Figures & data
Figure 1. Structure of the spore and life cycle.
a. The robust properties of the spore are due to its multi-layered structure, with each layer contributing to the overall resilience [Citation25]. The dense core is dehydrated due to the presence of up to 25% Ca-DPA, and the DNA is bound to and protected by the small acid-soluble proteins. Surrounding the core is an extremely impermeable inner membrane and the germ (primordial) cell wall. This thin layer of peptidoglycan has the same composition as in vegetative cells – and will become the nascent cell wall during germination. Around the germ cell wall is a much thicker layer of peptidoglycan, the cortex. Within the cortex peptidoglycan approximately 25% of the N-acetylmuramic acid moieties are modified to muramic-δ-lactam, and there are few crosslinks between adjacent N-acetylmuramic acid-N-acetylglucosamine polymers [Citation26]. This results in a much more flexible peptidoglycan structure, with a distinct chemical signature that allows specific degradation during germination, without risk of compromising the germ cell wall. The cortex is surrounded by a second membrane, derived from the mother cell during engulfment, and then finally, the protein coat. The coat is a lamellar structure consisting of a large number of often highly crosslinked proteins. The outermost layers of the coat in C. difficile appear less organized, with an amorphous structure, and vary in thickness. This layer has been described as exosporium but does not appear to have the same loose-attachment and hexameric organization seen in other spore-formers [Citation27]. Some of these structures can be seen in the transmission electron micrograph of a negative-stained, thin-sectioned spore on the right. b. When conditions are favorable, a C. difficile cell will normally be divided by binary fission. However, when environment conditions are less than ideal, most likely due to nutrient limitation, the cell can enter the sporulation pathway instead. Upon initiation of sporulation, the cell first undergoes asymmetric septation, producing the mother cell compartment and smaller forespore. A copy of the genome is transferred into the nascent spore, and the forespore is then engulfed by the mother cell - a phagocytosis-like event that results in an immature spore, bounded by two membranes, free in the cytoplasm of the mother cell. The spore then undergoes a maturation process whereby the DNA is compacted by the small acid-soluble proteins, Ca-DPA is synthesized, the core is dehydrated, and cortex and protein coats are synthesized. The final mature spore is released by lysis of the mother cell.

Table 1. Virulence factors of C. difficile.
Figure 2. The pathogenicity locus and toxin mode of action. .
a. The pathogenicity locus (PaLoc) is composed of 5 genes: tcdA and tcdB, encoding toxins A and B respectively; tcdR, encoding an alternative sigma factor and likely positive regulator of the PaLoc (regulation shown above in green); tcdE, encoding a holin-like protein putatively involved in toxin secretion; and tcdC, an anti-sigma factor and negative regulator of the PaLoc genes (regulation shown above in red). Toxins A and B both consist of a broadly similar four-domain structure. At the N-terminal, the glucosyltransferase domain (GTD) is the active toxin moiety which inactivates members of the Rho GTPase family. A cysteine protease domain is next to the GTD, and is involved in auto-processing and release of the GTD. The next domain, often called the Delivery and Receptor Binding Domain (DRBD), contains a hydrophobic region and is thought to be involved in translocation of the GTD from the lumen of endocytic vesicles into the host cell cytoplasm. The final C-terminal receptor-binding domain (also known as C-terminal combined repetitive oligopeptides (CROPS) domain) binds to a range of cellular receptors.b. Toxin mode of action [Citation65]. The toxins bind to various cellular receptors via the C-terminal CROPs domain, triggering clathrin-dependent endocytosis (1) followed by acidification of the resulting vesicle (2). The drop in pH triggers a conformational change in the delivery domain which inserts into, and forms a pore in, the vesicle membrane, through which the GTD transits into the host cytoplasm (3). The GTD is then released by a cleavage event mediated by the cysteine protease domain, in a process that is dependent on host inositol hexakisphosphate (4). The GTD is then able to glucosylate and inactive members of the small Rho GTPase family, including Rho, Rac, and Cdc42. Inactivation of Rho GTPases results in multi-level cellular disruption, including dysregulation of actin depolymerization, which causes disruption of tight junctions and loss of intestinal barrier function, induction of proinflammatory cytokines and activation of programmed cell death.
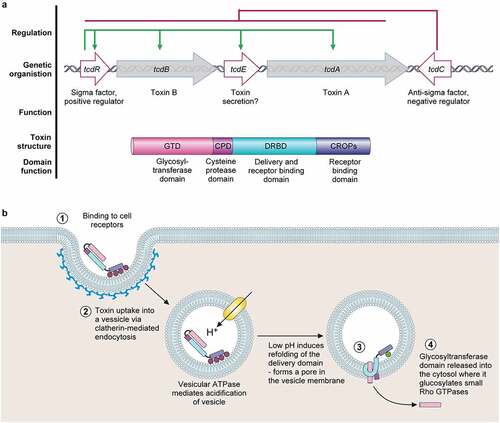
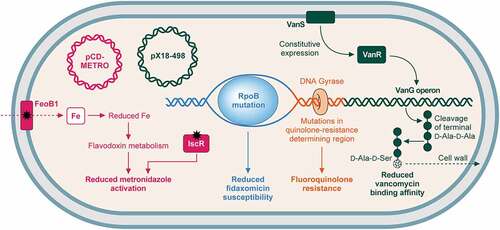
Figure 3. Mechanisms of resistance to commonly used antibiotics. .
Mechanisms of C. difficile resistance to antibiotics commonly used to treat CDI (vancomycin, fidaxomicin, and metronidazole) and fluoroquinolones. (i) metronidazole (pink): resistance can be gained via the plasmid pCD-METRO [Citation162]. Metronidazole resistance may also be gained through mutation of either FeoB1, which reduces intracellular iron, reducing flavodoxin metabolism and metronidazole activation; or IscR, which also reduces metronidazole activation [Citation163]. (ii) fidaxomicin (blue): mutations in RpoB reduce fidaxomicin susceptibility [Citation164]. (iii) vancomycin (green): Mutations in vanSR two-component system allow constitutive expression of the vanG-like operon, which aids vancomycin resistance through replacement of the terminal d-alanine in peptidoglycan pentapeptide sidechains with d-serine, reducing vancomycin binding affinity [Citation104,Citation165]. Plasmid pX18-498 has also recently been associated with resistance, although the mechanism is not understood [Citation166]. (iv) fluoroquinolones (orange): mutations in the genes encoding DNA gyrase, particularly gyrA results in fluoroquinolone resistance [Citation167].