
Abstract
Atherosclerosis is an aging disease in which increasing age is a risk factor. Modified low-density lipoprotein (LDL) is a well-known risk marker for cardiovascular disease. High-plasma LDL concentrations and modifications, such as oxidation, glycosylation, carbamylation and glycoxidation, have been shown to be proatherogenic experimentally in vitro and in vivo. Atherosclerosis results from alterations to LDL in the arterial wall by reactive oxygen species (ROS). Evidence suggests that common risk factors for atherosclerosis raise the likelihood that free ROS are produced from endothelial cells and other cells. Furthermore, oxidative stress is an important factor in the induction of endothelial senescence. Thus, endothelial damage and cellular senescence are well-established markers for atherosclerosis. This review examines LDL modifications and discusses the mechanisms of the pathology of atherosclerosis due to aging, including endothelial damage and oxidative stress, and the link between aging and atherosclerosis.
Evidence from the past several decades has suggested that modification of low-density lipoprotein (LDL) – especially oxidative changes – mediates the pathogenesis of atherosclerosis in humans and animals (Citation1, Citation2). Although aging is a major risk factor that precipitates atherosclerosis, there are many other factors that can cause the disease (Citation3, Citation4). The major risk factors for atherosclerosis are serum lipid concentrations, smoking, and hypertension (Citation5).
Gender appears to be another determinant (Citation6). Men are approximately twice as likely to develop atherosclerosis compared with age-equivalent women. Yet, merely half of the variability in the incidence of atherosclerosis and coronary heart disease is because of these factors. Genetics might have some influence, but age-related conditions might have a more prominent function for the development of the disease (Citation7). Furthermore, excess food intake affects obesity and diabetes, both of which are well-known independent risk factors of atherosclerosis and growing epidemics in an aging population (Citation8).
The presence of multiple risk factors can accelerate the progression of atherosclerosis. Modified LDL has a significant function in the development of endothelial dysfunction (Citation9), which is considered an early marker of atherosclerosis (Citation10). More extensive modification of LDL induces oxidative stress and accelerates senescence in human endothelial progenitor cells and endothelial cells (Citation11, Citation12). The hallmark of endothelial dysfunction is impaired endothelium-dependent vasodilatation, which is mediated by nitric oxide (NO). A defect in NO production or activity has been proposed as a significant mechanism of endothelial dysfunction and a contributor to atherosclerosis (Citation10).
The focus of this study is to discuss the function of protein modifications, particularly those to LDL, in aging-induced atherosclerosis, and how these molecules mediate senescence-related signaling and endothelial damage during atherosclerosis.
Chemical modifications to LDL related to atherosclerotic processes
Cardiovascular disease (CVD) remains the leading cause of mortality in developed countries, and LDL analysis is one of the most widely used diagnostic indices to evaluate and predict atherosclerosis risk (Citation2). The levels of LDL and other lipoproteins, however, usually have limited predictive value because many factors affect LDL concentrations in the arterial wall, the rate and extent of LDL modifications, and the accumulation of LDL in vascular cells and disease progression (Citation13).
Several chemically modified LDL species have been reported since the 1970s, including oxidized LDL (oxLDL) (Citation14, Citation15), acetylated LDL (acLDL) (Citation16, Citation17), ethylated (Citation17), methylated (Citation17), and glycated LDL (gLDL) (Citation18). Based primarily on studies of oxLDL and acLDL, endothelial cell injury by modified LDLs has been commonly accepted to initiate atherosclerotic processes (Citation19). These LDLs also promote vascular injury by increasing oxidative stress and accelerating senescence of endothelial progenitor cells through modifications and damage to DNA (Citation12).
It is widely believed that the modification of various components of LDL alters the properties that contribute to its atherogenic effects when interacting with cells of the arterial wall. Specifically, changes in apoliprotein B (apoB; the surface protein of LDL) destroy the ability of LDL to bind LDL receptor (Citation20). LDL modifications and especially oxidation might mediate the induction of atherogenesis through scavenger receptors (SRs) on macrophages and endothelial cells (Citation21).
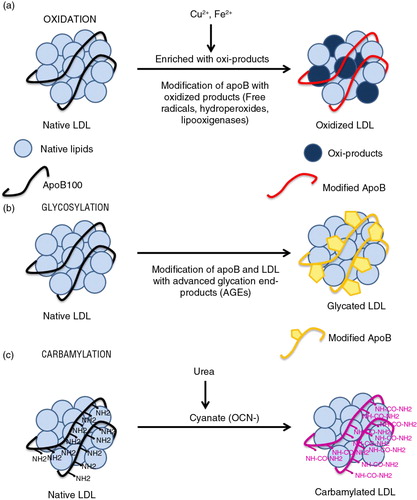
Several pathways and processes lead to harmful modification of LDL ().
Fig. 1 LDL modifications. (a) Oxidation: oxidized product-induced native LDL oxidation and modifications of apoB amino acids. (b) Glycosylation: modification of LDL and apoB by advanced glycosylation end-products (AGEs). (c) Carbamylation: cyanate from urea, which binds to NH2 groups in proteins – inducing their carbamylation – is generated by spontaneous dissociation from urea.
LDL oxidation
In 1981, Henriksen et al. (Citation22) discovered that native LDL, which is incubated overnight with cultured endothelial cells, converts to a form (endothelial cell-modified LDL) that is recognized specifically by peritoneal macrophages with high affinity. They proposed that this endothelium-induced modification is the step that permits rapid LDL uptake and foam cell formation. Later studies reported that during its incubation with endothelial cells (and with several cell types), LDL undergoes oxidative changes (Citation23), constituting the basis of the oxidative modification hypothesis of atherogenesis.
Oxidative modification of lipid and proteins occurs frequently in many pathophysiological processes in vivo, and it is well established that LDL undergoes oxidative alterations that confer atherogenic properties to it (Citation24).
Under oxidative stress (free radicals, hydroperoxides, and lipooxigenases), lipid molecules that contain native lipids in LDL are easily oxidized. A variety of lipid oxidation products is formed, and subsequently, apoB is covalently modified by these oxidized lipids (Citation25) (a).
OxLDL is taken up by SRs on macrophages, which then become lipid-laden foam cells, the pathological hallmark of early atherosclerotic lesions (Citation26). The concentration of LDL that is needed to induce foam cell formation (2 mg/ml) is 40-fold greater than that of oxLDL (50 μg/ml) (Citation27).
OxLDL has a wide range of properties that are expected to be proatherogenic, many of which are affected by oxidized phospholipids in oxLDL (Citation28). Oxidized phospholipid products are also the principal epitopes that are recognized by autoantibodies as oxLDL and the major structural feature by which SRs recognize oxLDL as a ligand.
Recently, oxLDL has been associated with changes in endothelial cell homeostasis through the suppression of important endothelial microRNAs (miRNAs) (Citation29). Further, the miRNAs are a link between endothelial injury and inflammation and can mediate inflammatory activation and lipid accumulation in macrophages during atherosclerosis (Citation29).
LDL glycosylation
Clinical studies have demonstrated increased levels of advanced glycosylation end products (AGEs) on LDL from diabetics compared to normal individuals (Citation30). AGEs accumulate continuously on long-lived vessel wall proteins with aging and at higher rates in diabetes (Citation31). One of the mechanisms of accelerated atherosclerosis in diabetes is the non-enzymatic reaction between glucose and proteins or lipoproteins in arterial walls. The degree of non-enzymatic glycation is determined primarily by glucose concentrations and the time of exposure (Citation31). Although non-enzymatic glycosylation of LDL occurs in all subjects, it has more adverse effects in people with diabetes mellitus.
Glycosylation of LDL apoB (b) occurs mainly on positively charged lysine residues in the putative LDL receptor-binding domain, which is essential for the specific recognition of LDL by LDL receptor (Citation32). This modification leads to a loss of electropositive charges on gLDL, decreasing its affinity toward LDL receptor and consequently increasing its mean lifetime in plasma (Citation33). Greater LDL glycosylation correlates with glucose levels, and AGE–apoB levels are up to fourfold higher in diabetic patients (Citation30). Once formed, AGE-protein adducts are stable and virtually irreversible.
Glycosylation of apoB results in significant impairing of LDL receptor-mediated uptake, decreasing the in vivo clearance of LDL compared with native LDL (Citation34). Thus, gLDL is poorly recognized by LDL receptor and binds preferentially to SRs on human macrophages. As LDL glycosylation enhances its uptake by human aortic intimal cells (Citation30) and monocyte-derived macrophages (Citation35) on stimulation of foam cell formation, the recognition of gLDL by the SR pathway is believed to promote intracellular accumulation of cholesteryl esters and atherosclerosis.
Prolonged hyperglycemia is now recognized to be the primary casual factor in the pathogenesis of diabetic complications (Citation36, Citation37). Hyperglycemia induces a large number of alterations in vascular tissue that potentially accelerate atherosclerosis. Two major mechanisms have emerged that account for most of the pathological alterations observed in the vasculature of diabetic animals and humans: [1] non-enzymatic glycosylation of proteins and lipids and [2] oxidative stress. Notably, these mechanisms are not independent (Citation38).
Another atherogenic effect of glycation is the increased susceptibility of gLDL to oxidative modification (Citation39) (). Glycosylation process occurs on the apoB (Citation40) and phospholipid (Citation30) components of LDL. Therefore, glycation raises the susceptibility of LDL to oxidative modification (Citation41), which is considered a critical step in its atherogenicity. Doubly modified LDL might have greater pro-atherosclerotic potential compared with gLDL. A number of reports from different groups have described shortened oxidative lag periods during Cu2+-mediated oxidation of gLDL and LDL in diabetic patients (Citation42, Citation43). Thus, glycosylation of LDL is not only noxious per se but also it promotes oxidation of LDL.
Fig. 2 LDL double modification. The glycation of LDL particles renders it more prone to oxidation.

LDL carbamylation
Carbamylation is a post-translational and non-enzymatic modification in which amine-containing residues react with cyanate, a compound that derives from urea or thiocyanate. The carbamylation of LDL occurs due to spontaneous, non-enzymatic chemical modification of apoB by urea-derived cyanate. Urea dissociates spontaneously to cyanate and ammonia in aqueous solutions, elevating cyanate levels (OCN-) (Citation44) (c). The active form of cyanate, isocyanic acid, reacts irreversibly with the NH2 and N-terminal groups of amino acids (Citation45). When a molecule of cyanate is removed by carbamylation, a new molecule of cyanate is formed to restore the equilibrium between urea and cyanate. Carbamylation of a protein is usually associated with a partial loss of function (Citation46, Citation47), but carbamylated proteins usually have no positive effect in humans or animals and are dispensable for normal metabolism.
Protein carbamylation is frequent in patients with chronic renal failure (CRF) and heavy smokers (Citation48). CVD rates are up to 30 times higher in patients with CRF compared with the general population, and morbidity and mortality rates rise, even in the initial stages of the disease (Citation48, Citation49). In particular, the incidence of atherosclerosis is high in CRF patients with uremia, but the pathogenic events that contribute to uremic atherosclerosis are poorly understood (Citation50). As suggested above, modified LDLs are significant atherogenic factors (Citation51, Citation52), and carbamylated LDL (cLDL) has been recognized to be a type of modified LDL (Citation12). Essentially, carbamylation of LDL could be is an important mechanism that impacts high-risk atherosclerotic individuals with increased urea (renal insufficiency) or thiocyanate (tobacco smoking). LDL carbamylation is more extensive in patients with end-stage kidney disease, especially those with atherosclerosis (Citation45, Citation53).
It remains unknown whether protein carbamylation induces atherosclerosis and whether the atherogenic process proceeds through LDL or other targets. Notwithstanding, there is a report that demonstrates the role of carbamylated plasma proteins have been implicated in the development of cardiovascular complications (Citation54). For animals, uremic mice with high-plasma cLDL have more severe atherosclerosis (Citation3). However, future therapies might be aimed at reducing cLDL and its effects.
Endothelial damage and atherosclerosis
Endothelial dysfunction of large- and medium-sized arteries is characterized by impaired NO-mediated vasodilatation and appears to play a pivotal role in atherosclerosis; both endothelial dysfunction and atherosclerosis are induced by coronary artery disease risk factors, such as cigarette smoking, hypertension, diabetes mellitus, hyperhomocysteinemia, serum lipid concentrations, and hypercholesterolemia (Citation55). Several processes, such as senescence and increased oxidative stress, affect endothelial dysfunction and the development of atherogenesis (Citation56, Citation57).
Aging endothelial cells
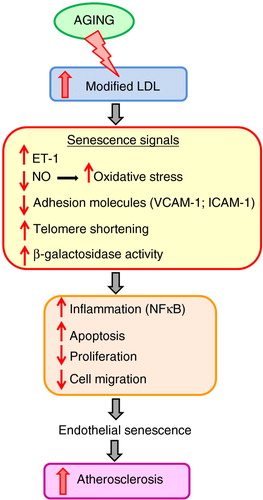
Aged endothelial cells become flatter and more enlarged and have an increasingly polypoid nucleus – all of which are associated with cellular senescence (Citation58, Citation59). These changes are accompanied by alterations in cytoskeleton integrity, proliferation, angiogenesis, and cell migration. Senescent endothelial cells produce less NO (Citation60) and release more endothelin-1 (ET-1) (Citation61). Late-passage endothelial cells also downregulate adhesion molecules, vascular cell adhesion protein 1 (VCAM-1), and intracellular adhesion molecule-1 (ICAM-1); show increased activation of nuclear factor (NF)-κB; and experience greater susceptibility to apoptosis (Citation59). In addition, NF-κB activation enhances endothelial cell senescence and can, therefore, reduce endothelial regeneration at sites that are prone to atherosclerosis (Citation62).
Furthermore, there are marked age-associated changes in function and activity (Citation63). Thus, endothelial cell senescence is associated with a loss of endothelial cell function and a shift toward a pro-inflammatory and pro-apoptotic state – which are predicted to enhance monocyte migration into the vessel wall (Citation64). In addition, it has been shown that both oxLDL and LDL induce accelerated senescence, as evidenced by telomere shortening and β-galactosidase activity (Citation12). All of these processes correlate with increasing severity of atherosclerosis ().
Fig. 3 Mechanisms of endothelial cell senescence during aging, initiating the atherogenic process.
Oxidative stress in endothelial cells
NO is a free radical signaling molecule that is produced by nitric oxide synthase enzymes (Citation65, Citation66). Some of the beneficial properties of NO include vasodilatation, promotion of endothelial cell survival, and inhibition of cell proliferation and migration (Citation67), which might protect against atherosclerosis. Several studies have reported a decrease in nitric oxide activity with aging.
Oxidative stress might be involved more broadly in atherogenesis. Reactive oxygen species (ROS) are produced in the endothelium, smooth muscle cells, and adventitia. There is a significant amount of evidence that implicates them in vasomotor activity, smooth muscle cell growth, expression of adhesion molecules, apoptosis, activation of metalloproteinases and, of course, lipid oxidation (Citation68). These oxidation-related processes can accompany oxidation of LDL or occur independently of it. Indeed, lipid peroxidation seems to be an important mechanism in the development of endothelial dysfunction in certain pathologies (Citation69, Citation70).
Aging and atherosclerosis
Age is a non-modifiable risk factor of atherosclerosis. Aging is not simply wear and tear but is an active process, like atherosclerosis, with which it shares mechanisms, such as endothelial dysfunction. Several studies have shown that endothelial cell function is compromised with aging and that endothelial cell senescence mediates the evolution of age-associated CVDs, such as atherosclerosis (Citation71, Citation72). Older primates and rodents develop more extensive atherosclerosis than younger animals when both groups are fed an atherogenic diet (Citation73).
Age-accelerated vascular injury is commonly considered to result from increased oxidative stress, leading to inflammation and endothelial dysfunction, but no definite mechanisms have been identified (Citation74). The accumulation of oxidative damage is believed to contribute to aging and its associated diseases (Citation75). Tissues from aged animals show an increased generation of ROS, leading to altered mitochondrial function, damage to vascular cells with age-associated remodeling, and oxidation of lipids, rendering them more atherogenic (Citation76).
Age and other atherosclerotic risk factors upregulate pathways that increase ROS production, whereas antioxidant mechanisms are enhanced and decrease in aging (Citation77, Citation78). Caloric restriction attenuates the increases in inflammation, oxidative stress, and endothelial dysfunction that accompany aging and extend the median lifespan in several animals (Citation79, Citation80). Conversely, caloric excess that leads to obesity worsens these factors and promotes insulin resistance, metabolic syndrome, and diabetes, all of which are frequently associated with aging and are established contributors to the accelerated atherosclerosis (Citation81, Citation82). Hypertensive patients are more prone to atherosclerotic lesions and acute ischemic events than normotensive individuals (Citation83).
The risk factors of atherosclerosis are well known, including hypertension (Citation84, Citation85), diabetes (Citation86, Citation87), serum total LDL cholesterol (Citation88–Citation90), smoking (Citation91, Citation92), and obesity (Citation93, Citation94). Increasing evidence indicates that aging is also an important risk factor for atherosclerosis and persists as an independent contributor when all other factors are controlled for. Premature or accelerated vascular aging can be promoted by cardiovascular risk factors (Citation95, Citation96), and cellular senescence is observed in patients with atherosclerosis. Atherosclerosis is thus a disease of organismal aging and cellular senescence.
Atherosclerosis is a condition caused by lipid-induced inflammation of the vessel wall that is orchestrated by the complex interplay of various types of cells, such as endothelial cells, smooth muscle cells, and macrophages. Hypercholesterolemia, especially high concentrations of serum LDL cholesterol – is considered a major factor of atherosclerosis. However, oxidation of LDL appears to have significant function in the early development of atherosclerosis through the formation of macrophage-derived foam cells on the arterial wall (Citation97, Citation98). Macrophages bind and take up oxLDL particles – but not nonoxidized, native LDL particles – via SRs (Citation99).
Aged vessels undergo several characteristic pathological processes, many of which are also seen in atherosclerosis (Citation100, Citation101).
Conclusions
CVD remains one of the most significant chronic diseases worldwide with regard to morbidity and mortality. Although CVD is associated with aging, accumulating evidence supports that CVD is linked to vascular cell senescence; considering that CVD occurs in aged people when vascular cells undergo replicative senescence and in patients whose risk factors promote premature senescence. In particular, senescence in endothelial cells seems to be the initial step in the cascade of events that lead to CVD. Thus, the factors that affect senescence in endothelial cells and the mechanisms by which they do so must be identified to develop new biomarkers and therapeutic tools against CVD. In this regard, determining how endothelial cells are affected by structural changes in lipoproteins, such as LDL, is a promising approach for future studies on CVD.
Authors' contributions
RR chose the topic of the review and enlisted MA, CL, and JC as coauthors. RR, JC, and MA defined the article topic and outline. MA, RR, and JC drafted the paper, which was subsequently edited by the other authors, all of whom have approved the final version.
Conflict of interest and funding
The authors declare no conflict of interest.
Acknowledgements
This work was supported by Plan Nacional Proyectos de Investigación en Salud of Instituto de Salud Carlos III (ISCIII) Fondos Feder Grants (PI11/01536, PI12/01489 and PI14/00806), Junta de Andalucía Grants (JA0797-2010, P010-CTS-6337, P11-CTS-7352), and Fundación Nefrológica. JC was supported by a contract from Fundación de Investigaciones Biomédicas de Córdoba (Servicio Andaluz de Salud, Programa Nicolás Monardes). CL is a fellow of Consejería de Innovacion, Ciencia y Empresa, Junta de Andalucía (CTS-6337). MA is a fellow of the ‘Ayuda Postdoctoral Programa Propio’ program, Universidad de Alcala, Madrid, Spain.
References
- Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med. 2000; 28(12): 1815–26.
- Itabe H. Oxidative modification of LDL: its pathological role in atherosclerosis. Clin Rev Allergy Immunol. 2009; 37(1): 4–11.
- Apostolov EO, Ray D, Savenka AV, Shah SV, Basnakian AG. Chronic uremia stimulates LDL carbamylation and atherosclerosis. J Am Soc Nephrol. 2010; 21(11): 1852–7.
- Apostolov EO, Basnakian AG, Ok E, Shah SV. Carbamylated low-density lipoprotein: nontraditional risk factor for cardiovascular events in patients with chronic kidney disease. J Ren Nutr. 2012; 22(1): 134–8.
- Berenson GS, Srinivasan SR, Bao W, Newman WP, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998; 338(23): 1650–6.
- Lansky AJ, Ng VG, Maehara A, Weisz G, Lerman A, Mintz GS, etal. Gender and the extent of coronary atherosclerosis, plaque composition, and clinical outcomes in acute coronary syndromes. JACC Cardiovasc Imaging. 2012; 5(3 Suppl): S62–72.
- Lusis AJ, Fogelman AM, Fonarow GC. Genetic basis of atherosclerosis: part I: new genes and pathways. Circulation. 2004; 110(13): 1868–73.
- Najemnik C, Sinzinger H, Kritz H. Endothelial dysfunction, atherosclerosis and diabetes. Acta Med Austriaca. 1999; 26(5): 148–53. [PubMed Abstract].
- Paoletti R, Gotto AM, Hajjar DP. Inflammation in atherosclerosis and implications for therapy. Circulation. 2004; 109(23 Suppl 1): III20–6. [PubMed Abstract].
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109(23 Suppl 1): III27–32. [PubMed Abstract].
- Ehara S, Ueda M, Naruko T, Haze K, Itoh A, Otsuka M, etal. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 2001; 103(15): 1955–60.
- Carracedo J, Merino A, Briceño C, Soriano S, Buendía P, Calleros L, etal. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. 2011; 25(4): 1314–22.
- Millán J, Pintó X, Muñoz A, Zúñiga M, Rubiés-Prat J, Pallardo LF, etal. Lipoprotein ratios: physiological significance and clinical usefulness in cardiovascular prevention. Vasc Health Risk Manag. 2009; 5: 757–65.
- Schuh J, Fairclough GF, Haschemeyer RH. Oxygen-mediated heterogeneity of apo-low-density lipoprotein. Proc Natl Acad Sci USA. 1978; 75(7): 3173–7.
- Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, etal. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997; 386(6620): 73–7.
- Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA. 1979; 76(1): 333–7.
- Steinbrecher UP, Fisher M, Witztum JL, Curtiss LK. Immunogenicity of homologous low density lipoprotein after methylation, ethylation, acetylation, or carbamylation: generation of antibodies specific for derivatized lysine. J Lipid Res. 1984; 25(10): 1109–16. [PubMed Abstract].
- Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev. 2001; 17(6): 436–43.
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999; 340(2): 115–26.
- Avogaro P, Cazzolato G, Bittolo-Bon G. Some questions concerning a small, more electronegative LDL circulating in human plasma. Atherosclerosis. 1991; 91(1–2): 163–71.
- Orekhov AN, Bobryshev YV, Sobenin IA, Melnichenko AA, Chistiakov DA. Modified low density lipoprotein and lipoprotein-containing circulating immune complexes as diagnostic and prognostic biomarkers of atherosclerosis and type 1 diabetes macrovascular disease. Int J Mol Sci. 2014; 15(7): 12807–41.
- Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc Natl Acad Sci USA. 1981; 78(10): 6499–503.
- Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA. 1984; 81(12): 3883–7.
- Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991; 88(6): 1785–92.
- Itabe H. Oxidized phospholipids as a new landmark in atherosclerosis. Prog Lipid Res. 1998; 37(2–3): 181–207.
- Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002; 8(11): 1211–17.
- Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, etal. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005; 280(3): 2352–60.
- Leitinger N. Oxidized phospholipids as triggers of inflammation in atherosclerosis. Mol Nutr Food Res. 2005; 49(11): 1063–71.
- Schober A, Nazari-Jahantigh M, Weber C. MicroRNA-mediated mechanisms of the cellular stress response in atherosclerosis. Nat Rev Cardiol. 2015; 12(6): 361–74.
- Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci USA. 1993; 90(14): 6434–8.
- Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988; 318(20): 1315–21.
- Bucala R, Makita Z, Vega G, Grundy S, Koschinsky T, Cerami A, etal. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. 1994; 91(20): 9441–5.
- Witztum JL, Mahoney EM, Branks MJ, Fisher M, Elam R, Steinberg D. Nonenzymatic glucosylation of low-density lipoprotein alters its biologic activity. Diabetes. 1982; 31(4 Pt 1): 283–91.
- Steinbrecher UP, Witztum JL. Glucosylation of low-density lipoproteins to an extent comparable to that seen in diabetes slows their catabolism. Diabetes. 1984; 33(2): 130–4.
- Klein RL, Laimins M, Lopes-Virella MF. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes. 1995; 44(9): 1093–8.
- Laakso M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes. 1999; 48(5): 937–42.
- Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, etal. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999; 100(10): 1134–46.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, etal. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404(6779): 787–90.
- Sobal G, Menzel J, Sinzinger H. Why is glycated LDL more sensitive to oxidation than native LDL? A comparative study. Prostaglandins Leukot Essent Fatty Acids. 2000; 63(4): 177–86.
- Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J Biol Chem. 1995; 270(18): 10828–32.
- Bowie A, Owens D, Collins P, Johnson A, Tomkin GH. Glycosylated low density lipoprotein is more sensitive to oxidation: implications for the diabetic patient?. Atherosclerosis. 1993; 102(1): 63–7.
- Kobayashi K, Watanabe J, Umeda F, Nawata H. Glycation accelerates the oxidation of low density lipoprotein by copper ions. Endocr J. 1995; 42(4): 461–5.
- Rabini RA, Fumelli P, Galassi R, Dousset N, Taus M, Ferretti G, etal. Increased susceptibility to lipid oxidation of low-density lipoproteins and erythrocyte membranes from diabetic patients. Metabolism. 1994; 43(12): 1470–4.
- Nilsson L, Lundquist P, Kågedal B, Larsson R. Plasma cyanate concentrations in chronic renal failure. Clin Chem. 1996; 42(3): 482–3.
- Kraus LM, Kraus AP. Carbamoylation of amino acids and proteins in uremia. Kidney Int Suppl. 2001; 78: S102–7.
- Park KD, Mun KC, Chang EJ, Park SB, Kim HC. Inhibition of erythropoietin activity by cyanate. Scand J Urol Nephrol. 2004; 38(1): 69–72.
- Ramirez R, Carracedo J, Nogueras S, Buendia P, Merino A, Cañadillas S, etal. Carbamylated darbepoetin derivative prevents endothelial progenitor cell damage with no effect on angiogenesis. J Mol Cell Cardiol. 2009; 47(6): 781–8.
- Reis SE, Olson MB, Fried L, Reeser V, Mankad S, Pepine CJ, etal. Mild renal insufficiency is associated with angiographic coronary artery disease in women. Circulation. 2002; 105(24): 2826–9.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998; 32(5 Suppl 3): S112–19.
- Chade AR, Lerman A, Lerman LO. Kidney in early atherosclerosis. Hypertension. 2005; 45(6): 1042–9.
- Steinbrecher UP, Zhang HF, Lougheed M. Role of oxidatively modified LDL in atherosclerosis. Free Radic Biol Med. 1990; 9(2): 155–68.
- Jialal I, Fuller CJ. Oxidatively modified LDL and atherosclerosis: an evolving plausible scenario. Crit Rev Food Sci Nutr. 1996; 36(4): 341–55.
- Ok E, Basnakian AG, Apostolov EO, Barri YM, Shah SV. Carbamylated low-density lipoprotein induces death of endothelial cells: a link to atherosclerosis in patients with kidney disease. Kidney Int. 2005; 68(1): 173–8.
- Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Hörkkö S, Barnard J, etal. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007; 13(10): 1176–84.
- Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003; 107(3): 416–21.
- Imanishi T, Tsujioka H, Akasaka T. Endothelial progenitor cells dysfunction and senescence: contribution to oxidative stress. Curr Cardiol Rev. 2008; 4(4): 275–86.
- Higashi Y, Kihara Y, Noma K. Endothelial dysfunction and hypertension in aging. Hypertens Res. 2012; 35(11): 1039–47.
- Shi Q, Aida K, Vandeberg JL, Wang XL. Passage-dependent changes in baboon endothelial cells – relevance to in vitro aging. DNA Cell Biol. 2004; 23(8): 502–9.
- Khaidakov M, Wang X, Mehta JL. Potential involvement of LOX-1 in functional consequences of endothelial senescence. PLoS One. 2011; 6(6): e20964.
- Sato I, Morita I, Kaji K, Ikeda M, Nagao M, Murota S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem Biophys Res Commun. 1993; 195(2): 1070–6.
- Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K, etal. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009; 297(1): H425–32.
- Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, etal. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012; 125(9): 1122–33.
- Zhou X, Perez F, Han K, Jurivich DA. Clonal senescence alters endothelial ICAM-1 function. Mech Ageing Dev. 2006; 127(10): 779–85.
- Carracedo J, Buendía P, Merino A, Soriano S, Esquivias E, Martín-Malo A, etal. Cellular senescence determines endothelial cell damage induced by uremia. Exp Gerontol. 2013; 48(8): 766–73.
- Sessa WC, Harrison JK, Barber CM, Zeng D, Durieux ME, D'Angelo DD, etal. Molecular cloning and expression of a cDNA encoding endothelial cell nitric oxide synthase. J Biol Chem. 1992; 267(22): 15274–76.
- Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, etal. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 1992; 256(5054): 225–8.
- Cooke JP. NO and angiogenesis. Atheroscler Suppl. 2003; 4(4): 53–60.
- Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003; 91(3A): 7A–11A.
- Tejovathi B, Suchitra MM, Suresh V, Reddy VS, Sachan A, Srinivas Rao PV, etal. Association of lipid peroxidation with endothelial dysfunction in patients with overt hypothyroidism. Exp Clin Endocrinol Diabetes. 2013; 121(5): 306–9.
- Haribabu A, Reddy VS, Pallavi C, Sachan A, Pullaiah P, Suresh V, etal. Evaluation of protein oxidation and its association with lipid peroxidation and thyrotropin levels in overt and subclinical hypothyroidism. Endocrine. 2013; 44(1): 152–7.
- Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF, etal. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA. 2006; 103(45): 17018–23.
- Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ, Kim JH, etal. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell. 2007; 18(11): 4543–52.
- Spagnoli LG, Orlandi A, Mauriello A, Santeusanio G, de Angelis C, Lucreziotti R, etal. Aging and atherosclerosis in the rabbit. 1. Distribution, prevalence and morphology of atherosclerotic lesions. Atherosclerosis. 1991; 89(1): 11–24.
- McEwen JE, Zimniak P, Mehta JL, Shmookler Reis RJ. Molecular pathology of aging and its implications for senescent coronary atherosclerosis. Curr Opin Cardiol. 2005; 20(5): 399–406.
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000; 408(6809): 239–47.
- Zhang X, Qi R, Xian X, Yang F, Blackstein M, Deng X, etal. Spontaneous atherosclerosis in aged lipoprotein lipase-deficient mice with severe hypertriglyceridemia on a normal chow diet. Circ Res. 2008; 102(2): 250–6.
- Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, etal. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007; 6(6): 783–97.
- Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 2008; 389(3): 233–41.
- Ferguson M, Rebrin I, Forster MJ, Sohal RS. Comparison of metabolic rate and oxidative stress between two different strains of mice with varying response to caloric restriction. Exp Gerontol. 2008; 43(8): 757–63.
- Huffman DM, Moellering DR, Grizzle WE, Stockard CR, Johnson MS, Nagy TR. Effect of exercise and calorie restriction on biomarkers of aging in mice. Am J Physiol Regul Integr Comp Physiol. 2008; 294(5): R1618–27.
- Meyers MR, Gokce N. Endothelial dysfunction in obesity: etiological role in atherosclerosis. Curr Opin Endocrinol Diabetes Obes. 2007; 14(5): 365–9.
- Segura J, Ruilope LM. Obesity, essential hypertension and renin-angiotensin system. Public Health Nutr. 2007; 10(10A): 1151–5.
- MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, etal. Blood pressure, stroke, and coronary heart disease. Part 1, prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet. 1990; 335(8692): 765–74.
- Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis?. Circulation. 2004; 109(21 Suppl 1): II27–33.
- Chang HJ, Chung J, Choi SY, Yoon MH, Hwang GS, Shin JH, etal. Endothelial dysfunction in patients with exaggerated blood pressure response during treadmill test. Clin Cardiol. 2004; 27(7): 421–5.
- Higashi Y, Yoshizumi M. [Endothelial function]. Nihon Rinsho. 2003; 61(7): 1138–44.
- Järvisalo MJ, Raitakari M, Toikka JO, Putto-Laurila A, Rontu R, Laine S, etal. Endothelial dysfunction and increased arterial intima-media thickness in children with type 1 diabetes. Circulation. 2004; 109(14): 1750–5.
- Maggi FM, Raselli S, Grigore L, Redaelli L, Fantappiè S, Catapano AL. Lipoprotein remnants and endothelial dysfunction in the postprandial phase. J Clin Endocrinol Metab. 2004; 89(6): 2946–50.
- Saini HK, Arneja AS, Dhalla NS. Role of cholesterol in cardiovascular dysfunction. Can J Cardiol. 2004; 20(3): 333–46.
- Liu L, Zhao SP, Gao M. [Influence of postprandial hypertriglyceridemia on the endothelial function in elderly patients with coronary heart disease]. Hunan Yi Ke Da Xue Xue Bao. 2002; 27(3): 259–62.
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004; 43(10): 1731–7.
- Poreba R, Skoczyńska A, Derkacz A. [Effect of tobacco smoking on endothelial function in patients with coronary arteriosclerosis]. Pol Arch Med Wewn. 2004; 111(1): 27–36.
- Lyon CJ, Law RE, Hsueh WA. Mini review: adiposity, inflammation, and atherogenesis. Endocrinology. 2003; 144(6): 2195–200.
- Mitu F, Mitu M. [Physical exercise and vascular endothelium]. Rev Med Chir Soc Med Nat Iasi. 2003; 107(3): 487–93.
- Farhat N, Thorin-Trescases N, Voghel G, Villeneuve L, Mamarbachi M, Perrault LP, etal. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can J Physiol Pharmacol. 2008; 86(11): 761–9.
- Niemann B, Chen Y, Teschner M, Li L, Silber RE, Rohrbach S. Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J Am Coll Cardiol. 2011; 57(5): 577–85.
- Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009; 50(Suppl): S376–81.
- Itabe H. Oxidized low-density lipoproteins: what is understood and what remains to be clarified. Biol Pharm Bull. 2003; 26(1): 1–9.
- Hevonoja T, Pentikäinen MO, Hyvönen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim Biophys Acta. 2000; 1488(3): 189–210.
- Virmani R, Avolio AP, Mergner WJ, Robinowitz M, Herderick EE, Cornhill JF, etal. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am J Pathol. 1991; 139(5): 1119–29.
- O'Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a clinical perspective. J Am Coll Cardiol. 2007; 50(1): 1–13.