic activity of the proteasome complex. Inhibition of proteasomes exerts several toxic effects, including accumulation of unfolded and damaged proteins as well as elevated ROS levels [Citation5]. In small-cell lung cancer, two proteasome inhibitors, bortezomib and siomycin A, successfully suppressed Forkhead box protein M1 (FOXM1) and NF-κB activity, resulting in significant cell cycle arrest and apoptosis induction in line with reduced cell proliferation. In particular, these effects were further strengthened by the supplementation of conventional chemotherapy with bortezomib [Citation61]. Administration of 5-azacytidine combined with bortezomib in multiple myeloma (MM) may present a novel therapeutic strategy to delay recurrence by potentiating bortezomib-mediated apoptosis and quiescence stability [Citation62]. Therefore, integration of proteasome inhibitors into conventional therapeutic strategies may present a promising approach for the treatment of multiple cancer types.
MG132 has been shown to enhance irradiation-induced apoptosis by suppressing activation of NF-κB and tumor growth [Citation57]. However, other reports suggest that proteasome inhibition with MG-132 or bortezomib induces G0–G1 phase arrest in MM cells, resulting in lower sensitivity of tumor cells to treatment and survival [Citation5,Citation63]. Recent experiments have demonstrated that MG132-treated proliferating fibroblasts undergo rapid apoptosis whereas MG132-treated quiescent cells mostly retain viability through induction of a variety of alternative protective mechanisms (including ROS-detoxifying pathways, clearance of ubiquitinated protein aggregates, and the Baf-sensitive autophagy/lysosomal pathway) to escape proteasome inhibition-mediated cell death. Hence, increasing cellular superoxide concentrations with 2-methoxyestradiol or inhibition of autophagy/lysosomal pathways with bafilomycin A1 could sensitize quiescent fibroblasts to MG132-mediated apoptosis [Citation5]. Furthermore, MM cells that survive bortezomib treatment exhibit a GRP78high/p21high/CDK6low/pRblow profile, which may distinguish quiescent MM cells capable of spawning tumor recurrence [Citation62]. Upregulation of the pro-survival chaperone, BiP/Grp78, an unfolded protein response (UPR) survival factor, facilitates prolonged survival of quiescent cancer cells, which may serve as a reliable target to eradicate residual tumor cells [Citation62,Citation64]. Subtilase cytotoxin, a bacterial AB5 toxin, specifically downregulates GRP78 and induces death of bortezomib-surviving MM cells [Citation62]. Moreover, the Hsp90 inhibitor, IPI-504, increases bortezomib sensitivity in mantle cell lymphoma cultures and tumors via BiP/Grp78 downregulation and UPR inhibition [Citation64]. Several studies have shown that YM155, a small-molecule inhibitor of survivin, exerts a strong cytotoxic effect on quiescent MM and bortezomib-resistant cells by suppressing survivin and Mcl-1 [Citation65]. Additionally, maintaining eukaryotic initiation factor 2α in a hyperphosphorylation state potentiates bortezomib efficiency and eliminates residual quiescent MM cells after proteasome inhibition [Citation63]. In summary, strategies that aid in overcoming proteasome inhibitor resistance or eliminating surviving quiescent tumor cells after proteasome inhibition may ultimately contribute to reducing residual disease and recurrence in various types of cancer.
Bisacodyl
The laxative bisacodyl/DDPM is the first small molecule identified displaying cytotoxicity to slow-growing cancer cells [Citation3]. Accumulating evidence indicates that DDPM, the active metabolite of bisacodyl, induces necrosis of quiescent human glioblastoma stem-like cells (GSCs) in vivo and in vitro without exerting deleterious effects on non-cancer neural cells [Citation5,Citation66]. In particular, microenvironment acidification of cultured GSCs induces cell cycle arrest and sensitization to DDPM [Citation66,Citation67]. Further reports have shown that bisacodyl/DDPM triggers necrosis in quiescent GSCs by inhibiting InsP3-induced Ca2+ release [Citation66]. Moreover, DDPM endows cytotoxic effects by suppressing the activity of a kinase cascade composed of WNK1 and its upstream regulators, AKT and SGK1, triggering a subsequent increase in NBC family Na+/HCO3– cotransporter activity [Citation67]. These findings support the utility of DDPM as a potential therapeutic agent for specifically targeting resistant quiescent cancer stem-like cells.
Other methods
An earlier study on pancreatic cancer demonstrated that a subpopulation of dormant cells surviving K-RAS oncogene ablation is dependent on oxidative phosphorylation for survival. Thus, the surviving cells showed high sensitivity to oligomycin (an oxidative phosphorylation inhibitor), highlighting mitochondrial respiration as an attractive druggable target to eliminate dormant cells [Citation31,Citation68]. VLX600 is reported to decrease mitochondrial oxidative phosphorylation, especially the rate of uncoupled respiration [Citation69]. Earlier research suggests that VLX600 is preferentially active against quiescent cancer cells. Exposure to VLX600 resulted in increased cytotoxic activity under conditions of nutrient starvation and tumor growth inhibition in vivo [Citation70]. Therefore, induction of mitochondrial dysfunction may present a strategy for targeting tumor cells in metabolically compromised microenvironments. Moreover, antineoplastic polymers have been designed to kill dormant prostate cancer cells. Takahashi et al. reported that anticancer peptide mimetic polymers exert cytotoxicity not only to proliferating prostate cancer but also dormant cancer cells inherently resistant to docetaxel [Citation71]. Moreover, expression of PKR-like endoplasmic reticulum kinase (PERK) is reported as abnormally high in dormant cancer cells. Early studies in mice have demonstrated cell killing activity of a PERK inhibitor but little is known about the underlying molecular mechanisms [Citation72]. Autocrine IGF1/AKT signaling manipulates pancreatic cancer cell dormancy in the absence of oncogenic drivers (such as mutant KRAS and c-MYC). IGF1-R inhibition decreases the burden of residual disease and tumor recurrence, indicating this molecular pathway is essential for the survival of cancer cells without primary oncogenic drivers [Citation73]. Photodynamic therapy (PDT), which uses 5-aminolevulinic acid (ALA) to drive generation of the intracellular photosensitizer, protoporphyrin IX (PpIX), is commonly used in clinical treatments. Several studies have shown that dormant cancer cells accumulate high PpIX levels and are sensitive to ALA-PDT. Furthermore, efficacy can be strengthened by combining with G0/G1 phase arrestors [Citation74]. These studies collectively indicate that development of new drugs that directly target and kill dormant cells may be an alternative to awakening dormant cells, which potentially represents a more translational strategy.
Radiation response of quiescent tumor cells
Radiotherapy, one of the main approaches in cancer therapy, is a considerable focus of research. Overall, ∼70% of cancer patients have been administered radiotherapy to date. However, little is known about the effects of radiotherapy on quiescent cells. Emerging evidence has shown that quiescence, at least in normal fibroblasts, does not affect the irradiation response of key proteins involved in stress and DNA damage [Citation75]. Moreover, the impact of radiotherapy on cancer cells is reported to be affected by cell cycle status, oxygenation, and nature of radiation [Citation76].
Effects of oxygenation and radiation properties on quiescent tumor cell radiosensitivity
Hypoxia, a consequence of uncontrollable tumor cell proliferation accompanied by the inability of the surrounding vasculature to meet increased oxygen and nutrient demands, is a common phenomenon in solid tumors closely correlated with decreased sensitivity to cancer therapy [Citation77]. The oxygen level in hypoxic tumor tissues is poorer than that in normal tissues (an average of 1–2% O2) [Citation76]. Hypoxia confers therapeutic resistance of cancer cells partly via inducing entry into quiescence. Deregulation of the PI3K/AKT/mTOR pathway through hypoxia-inducible factor 1α (HIF-1α) is considered essential for CSC quiescence and maintenance by inhibiting CSC metabolism and growth through mTOR and promoting survival via AKT signaling [Citation78]. In addition, reports have shown that the “peri-necrotic niche” harboring HIF-1α-expressing quiescent stem-like GSCs can be emerged in an intra-tumoral hypoxia gradient and is related to enhanced tumorigenic ability, which may present a promising target to eliminate GSCs and improve survival prospects of patients [Citation79].
Tumor hypoxia results from either limited oxygen diffusion (chronic hypoxia) or limited perfusion (acute hypoxia) [Citation14,Citation80]. Nicotinamide, a vitamin B3 analogue, prevents temporary fluctuations in tumor blood flow that results in relief of acute hypoxia, which tends to decrease the extent of tumor metastasis. Moreover, mild temperature hyperthermia (MTH) is proposed to improve the tumor response to radiation by elevating tumor blood flow, leading to increased tumor oxygenation and thereby overcoming chronic hypoxia [Citation80,Citation81]. Manipulation of hypoxia during high-dose rate (HDR) and reduced-dose rate (RDR) irradiation affects the radiosensitivity of the tumor, especially to γ-rays [Citation82]. HDR irradiation, nicotinamide and MTH have been shown to enhance the radiosensitivity of total and quiescent cells. Notably, upon administration of γ-rays in combination with nicotinamide or MTH, in particular, the former agent, HDR irradiation reduced the number of metastases more markedly than RDR irradiation [Citation14]. Hence, nicotinamide combined with HDR may present a promising strategy for reducing lung metastases. Moreover, bevacizumab, a vascular endothelial growth factor inhibitor, has the potential to significantly enhance the radiosensitivity of acute hypoxia-rich total cells to γ-rays and reduce the hypoxic fraction (HF) [Citation83]. Other studies have reported that reduction of HF induced by a combination of bevacizumab and MTH is more significant than that achieved with bevacizumab plus nicotinamide in both total and quiescent cells [Citation80]. In summary, the results to date indicate that control of chronic hypoxia-rich quiescent cells and acute hypoxia-rich total cells in primary tumors has the potential to affect local tumors and lung metastases, respectively.
The tumor oxygenation status after irradiation is vital for manipulating recovery following radiation-induced damage [Citation84]. Hypoxia-specific cytotoxin tirapazamine (TPZ) treatment after irradiation not only inhibited recovery from radiation-mediated damage but also enhanced radiosensitivity both in total and quiescent tumor cells. Among the pimonidazole-unlabeled fractions of both total and quiescent cells, TPZ was shown to inhibit the decrease in radiosensitivity more effectively than MTH or metformin (Met) without radiosensitization [Citation85]. Wortmannin is proposed to prevent nonhomologous end-joining repair by suppressing a catalytic subunit of DNA-dependent protein kinase. Combination of Wortmannin and γ-ray irradiation treatment effectively inhibited recovery from radiation-induced damage in pimonidazole-unlabeled quiescent cancer cells [Citation86]. Additionally, radiosensitivity and recovery ability from radiation-induced damage in pimonidazole-unlabeled intratumor quiescent cells is reported to be dependent on the status of p53 [Citation87]. Radiosensitivity of the pimonidazole-unlabeled cell population further depends on the quality of radiation delivered. Previous studies have shown that following γ-ray irradiation, radiosensitivity of pimonidazole-unlabeled cancer cells in the quiescent state is significantly higher than that of total cells [Citation11,Citation85]. However, using a delayed assay or decreased radiation dose rates, a more significant decrease in radiosensitivity in pimonidazole nonlabeled cells was observed in quiescent than total cells. These changes in radiosensitivity were largely inhibited using carbon ion beams, especially with higher liner energy transfer (LET) [Citation11]. In summary, targeting the pimonidazole nonlabeled subfraction of quiescent cancer cells representing oxygenation status may a provide novel therapeutic approach.
Strategies to improve radioresistance of quiescent tumor cells
Quiescence-associated radioresistance may be a primary reason for the failure of cancer therapy. Earlier studies have reported that FOXM1 expression is significantly decreased in quiescence cells and conversely increased in irradiated quiescence cells. Inhibition of FOXM1 expression leads to a significant downregulation of glucose-6-phosphate dehydrogenase, resulting in exacerbation of radiation-induced toxicity in quiescent cells [Citation88]. Accordingly, targeting of FOXM1 may be used to enhance the radiosensitivity of quiescent tumor cells. Rac family small GTPase 2 (RAC2) is a ∼21 kDa GTPase containing the catalytic subunit of NADPH oxidase, the main component of the respiratory chain. RAC2 induces aberrant proliferation of quiescent non-small cell lung cancer cells (NSCLC) via promoting JUNB expression through the MAL-SRF pathway [Citation89]. Moreover, earlier studies have reported that low expression of RAC2 in G0 cells accounts for cellular radioresistance through suppression of NAPDH oxidase activity, which results in low yields of ROS and high antioxidant activity [Citation13]. P38 MAPK interactions with RAC2 induced a decrease in functional RAC2. Phosphorylation of P38 MAPK enhanced cellular radioresistance. However, excessive ROS production led to P38 MAPK dephosphorylation, suggesting that the ROS/RAC2/P38 MAPK feedback loop generates quiescent cells more resistant to ionizing radiation (IR) [Citation13]. These findings reveal a new paradigm for understanding radioresistance of G0 cells and support the potential application of RAC2 inhibitors as radiation protectants. Other researchers have reported that SNS-032, a small-molecule CDK inhibitor, sensitizes quiescent and hypoxic NSCLC to IR via regulating double-strand break (DSB) repair [Citation90]. Cantharidin, an inhibitor of PP2A, induces pancreatic cancer cells to exit the quiescent G0/G1 phase and blocks the cell cycle in the G2/M phase, thereby enhancing irradiation-induced DNA damage, which may involve several mechanisms, including cell cycle regulation, DNA damage enhancement, and DNA damage repair inhibition [Citation91]. In view of the importance of radiation resistance in the failure of solid tumor therapy, these collective findings provide a basis for improvement of clinical treatment regimens.
Application of BNCT in quiescent tumor cells
Boron neutron capture therapy (BNCT) is a novel targeted radiotherapy that selectively kills tumor cells. After introduction into the human body, Boron (10B) is enriched in tumor cells and reacts with neutrons. The reaction generates high LET α particles (4He) and recoiling 7Li nuclei, which result in the induction of DSBs with strong biological effectiveness. Since the path lengths of these particles are almost equal to cell diameter size, only 10B-containing cancer cells are theoretically destroyed without causing serious radiation injury to surrounding normal tissue [Citation92,Citation93]. The cellular distribution of 10B from L-para-boronophenylalanine-10B (BPA) is believed to be largely dependent on the capability of the cells to take up 10B whereas that from sodium mercaptoundecahydrododecaborate-10B (BSH) mainly relies on drug diffusion [Citation11]. Importantly, the use of a 10B-carrier in BNCT, especially BPA, not only effectively eliminates hypoxia and quiescent cells but also kills oxygenated and proliferative cells [Citation11,Citation94].
Radiosensitivity of tumor cells partially depends on the nature of the 10B-carrier used in BNCT. In particular, BPA carrier improved the radiosensitivity of pimonidazole-unlabeled cells to a greater extent in the quiescent than total cell group [Citation11]. Previous studies have validated that hypoxia-induced quiescent cancer cells take up lower quantities of BPA or BSH than proliferating cells. Notably, BPA-treated quiescent cells are less radiosensitive than BSH-treated quiescent cells [Citation11,Citation95]. BSH-BNCT combined with MTH is reported to improve local tumor control whereas a combination of BPA-BNCT and nicotinamide may reduce the number of lung metastases [Citation96]. Bevacizumab is additionally capable of effectively alleviating acute hypoxia, with greater sensitivity of total tumor cells reported following BPA-BNCT relative to BSH-BNCT treatment [Citation80]. Moreover, uptake of 10B-BPA in human glioblastoma cells is reported to be reduced in a linear manner to the decrease in oxygen levels, which could be attributed to decreased L-amino acid transporter expression [Citation95]. Hence, strategies aimed at overcoming local tumor hypoxia may provide a powerful tool to improve the efficacy of BNCT.
Summary and future perspectives
The presence of quiescent tumor cells is one of the critical factors underlying tumor recurrence. While numerous potential therapeutic targets and chemoradiotherapy sensitizers have been identified that effectively eliminate tumor cells from the quiescent phase or render them permanently dormant, many limitations remain. Firstly, quiescent tumor cells are not only well hidden but also have clonal heterogeneity and thus not all cells respond to treatments. Secondly, treatments often proceed throughout the lifetime of the patient. Thirdly, administration of targeting agents could result in unpleasant side effects due to their involvement in other biological processes. Therefore, the development of novel and effective curative treatments that can efficiently target all quiescent cells without damaging normal tissues remains an urgent medical requirement.
BNCT is a novel approach in the field of radiotherapy widely applied owing to its utility as tumor cell-targeted treatment, high efficiency and low toxicity, and considered ideal for a variety of quiescent tumor cells. Future research directions for effective quiescent tumor cell therapy include the development of more efficient boron-containing drugs and suitable neutron sources as well as the establishment of a more accurate boron dosage measurement system.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mohammad K, Dakik P, Medkour Y, et al. Quiescence entry, maintenance, and exit in adult stem cells. Int J Mol Sci. 2019;20:pii: E2158.
- Sagot I, Laporte D. Quiescence, an individual journey. Curr Genet. 2019;65:695–699.
- Zeniou M, Feve M, Mameri S, et al. Chemical library screening and structure-function relationship studies identify bisacodyl as a potent and selective cytotoxic agent towards quiescent human glioblastoma tumor stem-like cells. PLoS One. 2015;10:e0134793.
- Zhu L, Xing S, Zhang L, et al. Involvement of Polo-like kinase 1 (Plk1) in quiescence regulation of cancer stem-like cells of the gastric cancer cell lines. Oncotarget. 2017;8:37633–37645.
- Legesse-Miller A, Raitman I, Haley EM, et al. Quiescent fibroblasts are protected from proteasome inhibition-mediated toxicity. Mol Biol Cell. 2012;23:3566–3581.
- Yao M, Xie C, Kiang MY, et al. Targeting of cytosolic phospholipase A2α impedes cell cycle re-entry of quiescent prostate cancer cells. Oncotarget. 2015;6:34458–34474.
- Dai J, Miller MA, Everetts NJ, et al. Elimination of quiescent slow-cycling cells via reducing quiescence depth by natural compounds purified from Ganoderma lucidum. Oncotarget 2017;8:13770–13781.
- Cackowski FC, Eber MR, Rhee J, et al. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J Cell Biochem. 2017;118:891–902.
- Oki T, Nishimura K, Kitaura J, et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0–G1 transition. Sci Rep. 2015;4:4012.
- Sun D, Buttitta L. States of G0 and the proliferation-quiescence decision in cells, tissues and during development. Int J Dev Biol. 2017;61:357–366.
- Masunaga S, Sakurai Y, Tanaka H, et al. Radiosensitivity of pimonidazole-unlabeled intratumor quiescent cell population to γ-rays, accelerated carbon ion beams and boron neutron capture reaction. Br J Radiol. 2013;86:20120302.
- Prunier C, Baker D, Ten Dijke P, et al. TGF-β family signaling pathways in cellular dormancy. Trends Cancer. 2019;5:66–78.
- Pei H, Zhang J, Nie J, et al. RAC2-P38 MAPK-dependent NADPH oxidase activity is associated with the resistance of quiescent cells to ionizing radiation. Cell Cycle. 2017;16:113–122.
- Masunaga S, Matsumoto Y, Kashino G, et al. Significance of manipulating tumor hypoxia and radiation dose rate in terms of local tumor response and lung metastatic potential, referring to the response of quiescent cell populations. Br J Radiol. 2010;83:776–784.
- Brown JA, Yonekubo Y, Hanson N, et al. TGF-β-induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell Stem Cell 2017;21:650–664.
- Alekseenko LL, Shilina MA, Lyublinskaya OG, et al. Quiescent human mesenchymal stem cells are more resistant to heat stress than cycling cells. Stem Cells Int. 2018;2018:3753547.
- Sagot I, Laporte D. The cell biology of quiescent yeast – a diversity of individual scenarios. J Cell Sci. 2019;132:213025.
- Nakazawa N, Teruya T, Sajiki K, et al. The putative ceramide-conjugation protein Cwh43 regulates G0 quiescence, nutrient metabolism and lipid homeostasis in fission yeast. J Cell Sci. 2018;131:217331.
- Hayashi T, Teruya T, Chaleckis R, et al. S-adenosylmethionine synthetase is required for cell growth, maintenance of G0 phase, and termination of quiescence in fission yeast. iScience. 2018;5:38–51.
- Miles S, Li LH, Melville Z, et al. Ssd1 and the cell wall integrity pathway promote entry, maintenance and recovery from the quiescent state in budding yeast. Mol Biol Cell 2019;mbcE19040190. doi:10.1091/mbc.E19-04-0190
- Guidi M, Ruault M, Marbouty M, et al. Spatial reorganization of telomeres in long-lived quiescent cells. Genome Biol. 2015;16:206.
- Hoffman CS, Wood V, Fantes PA. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics. 2015;201:403–423.
- Sajiki K, Tahara Y, Uehara L, et al. Genetic regulation of mitotic competence in G0 quiescent cells. Sci Adv. 2018;4:eaat5685.
- Aono S, Haruna Y, Watanabe YH, et al. The fission yeast Greatwall-Endosulfine pathway is required for proper quiescence/G0 phase entry and maintenance. Genes Cells. 2019;24:172–186.
- Nakhaei-Rad S, Nakhaeizadeh H, Gotze S, et al. The role of embryonic stem cell-expressed RAS (ERAS) in the maintenance of quiescent hepatic stellate cells. J Biol Chem. 2016;291:8399–8413.
- Nobutani K, Shimono Y, Mizutani K, et al. Downregulation of CXCR4 in metastasized breast cancer cells and implication in their dormancy. PLoS One. 2015;10:e0130032.
- Arora M, Moser J, Phadke H, et al. Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 2017;19:1351–1364.
- Subramaniam S, Sreenivas P, Cheedipudi S, et al. Distinct transcriptional networks in quiescent myoblasts: a role for Wnt signaling in reversible vs. irreversible arrest. PLoS One 2013;8:e65097.
- Bartosh TJ, Ullah M, Zeitouni S, et al. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc Natl Acad Sci USA. 2016;113:E6447–E6456.
- Shimanuki M, Uehara L, Pluskal T, et al. Klf1, a C2H2 zinc finger-transcription factor, is required for cell wall maintenance during long-term quiescence in differentiated G0 phase. PLoS One 2013;8:e78545.
- Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. 2019;40:128–141.
- Sosa MS, Parikh F, Maia AG, et al. NR2F1 controls tumor cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun. 2015;6:6170.
- Soppa U, Schumacher J, Florencio Ortiz V, et al. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014;13:2084–2100.
- Becker W. A wake-up call to quiescent cancer cells - potential use of DYRK1B inhibitors in cancer therapy. FEBS J. 2018;285:1203–1211.
- Perucca P, Cazzalini O, Madine M, et al. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8:105–114.
- Cheedipudi S, Puri D, Saleh A, et al. A fine balance: Epigenetic control of cellular quiescence by the tumor suppressor PRDM2/RIZ at a bivalent domain in the cyclin a gene. Nucleic Acids Res. 2015;43:6236–6256.
- Spencer SL, Cappell SD, Tsai FC, et al. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013;155:369–383.
- Kumar MG, Patel NM, Nicholson AM, et al. Reactive oxygen species mediate microRNA-302 regulation of AT-rich interacting domain 4a and C-C motif ligand 5 expression during transitions between quiescence and proliferation. Free Radic Biol Med. 2012;53:974–982.
- Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227.
- Baell JB, Leaver DJ, Hermans SJ, et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumor growth. Nature. 2018;560:253–257.
- Huang F. New KAT6 inhibitors induce senescence and arrest cancer growth. Synth Syst Biotechnol. 2018;3:244–245.
- Herbert SP, Odell AF, Ponnambalam S, et al. Activation of cytosolic phospholipase A2-{alpha} as a novel mechanism regulating endothelial cell cycle progression and angiogenesis. J Biol Chem. 2009;284:5784–5796.
- Naini SM, Choukroun GJ, Ryan JR, et al. Cytosolic phospholipase A2α regulates G1 progression through modulating FOXO1 activity. FASEB J. 2016;30:1155–1170.
- Xu H, Sun Y, Zeng L, et al. Inhibition of cytosolic phospholipase A2 α increases chemosensitivity in cervical carcinoma through suppressing beta-catenin signaling. Cancer Biol Ther. 2019;20:912–921.
- Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. 2015;15:238–247.
- Elsberger B. Translational evidence on the role of Src kinase and activated Src kinase in invasive breast cancer. Crit Rev Oncol Hematol. 2014;89:343–351.
- El Touny LH, Vieira A, Mendoza A, et al. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest. 2014;124:156–168.
- Buczacki SJA, Popova S, Biggs E, et al. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J Exp Med. 2018;215:1891–1912.
- Yang JB, Khan M, He YY, et al. Tubeimoside-1 induces oxidative stress-mediated apoptosis and G0/G1 phase arrest in human prostate carcinoma cells in vitro. Acta Pharmacol Sin. 2016;37:950–962.
- Li A, Wang J, Wu M, et al. The inhibition of activated hepatic stellate cells proliferation by arctigenin through G0/G1 phase cell cycle arrest: persistent p27(Kip1) induction by interfering with PI3K/Akt/FOXO3a signaling pathway. Eur J Pharmacol. 2015;747:71–87.
- Laporte D, Jimenez L, Gouleme L, et al. Yeast quiescence exit swiftness is influenced by cell volume and chronological age. Microb Cell. 2017;5:104–111.
- Fujimaki K, Yao G. Crack the state of silence: Tune the depth of cellular quiescence for cancer therapy. Mol Cell Oncol. 2018;5:e1403531.
- Boichuk S, Parry JA, Makielski KR, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013;73:5120–5129.
- Kyle AH, Baker JH, Minchinton AI. Targeting quiescent tumor cells via oxygen and IGF-I supplementation. Cancer Res. 2012;72:801–809.
- Facompre ND, Harmeyer KM, Sole X, et al. JARID1B enables transit between distinct states of the stem-like cell population in oral cancers. Cancer Res. 2016;76:5538–5549.
- Hanna J, Waterman D, Boselli M, et al. Spg5 protein regulates the proteasome in quiescence. J Biol Chem. 2012;287:34400–34409.
- Zhu W, Liu J, Nie J, et al. MG132 enhances the radiosensitivity of lung cancer cells in vitro and in vivo. Oncol Rep. 2015;34:2083–2089.
- Shen M, Schmitt S, Buac D, et al. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets. 2013;17:1091–1108.
- Gu ZC, Wu E, Sailer C, et al. Ubiquitin orchestrates proteasome dynamics between proliferation and quiescence in yeast. Mol Biol Cell. 2017;28:2479–2491.
- Laporte D, Salin B, Daignan-Fornier B, et al. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J Cell Biol. 2008;181:737–745.
- Taromi S, Lewens F, Arsenic R, et al. Proteasome inhibitor bortezomib enhances the effect of standard chemotherapy in small cell lung cancer. Oncotarget. 2017;8:97061–97078.
- Adomako A, Calvo V, Biran N, et al. Identification of markers that functionally define a quiescent multiple myeloma cell sub-population surviving bortezomib treatment. BMC Cancer. 2015;15:444.
- Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2α dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009;69:1545–1552.
- Roue G, Perez-Galan P, Mozos A, et al. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood. 2011;117:1270–1279.
- Ookura M, Fujii T, Yagi H, et al. YM155 exerts potent cytotoxic activity against quiescent (G0/G1) multiple myeloma and bortezomib resistant cells via inhibition of survivin and Mcl-1. Oncotarget. 2017;8:111535–111550.
- Dong J, Aulestia FJ, Assad Kahn S, et al. Bisacodyl and its cytotoxic activity on human glioblastoma stem-like cells. Implication of inositol 1,4,5-triphosphate receptor dependent calcium signaling. Biochim Biophys Acta Mol Cell Res. 2017;1864:1018–1027.
- Chen W, Zebaze LN, Dong J, et al. WNK1 kinase and its partners Akt, SGK1 and NBC-family Na(+)/HCO3(–) cotransporters are potential therapeutic targets for glioblastoma stem-like cells linked to Bisacodyl signaling. Oncotarget. 2018;9:27197–27219.
- Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632.
- Zhang X, De Milito A, Demiroglu-Zergeroglu A, et al. Eradicating quiescent tumor cells by targeting mitochondrial bioenergetics. Trends Cancer 2016;2:657–663.
- Zhang X, Fryknas M, Hernlund E, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumor cells in metabolically compromised microenvironments. Nat Commun. 2014;5:3295.
- Takahashi H, Yumoto K, Yasuhara K, et al. Anticancer polymers designed for killing dormant prostate cancer cells. Sci Rep. 2019;9:1096.
- Ledford H. Cancer researchers target the dormant cells that seed tumors. Nature. 2018;558:355–356.
- Rajbhandari N, Lin WC, Wehde BL, et al. Autocrine IGF1 signaling mediates pancreatic tumor cell dormancy in the absence of oncogenic drivers. Cell Rep. 2017;18:2243–2255.
- Nakayama T, Otsuka S, Kobayashi T, et al. Dormant cancer cells accumulate high protoporphyrin IX levels and are sensitive to 5-aminolevulinic acid-based photodynamic therapy. Sci Rep. 2016;6:36478.
- Dai J, Itahana K, Baskar R. Quiescence does not affect p53 and stress response by irradiation in human lung fibroblasts. Biochem Biophys Res Commun. 2015;458:104–109.
- Muz B, de la Puente P, Azab F, et al. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92.
- Kuo P, Le QT. Galectin-1 links tumor hypoxia and radiotherapy. Glycobiology. 2014;24:921–925.
- Marhold M, Tomasich E, El-Gazzar A, et al. HIF1α regulates mTOR signaling and viability of prostate cancer stem cells. Mol Cancer Res. 2015;13:556–564.
- Ishii A, Kimura T, Sadahiro H, et al. Histological characterization of the tumorigenic “peri-necrotic niche” harboring quiescent stem-like tumor cells in glioblastoma. PLoS One 2016;11:e0147366.
- Masunaga SI, Sakurai Y, Tano K, et al. Effect of bevacizumab combined with boron neutron capture therapy on local tumor response and lung metastasis. Exp Ther Med. 2014;8:291–301.
- Masunaga SI, Sanada Y, Moriwaki T, et al. Significance of fractionated administration of thalidomide combined with γ-ray irradiation in terms of local tumor response and lung metastasis. World J Oncol. 2014;5:155–165.
- Masunaga S, Hirayama R, Uzawa A, et al. Influence of manipulating hypoxia in solid tumors on the radiation dose-rate effect in vivo, with reference to that in the quiescent cell population. Jpn J Radiol. 2010;28:132–142.
- Masunaga S, Liu Y, Tanaka H, et al. Reducing intratumor acute hypoxia through bevacizumab treatment, referring to the response of quiescent tumor cells and metastatic potential. Br J Radiol. 2011;84:1131–1138.
- Masunaga S, Hirayama R, Uzawa A, et al. The effect of post-irradiation tumor oxygenation status on recovery from radiation-induced damage in vivo: with reference to that in quiescent cell populations. J Cancer Res Clin Oncol. 2009;135:1109–1116.
- Masunaga SI, Tano K, Sanada Y, et al. Effect of tirapazamine, metformin or mild hyperthermia on recovery from radiation-induced damage in pimonidazole-unlabeled quiescent tumor cells. World J Oncol. 2017;8:137–146.
- Masunaga S, Sakurai Y, Tanaka H, et al. Wortmannin efficiently suppresses the recovery from radiation-induced damage in pimonidazole-unlabeled quiescent tumor cell population. J Radiat Res. 2013;54:221–229.
- Masunaga SI, Liu Y, Tanaka H, et al. Radiosensitivity and capacity to recover from radiation-induced damage in pimonidazole-unlabeled intratumor quiescent cells depend on p53 status. World J Oncol. 2011;2:1–9.
- Eckers JC, Kalen AL, Sarsour EH, et al. Forkhead box M1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat Res. 2014;182:420–429.
- Pei H, Guo Z, Wang Z, et al. RAC2 promotes abnormal proliferation of quiescent cells by enhanced JUNB expression via the MAL-SRF pathway. Cell Cycle 2018;17:1115–1123.
- Kodym E, Kodym R, Reis AE, et al. The small-molecule CDK inhibitor, SNS-032, enhances cellular radiosensitivity in quiescent and hypoxic non-small cell lung cancer cells. Lung Cancer. 2009;66:37–47.
- Xu MD, Liu SL, Zheng BB, et al. The radiotherapy-sensitization effect of cantharidin: Mechanisms involving cell cycle regulation, enhanced DNA damage, and inhibited DNA damage repair. Pancreatology. 2018;18:822–832.
- Okamoto E, Yamamoto T, Nakai K, et al. Detection of DNA double-strand breaks in boron neutron capture reaction. Appl Radiat Isot. 2015;106:185–188.
- Miyabe J, Ohgaki R, Saito K, et al. Boron delivery for boron neutron capture therapy targeting a cancer-upregulated oligopeptide transporter. J Pharmacol Sci. 2019;139:215–222.
- Sun T, Zhang Z, Li B, et al. Boron neutron capture therapy induces cell cycle arrest and cell apoptosis of glioma stem/progenitor cells in vitro. Radiat Oncol. 2013;8:195.
- Wada Y, Hirose K, Harada T, et al. Impact of oxygen status on 10B-BPA uptake into human glioblastoma cells, referring to significance in boron neutron capture therapy. J Radiat Res. 2018;59:122–128.
- Masunaga S, Sakurai Y, Tanaka H, et al. Effects of employing a 10B-carrier and manipulating intratumor hypoxia on local tumor response and lung metastatic potential in boron neutron capture therapy. Br J Radiol. 2012;85:249–258.