Abstract
Data mining (DM) based on Bayesian neural networks (BNN) is popular for exploring consistent patterns and/or systematic relationships of variables in chemometrics. A combination of the unsupervised principal component similarity (PCS) analysis with the random‐centroid optimization for site‐directed mutagenesis of amino acid sequences (RCG) is proposed to correlate the sequence data with functions of proteins. Principal component similarity based on similarity was superior to classifications based on dissimilarity represented by multidimensional distances. Important factors (independent variables) to be used in the optimization could be selected through PCS processing of data to improve reliability of function prediction. Dimensionality reduction using PCS by eliminating minor factors and/or trend‐line drawing on response surface maps in RCG to determine the direction of search shift toward the global optimum are useful for approximating the underlying response surfaces. Application of the sequence PCS was useful in elucidating usually unknown mechanisms of underlying functions of a protein based on its amino acid sequence. The functions in question may be predicted using a modern version of neural networks.
Introduction
Artificial neural networks (ANN) are a superb curve‐fitting technique, however there is no assurance that the obtained relationship of structure parameters with target functions is the true underlying mechanism. Furthermore, ANN usually requires a large volume of data to enhance reliability of the relations discovered, especially in multidimensional cases, i.e., curse of dimensionality.Citation[1] It is a general practice to apply principal component analysis (PCA), which is the most popular, unsupervised learning technique, to the original variables to avoid multicolinearity between independent variables. The selected PC scores are then used to find the true relationships with functions in question, thereby eliminating unimportant variables including errors, which is the dimensionality reduction property. Principal component analysis was used to pre‐process original data for removing noise and reducing dimensionality prior to neural network computation for data mining (DM).Citation[2] However for classification purposes, PCA is inefficient as plotting of PC scores cannot accommodate more than 3 PC scores because of the limit of illustration up to the maximum three‐dimensions (3D). It is, therefore, possible to restrict the accuracy of classification due to discarding potentially important factors despite possible minor contribution to the total variance of the original data. Selection of truly influential structural variables without interferences from irrelevant factors is extremely important in finding accurate information on the structure‐function relationships.
Unfortunately, information on all of those variables is frequently unavailable a priori and the classifications including unknown factors are categorized as unsupervised learnings. We propose, therefore in this study, to apply the unsupervised principal component similarity (PCS) for preliminary classification of structure‐relating variables (pre‐processing) and PC scores thus selected are then used for relationship study after eliminating less influential factors as well as outliers. Thereafter, ANN or its equivalent algorithm is used to find more reliable structure‐function relationships. When the best condition found from the functional relationship matches to the global optimum discovered by applying random‐centroid optimization (RCO), the best functions thus obtained could be finally justified as the most accurate approximation to the true underlying relationships.
The objective of this study was to justify the use of a combination of PCS and RCO in genetic modification of protein molecules followed by probability neural networks for quantitative structure‐activity relationship (QSAR) study. This process, therefore, can be categorized as unsupervised DM.
Supervised Data Mining
Data mining is a technique to extract useful information from established databases mostly in a supervised mode. General protocol of DM being used in combinatorial chemistryCitation[3] is (i) choose a similarity probe; (ii) calculate similarity; (iii) rank all compounds; (iv) select top compounds; and (v) estimate success rate. Data mining accepts “black box” approach, such as ANN, which can generate valid prediction but are not capable of identifying the interrelations between the variables. This is because prediction is one of the most important objectives of DM. There are a great variety of representations available for DM, including rule bases, decision trees, and ANN; and also there are many techniques for DM such as density estimation, classification, regression, and clustering. However, it is used in conjunction with statistical techniques and the graphical model derived from Bayesian neural networks (BNN) is advantageous for DM study.Citation[4] Among the four advantages of BNN that were listed by Heckerman, the most important advantage is probably the capability to allow one to learn about casual relationships due to two reasons. The process is efficient when we are trying to gain understanding about a problem domain. Then, the knowledge of casual relationships readily allows us to make predictions.
Compared to the maximum likelihood techniques equivalent to the minimization of an error function, Bayesian algorithm considers a probability distribution function over weight spaces, thereby representing the relative degrees of belief in different values for the weight vector. This function is initially set to some prior distribution. Once the data has been observed, it can be converted to a posterior distribution through the use of Bayes' theorem. The posterior distribution can subsequently be used to minimize the probability of misclassification as well as to evaluate the prediction of the trained networks for new values of the input variables.Citation[1] Bayesian neural networks was superior to partial least squares (PLS) regression as well as maximum likelihood NN in QSAR studies.Citation[5] However, it does not completely eliminate incorrect information once it is included into the data under investigation, although incorporation of prior knowledge into ANN computation may assist the effort to approach to the underlying relationship. If possible, it is desirable to select relevant variables only during dimensionality reduction prior to training.
Furthermore, ANN requires a large volume of data, at least 125 cases, preferably 250 cases for five input variables according to Statsoft.Citation[6] This is inconvenient in the life science study, such as mutation of protein molecules, due to high costs, labor and time. In addition, the supervised nature of dealing with known variables always may restrict our capacity of finding hidden variables, which may be playing important roles in functions of our interest.
Principal Components Similarity for Unsupervised Classification
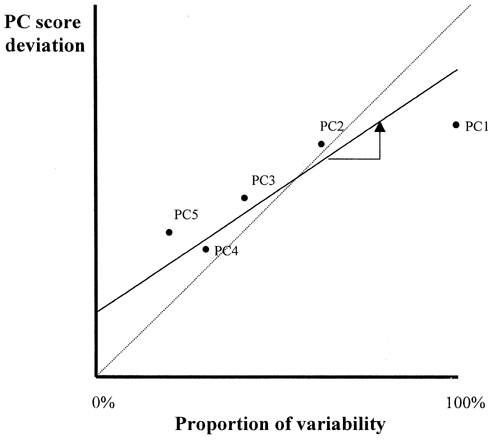
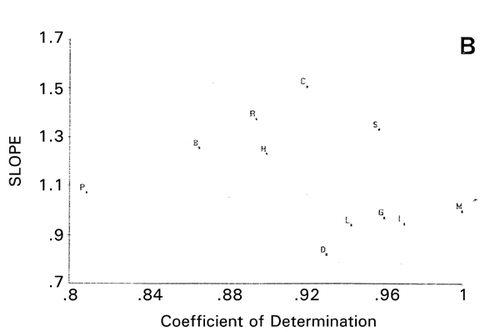
Principal component similarity plots slope (s) against coefficient of determination (r 2) obtained from linear regression analysis of deviations of sample PC scores from those of a reference sample (). An example of this plot is shown in the paper written by Vodovotz et al.Citation[7] If a sample is completely identical to the reference sample employed, the regression lines should be exactly on the 45° diagonal line with s = 1.0 and r 2 = 1.0.
Figure 1. Computation of slope and coefficient of determination for drawing PCS scattergram.
Advantages of PCS are:
| i. | More than three PC scores can be accommodated in 2D illustration; thus higher proportions (usually >85%) of the data variability can be accounted for than PCA using up to 3D plots. | ||||
| ii. | By rotating the reference sample, the classification ability of PCS can be more flexible than those based on distances, e.g., unsupervised cluster analysis and Kohonen net, which is used for converting ANN to a unsupervised mode. | ||||
| iii. | Because of the unsupervised nature of PCS, there is always a possibility of making new discovery by finding a previously unknown factor, which is playing an important role in QSAR as reported by Wang et al.Citation[8] | ||||
While ANN is ultimately a curve‐fitting technique, BNN, which is the currently a popular technique in DM, can add prior information to approach the underlying correlation. However, BNN cannot completely eliminate incorrect information once it is included in the original data; it seems that the effects of incorrect information are simply diluted during the BNN processing. In contrast, PCS can act as a scavenger of noises contaminated in the original data.
Recently, two approaches have been published for improving efficacy of PCA: the mean centered data matrix (MCDM) for information condensationCitation[9] and sequential projection pursuit (SPP) using genetic algorithms.Citation[10] The former is a pre‐processing of the original data prior to PCA computation. The MCDM can provide PC1 and PC2 together over 90% accumulated proportion of the total variance to be accounted for. However, it is risky to overload principal components as they may include effects of error variables more frequently than the regular PCA. The latter maximizes the entropy of the projection using genetic algorithm. The classification capacity of combinations of PP scores was compared with those of PC scores. For classification, finding of the best combinations of variables is more effective than improving the proportion of variances in the original data. At any rate, it is worth noting that the major function of PCA is dimensionality reduction and not classification.
Principal Component Similarity vs. Dissimilarity Based on Distances for Classification
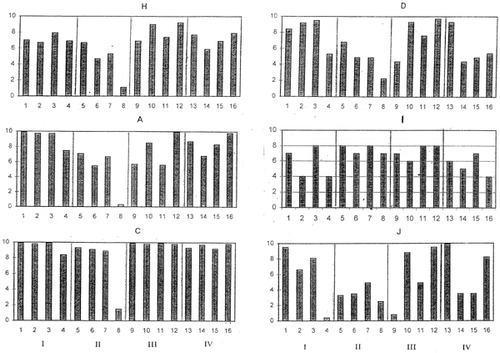
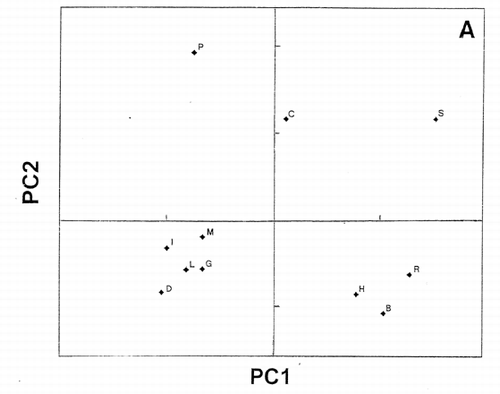
Importance of outliers has been already discussed in our previous publication.Citation[11] Definition of outliers for each group of objective functions would be useful in improving the accuracy of analytical data. An example was shown in panel evaluation of four attributes for samples I to IV of a meat product.Citation[12] Using panelist H as the reference, PCS classified panelists C, I, and J as outliers (). Meanwhile, when panelist A was chosen as the reference, different classification was obtained as shown in . Evaluation patterns made by the panelists are shown in . The evaluation scores of panelist I were intentionally assigned at random.
Figure 2. Principal component similarity scattergrams of panelists for evaluation of meat samples. A: Panelist H as the reference, B: Panelist A as the reference.
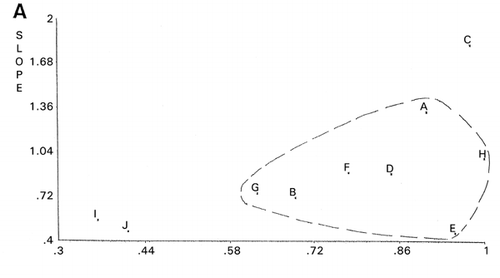
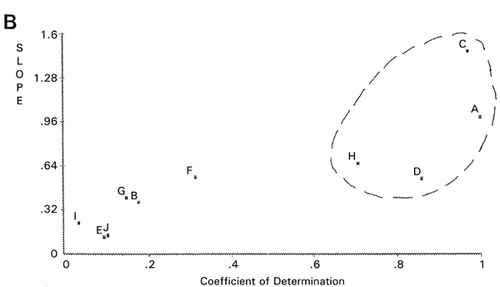
Figure 3. Evaluation patterns reported by selected panelists. Panelist H: Used as the reference, Panelists A and D: Good, Panelists C, I (random) and J: Outliers.
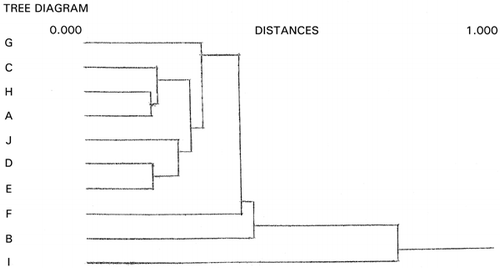
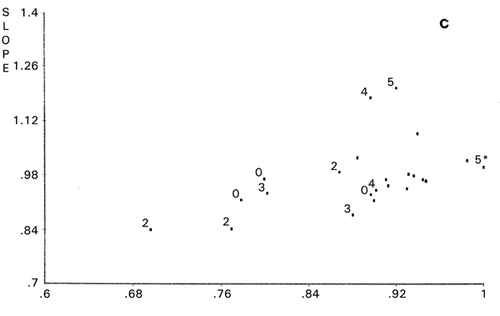
Dendrogram derived from cluster analysis of the above meat evaluation is shown in , which shows clearly that panelist I, a random scorer, ( and ) be an outlier. However, panelist J found to be an outlier in was not designated as an outlier by cluster analysis based on the distances (horizontal axes of ). Group ACDH found in was not classified in the same group in , in which ACH made a group. Furthermore, outlying behavior of B, F, and G in and grouping of J, D, and E were not observed in the PCS scattergrams (). Similar disagreement between cluster analysis and PCS was observed previously.Citation[7] Based on these observations, it may be concluded that distance‐based cluster analysis is unilateral compared to the more flexible, multilateral PCS.
Figure 4. Dendrogram of panelists.
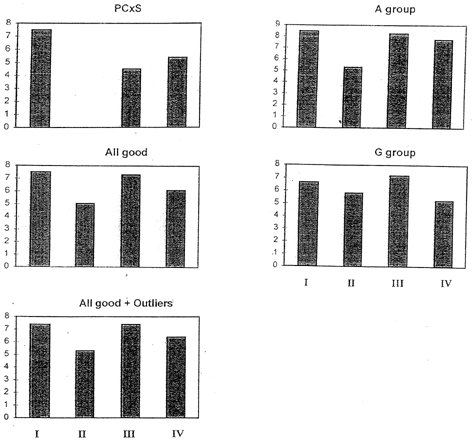
Ranking of samples by averaging the evaluation scores for four attributes are shown in . Averaging mitigates the effect of outliers as shown in “All good + Outliers” (= total) compared to “All good”. However, scores of outliers, for instance, show the rank IV > I > III > II by panelist J (), which is quite different from the rank computed by PC × S (PC score × proportion) for samples after eliminating scores made by outliers (all good panelists) as shown in . It was found that this PC × S rank was more realistic than other simply averaged ranks because sample II was so poorly evaluated that it was recalled from the market. Another aspect of outliers was revealed by Buydens et al.,Citation[13] who stated that an appropriate handling of ourliers was extremely important for DM of chemometrics. They elaborated that in knowledge discovery, these outliers may represent novelties, which should not be hastily disposed without careful attention.
Figure 5. Averages of evaluation scores reported for samples I–IV.
It is interesting to note that the currently popular classification methods are mostly based on distances, and use of similarity‐based classification is rather rare. Although in reality there have been many cases of similarity classifications in the literature, they have in most cases adhered to the familiar relationship:Citation[14]
Search for the Underlying Relationships
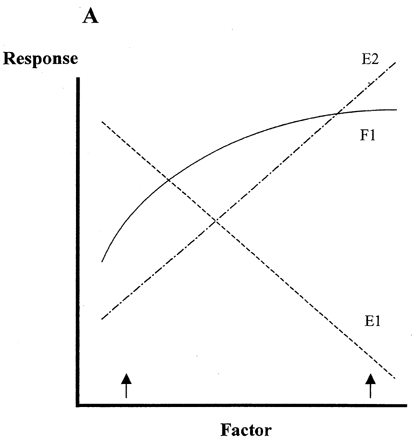
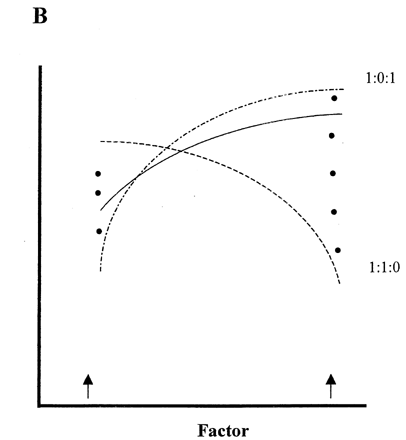
is a hypothetical illustration of the effects of linear error functions based on wrong information, which is accidentally included in measured data on the response surface of an independent variable (factor 1). We assume that there are two kinds of errors included in factor 1 (F1), namely negative error (E1) and positive error (E2) as shown in . As a result, the response surfaces may appear as shown in when ratios of true values (solid line) vs. error values are 1:1 (sums 1:1:0 and 1:0:1 of F1:E1:E2 in ). At lower ratios of errors' effects, the net response values will approach the true F1 values shown as solid circles at two factor levels at both ends of the factor scale (arrows on x‐axis). Since the same errors will not occur always with the same intensity, the response surface will slowly but infinitesimally approach to the true response surface (solid line in ) by increasing number of replications of experiments. However it will never reach the true values.
Figure 6. Effects of errors involved in the response surface of a factor. Dots at both ends in Fog. B show response values when errors mixing in true value are lower than 1.0 against 1.0 of true value (solid line).
Two approaches are proposed for searching for the true structure‐function relationships.
| i. | By discarding minor PC scores caused mostly by errors during PCS computation, the effects of noises may be eliminated, thus enabling approximation to the true relationships. | ||||
| ii. | Trend curves drawn on RCO maps by ignoring the effects of other factors may demonstrate the direction of search shift towards the true global optimum.Citation[15],Citation[16] | ||||
Random‐Centroid Optimization in Search for the Global Optimum
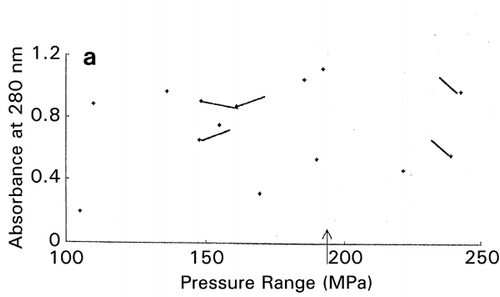
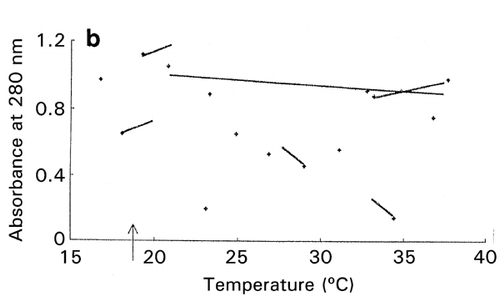
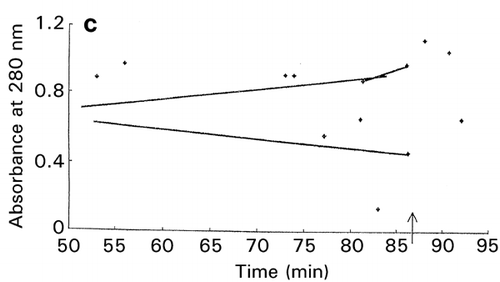
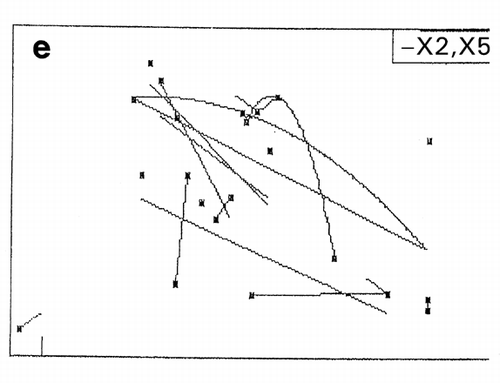
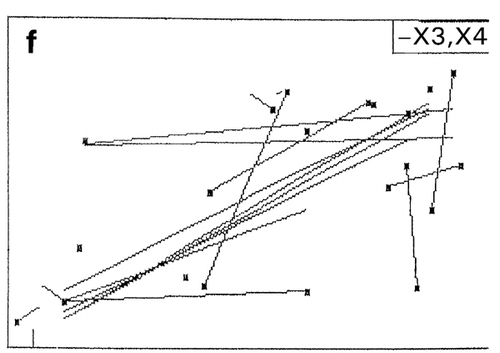
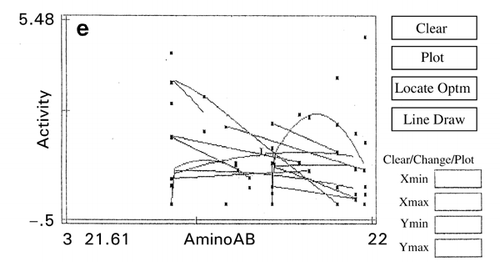
In RCO, at the end of each search cycle that combines searches using random design and centroid design, a mapping process approximates the response surfaces.Citation[15] The basic principle of response surface approximation on maps is to conduct quadratic curve fitting for data points or simply to link two data points with a straight line for a factor when other factors are within same ranges. However, due to a small number of data points available, especially in the early stage of optimization, lines drawn are usually excessively scars and speculative as shown on the left‐hand half (, and ) of the figure. To improve the quality of information about which direction the search should be shifted in the following cycle, one or two factors other than the factor of concern are intentionally ignored for computing data‐point linking to increase the number of trend curves as shown in the right‐hand half (, and ) of the figure. The trend curves thus drawn effectively improved the capacity of RCO to search for the global optimum in multimodal distributions despite a decline of credibility of the trend curves thus drawn.Citation[17] Information on the search direction toward the global optimum is an utmost necessity for global optimization by overwhelming the need for drawing accurate response surfaces.
Figure 7. Effect of trend‐curve drawing by ignoring factors in RCO mapping during RCO optimization of high‐pressure treatment of an enzyme.
Depending on the availability of information on the function in question, the mode of combination of PCS and RCO should be different during the search for the truth as follows. When a plenty of information already exist, PCS classification should precede RCO optimization to find whether the results obtained match one another. When less information available, RCO optimization should precede PCS classification, as the availability of adequate information is not a prerequisite of RCO because it is a random search.
If the error functions, especially when they are continually jeopardizing the true relation finding, this function is no longer errors; it is another distinct factor to be considered (). Therefore, our unsupervised DM using PCS is supposed to readily detect their existence. Conversely, if their effects are spontaneous, thereby working toward different directions inconsistently, i.e., both positive and negative errors occurring simultaneously, it could be genuine errors. These errors should gradually decrease their size within the total variance as the number of observations increases. This effect introduced by error variables may be almost completely eliminated by ignoring minor PC scores. Meanwhile, RCO can provide more direct evidence of the real relations and the truly influential factors should only be employed in search for the optimum by eliminating irrelevant factors to begin with or even during optimization progress of the search cycles. When near horizontal trend lines only appear on maps demonstrating no effect of changing this specific variable used for mapping, these uninfluential factors should be eliminated from the subsequent search cycle. This elimination would expedite search toward the global optimum.
Application of Principal Component Similarity Classification to Amino Acid Sequences of Antimicrobial Peptides
Principal component similarity analysis was applied to amino acid sequences of cationic antimicrobial peptides (CAP) to classify them based on physicochemical properties (hydrophobic, charge, helix and strand propensities and bulkiness) of amino acid residues. Amino acid residues in sequences were converted to scale values using different amino acid indices followed by PCS analysis. Using amino acid index employed, classification results can be explained for its principle underlying the classification.
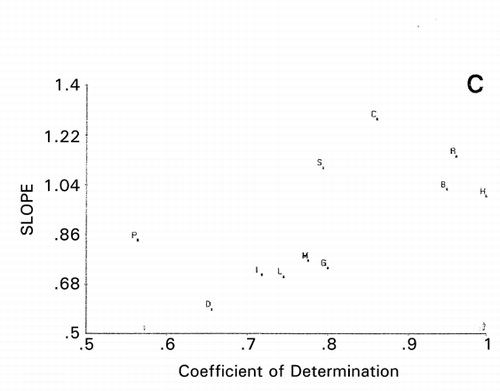
is the amino acid sequences of R‐type CAP, except for K‐type MSI‐95 (M). When peptide M was used as the reference, PCS scattergram as shown in was obtained using the charge index represented by isoelectric points of amino acids. To draw this scattergram, 10 PC scores were used with 85% variability accounted for, instead of only 53% and 40% using 3D and 2D () plots of PCA, respectively, implying that chances of missing important information are greater in PCA than PCS. Five protamines form a group separated from L (lactoferricin) and I (indolicidin), which have been reported that α‐helix is playing an important role in their antimicrobial activity.Citation[18],Citation[19] It is, therefore, likely that antimicrobial activity of protamines is due solely to their strong cationic properties, like polylysine and polyarginine. It is interesting to note that shorter salmon protamine is separated from longer protamines on the scattergrams. This can be explicitly seen in , in which protamines C, R, H, and G are clearly separated when S was used as the reference. As expected from , chicken protamine (C) is different from mammalian protamines H, R, and B (). This result shows that more accurate information can be obtained by using PCS than PCA.
Table 1. Sequences of major R‐type cationic antimicrobial peptides
Figure 8. Principal component similarity scattergram of cationic CAP using charge scale for amino acid residues in the sequences. S: Salmon protamine, H: Human protamine, B: Rabbit protamine, R: Rat protamine, C: Chicken protamine, P: PR39, L: Lactoferricin, M: MSI‐95, G: BNCP‐1, D: α1‐Defensin, I: Indolicidin.
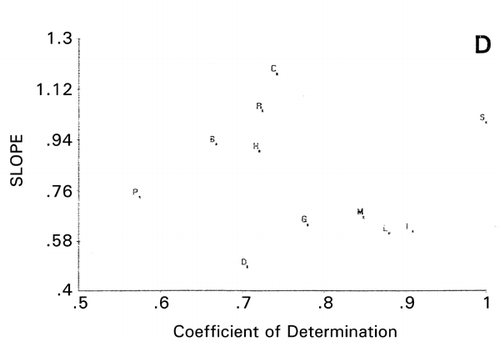
As lactoferricin derivatives, seven peptides consisted of 11 common amino acid residues: RWQWRMKKLGACitation[20] and eight peptides with 12–18 residuesCitation[18] were classified using PCS (the latter not shown). The most reasonable classification by comparing the minimum inhibitory concentration (MIC) against E. coli was obtained by using helix propensity of amino acids in the PCS computation. When seven peptides with 12–15 residuesCitation[18] excluding the sequences of very weak MIC were compared with the MIC against S. aureus (), a reasonable scattergram was obtained by using charge (). Stronger and weaker peptides in terms of antimicrobial activity are separated into data points in lower and higher slopes, respectively. Although this scattergram is based on charge, about the same classification was obtained when the helix scale was used in the PCS computation. These results corroborate the statement made by Rekdal et al.Citation[18] that helix region, net charge, asymmetry in charge distribution and chain‐length may be important factors for determining the antibiotic activity of lactoferricins.
Table 2. Antimicrobial activity of lactoferricin derivatives against S. aureus
Figure 9. Principal component similarity scattergram of lactoferricin derivatives using charge scale for amino acid residues in the sequences. Sequence numbers are the Derivative numbers shown in . *The reference used.
This method may be applied to any other homology patterns similar to in the future to classify sequences based on different chemical forces working between amino acid residues within the sequences. This finding may be important when relationships with a function are being investigated because the mechanisms underlying the function could be different between classes even though proteins are activated by the same function mechanism. Therefore, different QSAR should be investigated for proteins belonging to separate classes. In addition to the conventional homology analysis for searching for active site, a similarity analysis such as PCS can provide useful supplemental information to structure‐function study.
Random‐Centroid Optimization in Genetics Applied to Site‐Directed Mutagenesis
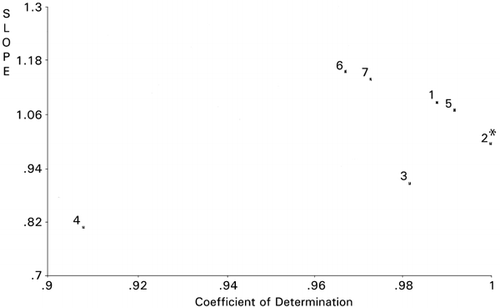
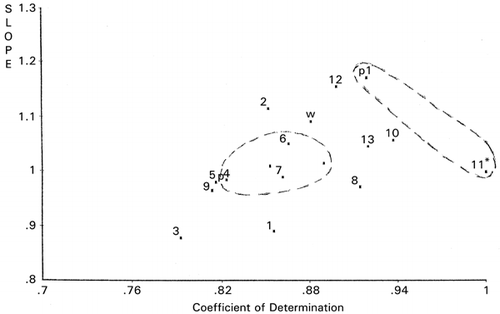
Site‐directed mutagenesis of the active site consisted of 16 amino acid residues in the sequence of B. stearothermophilus neutral protease was optimized by applying RCG.Citation[16] It was successful in elevating thermostability by 6.5°C from 68.3°C of the wild type protease in addition to a 32% hydrolyzing activity increase. Based on maps of RCG, it was concluded that lower hydrophobicity and bulkiness may be important for thermostability of the enzyme. The same data and the data from a proline substitution study in the active siteCitation[21] were combined and processed using PCS. shows that mutants 11 and p1 (a proline‐introduced mutant), which were more stable, and mutants 6, 7, 12, p3 and p4, which were less stable than the wild type (w) and other mutants make separate clusters. This PCS scattergram was drawn using bulkiness scale for amino acid residues. The fact that four other amino acid scales (helix, strand, hydrophobicity and charge) did not show better clustering in scattergrams may implicate the more effective demonstration by PCS of the importance of bulkiness in thermostability than the RCG maps as reported previously.Citation[16]
Figure 10. Principal component similarity scattergram based on bulkiness of neutral protease mutants at the enzyme active site. *The reference used.
Genetic optimization (RCG) was also carried out for the entire sequence of 120 amino acid residues of human cystatin C at two separate positions simultaneously, i.e., mutation zone I of position 1–35 (helix region) and mutation zone II of position 36–120 (strand region). The optimization has brought about a 5‐fold increase in papain‐inhibitory activity in a double mutant G12W/H86V and a 5°C improvement from 68.2°C of wild type cystatin in thermostability in a single mutant P13F.Citation[22] The trend lines appearing on the map in the first mutation zone indicate that residues near the N‐terminus around site 12 are responsible for increasing activity (). This is in agreement with the report that N‐terminal region nearby site Gly‐11 is important for the tight enzyme‐binding properties with papain.Citation[23]
Figure 11. Effect of amino acid scales on papain‐inhibitory activity and thermostability of human cystatin. a: mutation zone I, b: mutation zone II, c: helix in zone I, d: strand in zone II, e: bulkiness in zone I. Digit at the bottom between the terminal values of the abscissa is the value of maximal data point of each map.
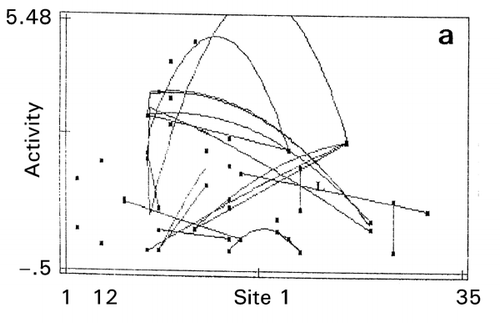
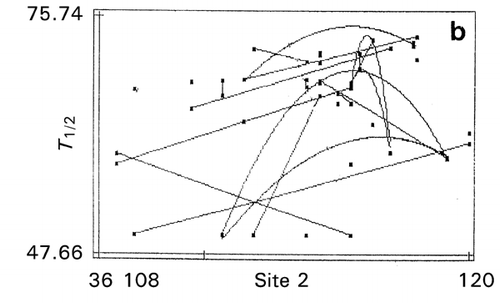
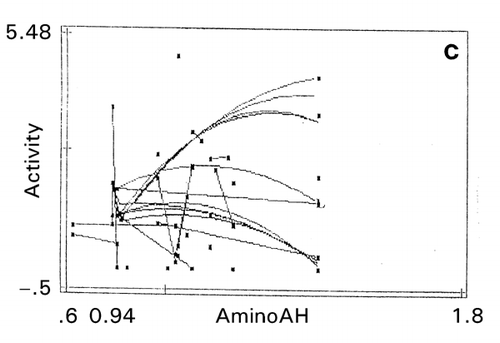
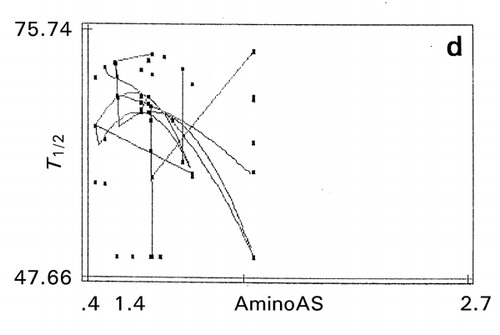
In the second mutation zone, the optimum thermostability exists near the site 108 that could be the substrate‐binding site affecting self‐association, thereby destroying the papain‐inhibitory activity. In the second zone, tends toward lower α‐helix propensity (opposite to the index values in ) and lower β‐strand propensity () were favorable for activity and thermostability, respectively. This is reasonable since conformational transition of α‐helix to β‐sheet is related to protein destabilization, which is caused by an amyloidosis.Citation[24] The map on bulkiness () indicates that the lower bulkiness in the first zone is effective in increasing bioactivity, as demonstrated by the trend lines in negative slopes. An exhaustive study of mutations at position 8–10 indicated that substitutions to glycine at these sites increased thermostability of cystatin.Citation[25]
By contrast, PCS scttergrams demonstrated the importance of bulkiness for zone I in activity based on the single site mutation (). Simultaneous two site mutations revealed that hydrophobicity and strand in zone II were playing important roles in thermostability and activity, respectively (). β‐Strand in zone II could, therefore, be a reason of amyloidosis of wild type cystatin, which encounter frequent inactivation or even insolubilization during its production by genetic fermentation. This strand/helix shift in zone II is supported by the sequence PCS scattergrams in by the appearance of multiples 3–5 in activity enhancement separately from multiple 0–2, which did not occur in single mutants (dots without labeling) in zone I. The improved thermostability and activity, especially the latter, might have been derived from the decreasing strands upon mutation. This important discovery was more clearly evidenced by PCS than RCG maps. Like in the case of single mutations, the PCS scattergrams demonstrated to be a useful supplement to RCG mapping also in the case of double mutations.
Figure 12. Sequence PCS scattergrams of human cystatin mutants. a: Single site mutants at zone I of human cystatin when bulkiness was used as amino acid scale. •: Highly active, ▴: Less active than wild type, b: Double site mutants at sites I and II of human cystatin when hydrophobicity was used as amino acid scale, •: Highly stable, ▴: Less stable than wild type. *The reference used.
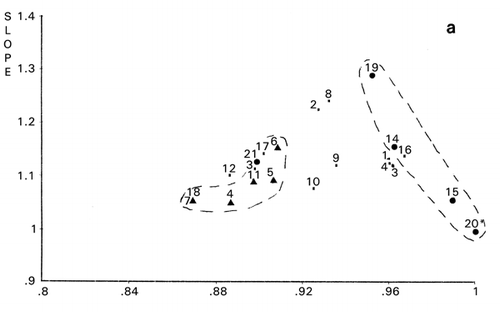
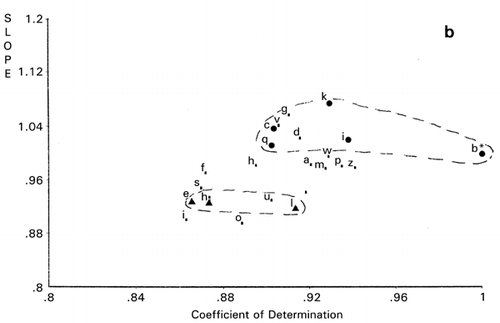
Figure 13. Sequence PCS scattergrams of double mutants of human cystatin using helix propensity (c) and strand propensity (d). Mutant 12W86V was used as the reference. Twelve and 11 PC scores were used in PCS computation to account for 88 and 85%, respectively, of the total variances. Digit labels show multipliers of the inhibitor activity compared to that of wild type after rounded off. Dots without labels are for single site mutants at Zone I. *The reference used.
RCG was again applied to a larger molecule of novel cyclodextran‐glucanotranferase (CITase) with 972 residues. Because of the greater size with relatively unknown molecule, a half‐space design (HSD) developed for large search space optimizationCitation[17] was applied to the first half (sites 1–441). Of the total of 81 mutants (one, two and 1–2 mixture of 27 two‐site mutations), up to about four‐fold activity increase but with almost no thermostability improvement was observed in the mutant A452N. It appeared that lower helix and strand similar to afore‐mentioned cystatin and higher charges were favorable for activity improvement. This result is in agreement with the general understanding that carboxylic amino acids are playing an important role at the active sites of glycoside hydrolases. A greater rate of complete inactivation during mutation (71% vs. less than 3% of cystatin) may be due to more severe amyloidosis of CITase.
Artificial Neural Networks for Quantitative Structure Activity Relationship Study
According to Burden and Winkler,Citation[5] BNN was superior to partial least squares regression (PLS) as well as maximum likelihood NN in QSAR. In our recent paper on ANN of bacterial growth,Citation[26],Citation[27] ANN was superior to the prediction made by the microbiological modeling approach. By using important PC scores or factors closely correlated to them as can be found from component loadings computed during ANN prediction, it is possible to obtain highly accurate QSAR prediction.
Since 1993 when our ANN prediction of foaming and emulsifying properties of food proteins was reported,Citation[28] ANN techniques and computer programs have been dramatically improved in capacity. An example is “Statistica” (brand name) Neural Networks (STNN) from StatsoftCitation[6] based mostly on Bishop,Citation[1] who added many important techniques such as density estimation, error functions, parameter optimization algorithms, data pre‐processing and incorporation with Bayes' theorem. Statistica neural network includes Bayesian NN and is capable to use PCA to select input variables for dimensionality reduction among many other choices, such as Kohonen nets and genetic algorithm, to train networks. When it fails, the autoassociative networkCitation[29] can be used for conducting nonlinear PCA. Furthermore, STNN can compute sensitivity to rank the importance of input variables and draw scatterplots as well as 3D response surfaces without relying on other algorithms. Another important feature of the STNN is an easy‐to‐use wizard for network creation including an automated network designing system. By maintaining the number of input variables and hidden layers to minimum, the well‐documented problem of overlearning in ANN can be avoided. However, it is worth noting that this treatment in STNN is different from the preliminary classification of data using PCS in our approach because PCS is superior to PCA as far as the classification capacity is concerned.
Despite irrelevance to DM in this study because of application of ANN to predict bacterial growth rate, the above ANN papersCitation[26],Citation[27] are examples of full exploitation of the revitalized modern ANN technology. Usually, protein functions cannot be clearly explained directly from the properties of the constituting amino acid residues as reported by Gromiha and Ponnuswamy.Citation[30] In addition, presence of the multicolinearity problem cannot be ignored in their paper because of too many input variables used for too small number of data. It is premature to anticipate the perfect application of the DM study to elucidate the behavior of macromolecules like proteins.
The protocol of our DM technique is: Upon classification of sequences using homology, it is recommended to carry out preliminary structure‐function study by applying PCS first (e.g., and ). Then together with function data assessed separately, PCS computation is repeated to find more reliable function‐related classification (e.g., and ). Subsequently, RCG optimization is carried out to confirm the results obtained in the preceding PCS study. Finally, ANN can be applied for finding the structure‐function relationships quantitatively for individual classes of sample found by PCS. In addition to PCS scattergrams, RCG maps could be useful in elucidating the mechanism of function in study. The importance of appropriate classification prior to DM should be emphasized, as the direct application of QSAR has to explain excessive number of factors involved in protein functions, so‐called curse of dimensionality. The most important, truly influencing factors should be singled out prior to DM for QSAR study.
Conclusion
Noises in data to be used in ANN computation for DM can be eliminated by employing the only major influential PC scores belonging to a group selected by PCS and/or using trend curves on RCO maps based on important factors directing toward the true optimum. This new strategy of unsupervised DM could correctly select the most effective factors in elucidating underlying mechanisms of protein functions. This approach is especially useful in the case when truly influencing factors are unknown beforehand. A definite advantage of this new protocol is potential of making unexpected new discovery. It is critical to eliminate uninfluential factors (noises) during DM computation to find underlying structure‐function relationships of protein molecules.
Acknowledgment
We are grateful for a financial support from the Natural Sciences Engineering Council of Canada.
References
- Bishop , C. M. 1995 . Neural Networks for Pattern Recognition 17 385 – 387 . Oxford , UK : Clarendon Press .
- Yuan , B. , Wang , X. Z. and Morris , T. 2000 . Software analyzer design using data mining technology for toxicity prediction of aqueous effluents . Waste Manag. (Oxford) , 20 : 677 – 686 .
- Tropsha , A. , Cho , S. J. and Zheng , W. 1999 . “New tricks for an old dog”: development and alication of novel QSAR methods for rational design of combinatorial chemical libraries and database mining ” . In Rational Drug Design Edited by: Parrill , A. L. and Reddy , M. R. 198 – 211 . Washington, DC : American Chemical Society .
- Heckerman , D. 1997 . Bayesian networks for data mining . Data Min. Knowl. Disc. , 1 : 79 – 119 .
- Burden , F. R. and Winkler , D. A. 2000 . A QSAR model for the acute toxicity of substituted benzenes to Tetrahymena pyriformis using Bayesian‐regulated neural networks . Chem. Res. Toxicol. , 13 : 436 – 440 .
- 1999 . Statistica Neural Networks & Addendum for Version 4 Tulsa , OK : Statsoft . Statsoft, www.statsoft.com
- Vodovotz , Y. , Arteaga , G. E. and Nakai , S. 1993 . Principal component similarity analysis for classification and its application to GC data of mango . Food Res. Int. , 26 : 355 – 363 .
- Wang , Z. H. , Dou , J. , Macura , D. , Durance , T. D. and Nakai , S. 1998 . Solid phase extraction for GC analysis of beany flavor in soymilk . Food Res. Int. , 30 : 503 – 511 .
- Kellner , R. , Mermet , J. M. , Otto , M. and Widner , H. M. 1998 . Analytical Chemistry 775 – 808 . Weinheim , , Germany : Wiley‐VCH .
- Guo , Q. , Wu , W. , Questier , F. , Massart , D. L. , Boucon , C. and de Jong , S. 2000 . Sequential projection pursuit using genetic algorithms for data mining of analytical data . Anal. Chem. , 72 : 2846 – 2855 .
- Nakai , S. , Amantea , G. , Nakai , H. , Ogawa , M. and Kanagawa , S. 2002 . Definition of outliers using unsupervised principal component similarity analysis for sensory evaluation of foods . Int. J. Food Properties , 5 : 289 – 306 .
- Nakai , S. , Dou , J. and Richards , J. F. 2000 . New multivariate strategy for panel evaluation using principal component similarity . Int. J. Food Properties , 3 : 149 – 164 .
- Buydens , L. M.C. , Reijmers , T. H. , Beckers , M. L.M. and Wehrens , R. 1999 . Molecular data‐mining: A challenge for chemometics . Chemom. Intell. Lab. Syst. , 49 : 121 – 133 .
- Krzanowski , W. J. 1988 . Principles of Multivariate Analysis: A User's Perspective Oxford , UK : Clarendon Press .
- Nakai , S. , Dou , J. , Lo , K. V. and Scaman , C. H. 1998a . Optimization of site‐directed mutagenesis. 1. A new random‐centroid optimization program for Windows useful in research and development . J. Agric. Food Chem. , 46 : 1642 – 1654 .
- Nakai , S. , Nakamura , S. and Scaman , C. H. 1998b . Ibid, 2. Application of random‐centroid optimization to one‐site mutation of B. sterothermophilus neutral protease for improving thermostability . J. Agric. Food Chem. , 46 : 1655 – 1661 . The RCO and RCG programs both can be downloaded from ftp://ftp.agsci.ubc.ca/foodsci/ and run on Windows
- Nakai , S. , Saeki , H. and Nakamura , K. 1999 . A graphical solution of multimodal optimization to improve food properties . Int. J. Food Properties , 2 : 277 – 294 .
- Rekdal , Ø , Andersen , J. , Vorland , L. H. and Sevendsen , J. S. 1999 . Construction and synthesis of lactoferrin derivatives with enhanced antibacterial activity . J. Peptide Sci. , 5 : 32 – 45 .
- Falla , T. J. and Hancock , R. E.W. 1997 . Improved activity of a synthetic indolicidin analog . Antimicrog. Agents Chemoth. , 41 : 771 – 775 .
- Kang , J. H. , Lee , M. K. , Kim , K. L. and Hahn , K.‐K. 1996 . Structure‐biological activity relationships of 11‐residue highly basic peptide segment of bovin lactoferrin . Int. J. Peptide Protein Res. , 48 : 357 – 363 .
- Nakamura , S. , Tanaka , T. , Yada , R. and Nakai , S. 1997 . Improving the thermostability of B. stearothermophilus neutral protease by introducing proline into the active site helix . Protein Eng. , 10 : 1283 – 1269 .
- Ogawa , M. , Nakamura , S. , Scaman , C. H. , Jing , J. , Kitts , D. D. , Dou , J. and Nakai , S. 2002 . Enhancement of proteinase inhibitory activity of recombinant human cystatin C using random‐centroid optimization . Biochim. Biophys. Acta , 1599 : 115 – 124 .
- Björk , I. , Brieditis , I. and Abrahamson , M. 1995 . Probing the functional role of the N‐terminal region of cystatins by equilibrium and kinetic studies of the binding of Gly‐11 variants of recombinant human cystatin C to target proteinases . Biochem. J. , 306 : 513 – 518 .
- Takahashi , Y. , Ueno , A. and Mihara , H. 1998 . Design of a peptide undergoing α‐β structure transition and amyloid fibrilogenesis by the introduction of a hydrophobic defect . Chem. Eur. J. , 4 : 2475 – 2483 .
- Hall , A. , Håkannson , K. , Mason , R. W. , Grubb , A. and Abramson , M. 1995 . Structural basis for the biological specificity of cystatin C . J. Biol. Chem. , 270 : 5115 – 5121 .
- Lou , W. and Nakai , S. 2001a . Artificial neural network‐based predictive model for bacterial growth in a simulated medium of modified atmosphere packed cooked meat products . J. Agric. Food Chem. , 49 : 1799 – 1804 .
- Lou , W. and Nakai , S. 2001b . Application of artificial neural networks for predicting thermal inactivation of bacteria. A combined effect of temperature, pH and water activity . Food Res. Int. , 34 : 573 – 579 .
- Arteaga , G. E. and Nakai , S. 1993 . Prediction of protein functionality using artificial neural networks: Foaming and emulsifying properties . J. Food Sci. , 58 : 1152 – 1156 .
- Kramer , M. A. 1991 . Nonlinear principal component analysis using autoassociative neural networks . AIChe J. , 37 : 233 – 243 .
- Gromiha , M. M. and Ponnuswamy , P. K. 1993 . Relationship between amino acid properties and protein compressibility . J. Theor. Biol. , 165 : 87 – 100 .