
ABSTRACT
Methyl-β-cyclodextrin (MβCD) is an effective agent for the removal of plasma membrane cholesterol. In this study, we investigated the modulating effects of MβCD on the antiproliferation induced by benzyl isothiocyanate (BITC), an ITC compound mainly derived from papaya seeds. We confirmed that MβCD dose-dependently increased the cholesterol level in the medium, possibly through its removal from the plasma membrane of human colorectal cancer cells. The pretreatment with a non-toxic concentration (2.5 mM) of MβCD significantly enhanced the BITC-induced cytotoxicity and apoptosis induction, which was counteracted by the cholesterol supplementation. Although BITC activated the phosphoinositide 3-kinase (PI3K)/Akt pathway, MβCD dose-dependently inhibited the phosphorylation level of Akt. On the contrary, the treatment of MβCD enhanced the phosphorylation of mitogen activated protein kinases, but did not potentiate their BITC-induced phosphorylation. These results suggested that MβCD might potentiate the BITC-induced anti-cancer by cholesterol depletion and thus inhibition of the PI3K/Akt-dependent survival pathway.
Abbreviations: CDs: cyclodextrins; MβCD: methyl-β-cyclodextrin; ITCs: isothiocyanates; BITC: benzyl isothiocyanate; PI3K: phosphoinositide 3-kinase; PDK1: phosphoinositide-dependent kinase-1; MAPK: mitogen activated protein kinase; ERK1/2: extracellular signal-regulated kinase1/2; JNK: c-Jun N-terminal kinase; PI: propidium iodide; FBS: fatal bovine serum; TLC: thin-layer chromatography; PBS(-): phosphate-buffered saline without calcium and magnesium; MEK: MAPK/ERK kinase; PIP2: phosphatidylinositol-4,5-bisphosphate; PIP3: phosphatidylinositol-3,4,5-trisphosphate
GRAPHICAL ABSTRACT
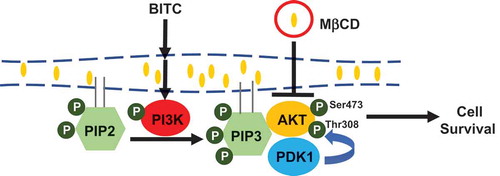
MβCD potentiated the BITC-induced antiproliferation through inhibition of the PI3K/AKT survival pathway
Cholesterol is one of the major lipid components in the plasma membrane and essential for human health [Citation1]. The plasma membrane cholesterol critically contributes to the maintenance of membrane fluidity and permeability and is of importance for membrane trafficking as well as the lipid and protein sorting [Citation2,Citation3]. Membrane microdomains, such as lipid rafts, containing a higher concentration of cholesterol, play regulating roles in transduction of the transmembrane receptor-mediated cell signaling [Citation1,Citation4]. On the other hand, the excessive accumulation of cholesterol in mammalian cells increases the risk of various diseases, such as coronary heart disease [Citation5], Alzheimer’s disease [Citation6] and several types of cancer [Citation7].
Cyclodextrins (CDs), comprising a family of cyclic oligosaccharides with exterior hydrophilic and interior hydrophobic cavities, are industrially used in pharmaceutical and allied applications to promote drug solubility, bioavailability and stability [Citation8]. CDs also act as potential sensitizers of chemotherapy, possibly through the increased permeability of mucosa epithelial cells [Citation9] and influence of cell signaling by lipid raft modification [Citation10]. Methyl-β-cyclodextrin (MβCD), a CD derivative, is one of the most effective agents for removal of plasma membrane cholesterol due to its high affinity for cholesterol [Citation11].
Isothiocyanates (ITCs), derived from cruciferous vegetables, are potential compounds that inhibit the development and proliferation of cancer cells in vitro and in vivo [Citation12]. Benzyl isothiocyanate (BITC), one of the ITCs, exerts the antiproliferative effects by inducing cell cycle arrest and apoptosis through related signaling pathways in various human cancer cells [Citation12–Citation15]. The phosphoinositide 3-kinase (PI3K)/phosphoinositide-dependent kinase-1 (PDK1)/Akt pathway mediates resistance against chemotherapy drugs and radiation therapy in a variety of cancer types [Citation16,Citation17]. Because BITC further activates the proliferative PI3K/Akt/FoxO pathway as well as the apoptosis-inducing pathway, the antiproliferative potential of BITC is not fully exerted in human colorectal cancer cells [Citation17]. Hence, it is important to enhance the anti-cancer effects of BITC without inducing side effects such as activation of the survival pathway.
The drug resistance could be overcome by a treatment with a low dose of drugs in combination with other compounds, which show enhancement of the cytotoxic effects or suppression of side effects. Thus, this study was initially designed to identify a component that can be effectively used in combination with BITC and to determine its molecular mechanism. We demonstrated that MβCD, successfully depleting cholesterol from the plasma membrane, significantly enhanced the BITC-induced antiproliferation and apoptosis induction in human colorectal cancer cells. These synergistic effects were cancelled by the supplementation of cholesterol. MβCD actually inhibited the BITC-induced Akt phosphorylation. These results provide evidence that the combination of BITC with MβCD might be a promising therapeutic strategy to overcome resistance against the PI3K/PDK/Akt activating anti-cancer agent.
Materials and methods
Materials
BITC was purchased from LKT Laboratories, Inc. (St. Paul, MN, USA). Antibodies against phospho-PI3K (Y458), phospho-Akt (S473), phospho-Akt (T308), PDK1, phospho-p38 mitogen activated protein kinase (MAPK, Thr180/Tyr182), phospho-p44/p42 MAPK (extracellular signal-regulated kinase1/2; ERK1/2, Thr202/Tyr204), phospho-SAPK/c-Jun N-terminal kinase (JNK, Thr183/Tyr185), ERK, p38, JNK, and Akt were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Antibodies against PI3K, actin and horseradish peroxidase-linked anti-rabbit and anti-mouse IgGs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Annexin-V-FLUOS stain kit was purchased from Roche. (Mannheim, Germany). Propidium iodide (PI) and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fatal bovine serum (FBS) was purchased from Nichirei Corporation (Tokyo, Japan). Bio-Rad Protein Assay was purchased from Bio-Rad Laboratories (Hercules, CA, USA). Chemi-Lumi One Super was purchased from Nakalai Tesque Inc. (Kyoto, Japan). Cholesterol and Cholesterol-Water Soluble were purchased from Sigma-Aldrich (St. Louis, MO, USA). The thick silica gel 60 plate for thin-layer chromatography was purchased from MERK (Darmstadt, Germany). All other chemicals including MβCD were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan).
Cell culture and treatments
HCT-116 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). HCT-116 cells were maintained in DMEM (Dulbecco’s modified Eagle’s medium, high glucose). The culture medium was supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. Cells were grown at 37°C in an atmosphere of 95% O2 and 5% CO2.
Cholesterol amount determination
Cells were treated by MβCD (0, 1, 2.5, and 5 mM) in DMEM medium (without FBS) for 1 h, then the medium was collected and centrifuged at 3,000 rpm for 5 min. The supernatant (3ml) was mixed with 1 ml of chloroform/methanol (2:1) and centrifuged at 8,000 rpm for 5 min. The upper aqueous phase liquid was aspirated and 1 ml chloroform/methanol/water (2:1:3) was added to the lower organic phase, then centrifuged at 8,000 rpm for 5 min. The lower phase was collected and analyzed by thin-layer chromatography (TLC). Five microliters of the samples were separated by one-dimensional TLC using the sequential solvent system: ethanol/chloroform/trimethylamine/water 8:7:7:2 up to 5 cm, then hexane/ethyl acetate 5:1 up to 10 cm. The dried plates were sprayed with chromogenic agent (20% H2SO4 in methanol) and heated at 180°C for 30 min. The plates were then scanned by CanoScan LiDE 120 and analyzed using the Image J Software Program (National Institutes of Health, Bethesda, MD, USA).
Measurement of intracellular BITC accumulation
The BITC level in the lysates was determined by the cyclocondensation assay with 1,2-benzenedithiol as previously reported [Citation18]. HCT-116 cells were suspended at a density of 5 × 106 cells on a 60-mm plate. After overnight preculture, the cells were treated with MβCD (2.5 mM) in DMEM (without FBS) for 1 h, then incubated with or without BITC (50 μM) in DMEM (without FBS) for 0.5, 1 and 3 h. After harvesting, the cells were homogenized in 200 μL of 100 mM potassium phosphate buffer (pH 8.5) with sonication. The lysates were centrifuged, and the protein concentration in the supernatant was determined by the Bio-Rad protein assay. Equal quantities of the protein samples (50 μg/200 μL in potassium phosphate buffer) were subjected to the assay. The samples were incubated at 65°C for 2 h with 200 μL of 20 mM 1,2-benzenedithiol dissolved in methanol. After centrifugation, the absorbance of the samples was measured at 365 nm. Quantification of BITC was carried out by comparing the absorbance from the experimental samples to its standard curve.
MTT assay
HCT-116 cells were suspended at a density of 4 × 104 cells per well in a 96-well plate. After overnight preculture, the cells were treated with MβCD (0, 1, 2.5, and 5 mM) in DMEM (without FBS) for 1 h, then incubated with or without BITC in DMEM with 1% FBS for 48 h. In the cholesterol supplementation experiment, the cells were treated with 2.5 mM MβCD, followed by 1 h cholesterol supplementation and treatment with BITC for 48 h. The cell viability was determined by an MTT assay. Ten microliters of the MTT solution (5 mg/mL) were added to each well, and the absorbance was measured by a microplate reader (Benchmarkplus, Bio-Rad laboratories, Hercules, CA, USA) at 570 nm according to the manufacturer’s instructions after incubation at 37°C for 2 h in a humidified CO2 incubator. The obtained values were compared to each of the controls incubated with only vehicle.
Apoptosis assay
The collected cells were washed with ice-cold phosphate-buffered saline without calcium and magnesium (PBS (-)). After centrifuge, cells were well suspended in Annexin-V-FLUOS stain kit solution and incubated in the dark at room temperature for 15 min as described in the kit manufacture. The stained HCT-116 cells were analyzed by a Tali™ image-based cytometer (Life Technologies, Carlsbad, CA, USA).
Separation of membrane and cytosol fractions
The total crude cell membranes were isolated as previously described [Citation19]. Briefly, the cells were homogenized in 1 mL of buffer containing 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 200 mM sucrose and protease inhibitor mix (Roche Diagnostics, Mannheim, Germany). The nuclei and cellular debris were removed by centrifugation at 900 g for 10 min at 4°C. The resulting supernatant was centrifuged at 110,000 g for 75 min at 4°C to obtain the crude membrane pellet. The crude membrane pellet was solubilized in buffer containing 10 mM Tris, pH 7.4, 1 mM EDTA, and 0.5% Triton ×-100 for 1 h on ice with intermittent vortexing, followed by centrifugation at 13,000g for 10 minutes at 4°C. The supernatant was considered as the membrane fraction.
Western blot analysis
The whole cell lysates were prepared in lysis buffer (20 mM Tri-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM NaH2PO4, 10 mM NaF, 2 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1% sodium dodecyl sulfate, 1% sodium deoxycholate and 1% Triton X-100) containing protease inhibitor cocktail and left on ice for 20 min. After sonication, the lysates were centrifuged, and the supernatant was used as the whole cell lysates. The protein concentration in the supernatant was determined by the Bio-Rad protein assay.
Equal quantities of the protein samples were subjected to SDS-PAGE and transferred to Immobilon-P membranes. The membranes were blocked, then incubated with the primary antibody overnight at 4°C followed by the appropriate secondary antibody. Secondary antibody binding was visualized using a Chemi-Lumi One Super (Nacalai Tesque). Densitometric analysis of the bands was carried out using the Image J Software Program.
Statistical analysis
All values were expressed as means ± SD. Statistical significance was analyzed by Student’s t-test or one-way ANOVA followed by Tukey’s HSD using XLSTAT software.
Results
Effect of MβCD treatment on the medium cholesterol content
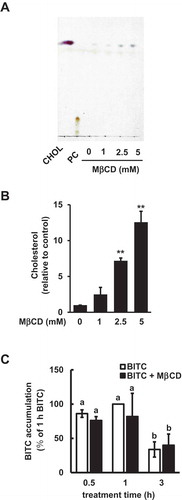
Since MβCD is reported to have the ability to deplete the membrane cholesterol [Citation20], we examined the effect of MβCD on the membrane cholesterol in human colorectal cancer HCT-116 cells, commonly used as a colorectal cancer model with gain-of-function mutations in PI3KCA (PI3K catalytic subunit, alpha isoform) to clarify the role of the PI3K/Akt survival pathway in the pathogenesis of colon cancer [Citation17,Citation21,Citation22]. As shown in ( the cholesterol content in the cell culture medium was increased by a 1-h incubation with MβCD in a dose dependent manner, supporting the idea that MβCD can extract cholesterol from the cell plasma membrane. We then initially examined the effect of MβCD on the intracellular BITC accumulation. As shown in ()), BITC was accumulated in HCT-116 cells 30 min after treatment, which reached a plateau at 1 h, then decreased. However, MβCD showed no significant effect on the intracellular level of BITC at each time point. These results suggested that 2.5 mM MβCD might be ineffective in the accumulation or elimination of the intracellular BITC.
Figure 1. Modulating effects of MβCD on the medium cholesterol and intracellular BITC levels. (a and b) Enhancing effect of MβCD on the medium cholesterol level. The cells were treated with MβCD (0, 1, 2.5, and 5 mM). The cholesterol level in the medium was determined by a TLC analysis. Representative chromatogram (a) and quantitative data (b). PC; phosphatidylcholine. (c) Effect of MβCD on the intracellular BITC level. The cells were treated by MβCD (2.5 mM) for 1 h, then incubated with or without BITC (50 μM) for the indicated periods. Equal quantities of protein samples were subjected the cyclocondensation assay. All values were expressed as means ± SD of three separate experiments (*p < 0.05, **p < 0.01 compared to negative control).
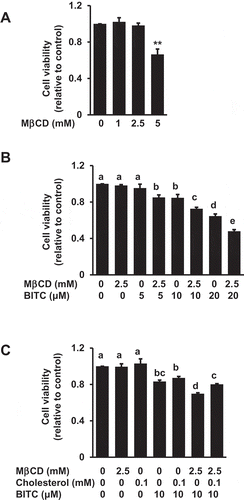
Enhancing effects of MβCD on the BITC-induced antiproliferation and apoptosis
Since the MβCD treatment significantly depleted cholesterol from the cells, we checked the effect of MβCD itself on the cell viability by an MTT assay. Since the non-toxic concentration of MβCD was found to be 2.5 mM ()), this concentration was selected as its maximal concentration to test. We next examined the effect of the combination of MβCD with BITC on the cell viability in human colorectal cancer HCT-116 cells. The pretreatment of 2.5 mM MβCD significantly potentiated the BITC-induced decrease in the cell viability ()). More interestingly, this enhancing effect was counteracted by the cholesterol supplementation ()).
Figure 2. Enhancing effect of MβCD on the BITC-induced antiproliferation. (a) HCT-116 cells were exposed to the indicated concentrations of MβCD for 1 h, then incubated in 1% FBS DMEM medium for 48 h. Cell viability was determined by an MTT assay. All values were expressed as means ± SD of three separate experiments (*p < 0.05, **p < 0.01 compared to negative control). (b) After the pretreatment with 2.5 mM MβCD, the cells were treated with BITC for 48 h. (c) After the pretreatment with 2.5 mM MβCD, the cells were exposed to cholesterol for 1 h, followed by the BITC treatment for 48 h. Cell viability was determined by an MTT assay. All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
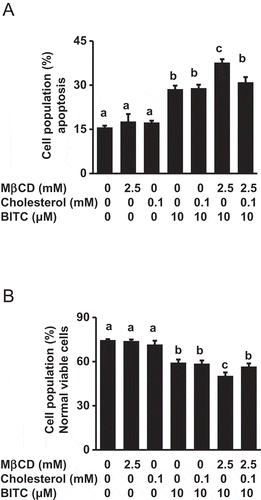
We next clarified the mechanism underlying the enhancement of the MβCD pretreatment on the BITC-induced antiproliferation. As shown in , the MβCD pretreatment significantly potentiated the apoptosis induced by BITC, whereas MβCD alone had no effects. The potentiation of apoptosis induction by MβCD was also diminished by the cholesterol supplementation ()). These results suggested that MβCD has a synergistic effect on the BITC-induced apoptosis, possibly through disturbance of the plasma membrane structure by cholesterol depletion.
Figure 3. Enhancing effect of MβCD on the BITC-induced apoptotic cell death. HCT-116 cells were pretreated with MβCD (2.5 mM) for 1 h and incubated with or without cholesterol (0.1 mM) for 1 h, followed by the treatment of BITC (10 μM) for 48 h. Apoptosis was detected by an Annexin-V-FLUOS stain kit and analyzed by a Tali™ image-based cytometer. (a) apoptotic cell population (Annexin V positive) and (b) viable cell population (Annexin V negative, propidium iodide negative). All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
Modulating effects of MβCD on the PI3K/Akt pathway
BITC has recently been reported to enhance the PI3K/Akt/FoxO pathway, even though it inhibits the proliferation in human colorectal cancer HCT-116 cells [Citation17]. We thus examined whether the MβCD pretreatment affects the PI3K/Akt pathway. As shown in ()), MβCD alone significantly inhibited the Akt phosphorylation at both Thr308 and Ser473, whereas it showed no significant effect on the PI3K phosphorylation. In the combination experiment, MβCD significantly inhibited the BITC-induced phosphorylation of Akt, but not that of PI3K ()). The inhibition of Akt phosphorylation by MβCD was also diminished by the cholesterol supplementation . These results suggested that MβCD inhibits the BITC-enhanced Akt phosphorylation, possibly through cholesterol depletion.
Figure 4. Modulating effect of MβCD on the PI3K/Akt cell survival pathway. (a) HCT-116 cells were pretreated with the indicated concentrations of MβCD for 1 h, then incubated in 1% FBS medium for 30 min. (b) After the pretreatment with MβCD (2.5 mM) for 1 h, the cells were treated with BITC (10 μM) for 30 min. The phosphorylated and total proteins of Akt and PI3K as well as actin were analyzed by Western blotting. All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
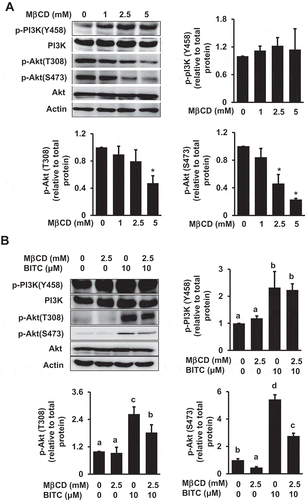
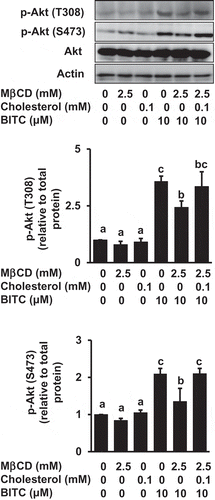
Figure 5. Impairment of the MβCD-induced inhibition of Akt phosphorylation by cholesterol supplementation. HCT-116 cells were pretreated with MβCD (2.5 mM) for 1 h and incubated with or without cholesterol (0.1 mM) for 1 h, followed by the treatment of BITC (10 μM) in the completed medium for 30 min. The phosphorylated and total proteins of Akt as well as actin were analyzed by Western blotting. All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
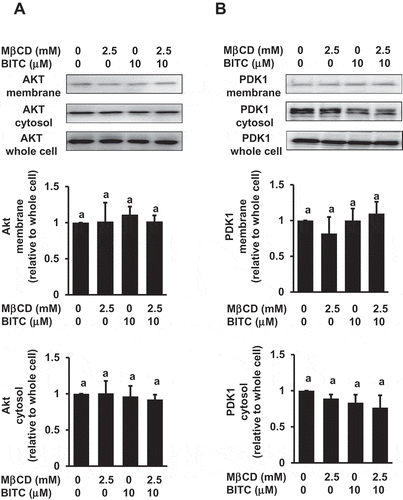
The signal transduction of the PI3K/Akt pathway mainly takes place in the plasma membrane, and Akt would be recruited to the membrane specific domain [Citation23,Citation24]. We thus examined the membrane distribution of Akt and PDK1, a protein kinase situated between PI3K and Akt. As shown in , the amounts of Akt and PDK1 in the cytosol or the membrane were not significantly changed. These results suggested that the MβCD pretreatment inhibits the PI3K/Akt survival pathway probably through suppression of the Akt phosphorylation.
Figure 6. No significant effects of MβCD on the membrane distribution of PDK1 and Akt. After the pretreatment with MβCD (2.5 mM) for 1 h, the cells were treated with BITC (10 μM) for 30 min. Separation of the cytosol and membrane fractions was performed as described in materials and methods section. The total proteins of Akt and PDK1 were analyzed by Western blotting. All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
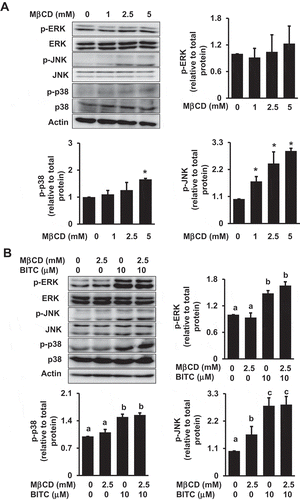
MAPKs are serine-threonine kinases that mediate intracellular signaling associated with a variety of cellular activities. Similar to the PI3K/Akt pathway, the MAPK/ERK kinase (MEK)/ERK signaling cascade plays a significant role in the cell survival and proliferation [Citation17]. The activation of JNK and/or p38 MAPK is related to the apoptotic response in various cancer cells [Citation25]. As shown in ()), MβCD alone did not affect the ERK phosphorylation level, whereas it shows a tendency to enhance the phosphorylation of JNK and p38 MAPK. In the combination experiment, MβCD did not change the BITC-induced phosphorylation of ERK, JNK or p38 ()). These results suggested that the MAPK pathways could be ruled out in the mechanism underlying the synergistic effect of MβCD on the BITC-induced antiproliferation.
Figure 7. Modulating effects of MβCD on the MAPK pathway. (a) HCT-116 cells were pretreated with the indicated concentrations of MβCD for 1 h, then incubated in 1% FBS medium for 30 min. (b) After the pretreatment with MβCD (2.5 mM) for 1 h, the cells were treated with BITC (10 μM) for 30min. The phosphorylated and total proteins of ERK, JNK, and p38 as well as actin were analyzed by Western blotting. All values were expressed as means ± SD of three separate experiments. Different letters above the bars indicate significant differences among the treatments for each condition (p < 0.05).
Discussion
In this study, we demonstrated that MβCD is a potential enhancer of the BITC-induced antiproliferation in human colorectal cancer cells. MβCD is accepted as an effective agent to remove the plasma membrane cholesterol [Citation20] and reported to induce cholesterol efflux in HCT-116 cells [Citation26], consistent with the present result . The non-toxic concentration of MβCD (2.5 mM) enhanced the BITC-induced antiproliferation ()) and apoptosis induction ()) without change in the BITC accumulation ()). This finding is supported by a previous report showing that MβCD enhances the cytotoxic effects of some anticancer drugs [Citation27]. The MβCD-induced cholesterol depletion is an acute process, in which the membrane reorganization may not be completed [Citation28]. Concordantly, cholesterol replenishment was able to counteract the enhancing effect of MβCD ()). These results suggested that the membrane cholesterol plays an important role in the cell survival or resistance against BITC, which can be modulated by MβCD.
Intrinsic or acquired drug resistance is a key event to limit the efficacy of chemotherapy. In addition to the increased drug efflux, PI3K is also a plausible molecule that mediates resistance to the chemotherapy drugs. The PI3K-mediated pathway is often activated in various human cancer cells including colorectal cancer cells [Citation29,Citation30] and influences cell survival and drug resistance [Citation16]. BITC further enhanced the PI3K/Akt pathway in human colorectal cancer HCT-116 cells having gain-of-function mutations in PI3KCA ()), consistent with our previous report [Citation17]. The enhanced PI3K/Akt survival pathway is considered to contribute to the resistance against BITC, which is supported by the observation that the antiproliferation by BITC was significantly enhanced by the PI3K inhibitors at concentrations sufficient for inhibition of the Akt activation [Citation17]. The MβCD pretreatment significantly inhibited the Akt phosphorylation both at Thr308 (the PDK1 phosphorylation site) and Ser473 (the site for full activation), but not that of PI3K (). PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3), which can bind to Akt and PDK1, recruit them to the plasma membrane, and activate this kinase cascade [Citation23]. The distribution of PDK1 or Akt between the plasma membrane and cytosol was not significantly changed (), suggesting that the cholesterol depletion by MβCD might not affect the PIP3 level in the plasma membrane. This finding is consistent with a previous report showing that MβCD inhibits the phosphorylation of Akt and its downstream targets, possibly through alteration of the integrity of lipid raft microdomains, but not by the PI3K inhibition and change in the membrane PIP3 level [Citation20]. These results suggested that MβCD probably interferes with the Akt phosphorylation by PDK1 through inhibition of the local molecular assembly and/or catalytic reaction.
Another essential signaling pathway involved in the cell survival and proliferation is the MEK/ERK signaling cascade [Citation25,Citation31]. BITC potentiated the ERK phosphorylation in HCT-116 cells, whereas MβCD treatment did not affect the BITC-enhanced ERK phosphorylation level (). The other MAPKs, JNK and p38 MAPK, are involved in the BITC-induced apoptosis [Citation13,Citation14]. The phosphorylation of JNK and p38 was increased by MβCD alone, whereas MβCD pretreatment did not show any additive or inhibitory effect on the phosphorylation of JNK or p38 in combination with BITC (). Taken together, these results suggested that the MAPK signaling cascades could be ruled out in the mechanism of the antiproliferation potentiated by MβCD.
In conclusion, we revealed that MβCD pretreatment synergistically potentiated the BITC-induced antiproliferation in human colorectal cancer cells, possibly through inhibition of the PI3K/Akt survival pathway. The present results provide evidence that the combined treatment with MβCD is a promising therapeutic strategy to overcome drug resistance against anticancer compounds activating the PI3K/Akt survival pathway. Future efforts will be concerned with further understanding the signaling transduction of the PI3K/Akt signaling pathway on the membrane specific domains such as the lipid raft.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110(5):597–603.
- Lundbaek JA, Andersen OS, Werge T, et al Cholesterol-induced protein sorting: an analysis of energetic feasibility. B Biophys J. 2003;84(3):2080–2089.
- Miersch S, Espey MG, Chaube R, et al Plasma membrane cholesterol content affects nitric oxide diffusion dynamics and signaling. J Biol Chem. 2008;283(27):18513–18521.
- Li YC, Park MJ, Ye SK, et al Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168(4):1107–1118.
- Genest J, McPherson R, Frohlich J, et al. Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult-2009 recommendations. Can J Cardiol. 2009;25(10):567–579.
- De Chaves EP, Narayanaswami V, Christoffersen C, et al Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008;3(5):505–530.
- Warner M, Gustafsson JA. On estrogen, cholesterol metabolism, and breast cancer. N Engl J Med. 2014;370(6):572–573.
- Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3(12):1023.
- Morrison PW, Connon CJ, Khutoryanskiy VV. Cyclodextrin-mediated enhancement of riboflavin solubility and corneal permeability. Mol Pharm. 2013;10(2):756–762.
- Reis-Sobreiro M, Roué G, Moros A, et al Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J. 2013;3(5):e118.
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta -Biomembr. 2007;1768(6):1311–1324.
- Nakamura T, Abe N, Nakamura Y. Physiological relevance of covalent protein modification by dietary isothiocyanates. J Clin Biochem Nutr. 2018;62(1):11–19.
- Nakamura Y, Miyoshi N. Electrophiles in foods: the current status of isothiocyanates and their chemical biology. Biosci Biotechnol Biochem. 2010;74(2):242–255.
- Miyoshi N, Uchida K, Osawa T, et al A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation. Cancer Res. 2004;64(6):2134–2142.
- Abe N, Hou DX, Munemasa S, et al Nuclear factor-kappaB sensitizes to benzyl isothiocyanate-induced antiproliferation in p53-deficient colorectal cancer cells. Cell Death Dis. 2014;5:e1534.
- Jian J, Xuan F, Qin F, et al Bauhinia championii flavone inhibits apoptosis and autophagy via the PI3K/Akt pathway in myocardial ischemia/reperfusion injury in rats. Drug Des Dev Ther. 2015;9:5933–5945.
- Liu X, Takano C, Shimizu T, et al Inhibition of phosphatidylinositide 3-kinase ameliorates antiproliferation by benzyl isothiocyanate in human colon cancer cells. Biochem Biophys Res Commun. 2017;491:209–216.
- Zhang Y, Wade KL, Prestera T, et al Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide and related thiocarbonyl compounds by cyclocondensation assay with 1,2-benzenedithiol. Anal Biochem. 1996;239(2):160–167.
- Henkhaus RS, Roy UKB, Cavallo-Medved D, et al Caveolin-1-mediated expression and secretion of kallikrein 6 in colon cancer cells. Neoplasia. 2008;10(2):140–148.
- Calay D, Vind-Kezunovic D, Frankart A, et al Inhibition of Akt signaling by exclusion from lipid rafts in normal and transformed epidermal keratinocytes. J Invest Dermatol. 2010;130(4):1136–1145.
- Yang F, Gao JY, Chen H, et al Targeted inhibition of the phosphoinositide 3-kinase impairs cell proliferation, survival, and invasion in colon cancer. Onco Targets Ther. 2017;10:4413–4422.
- Li XL, Oduola WO, Qian L, et al. Integrating multiscale modeling with drug effects for cancer treatment. Cancer Inform. 2016;14(Suppl 5):21–31.
- Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27(50):6473–6488.
- Arcaro A, Aubert M, Mee DH, et al Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell Signal. 2007;19(5):1081–1092.
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405.
- Elamin KM, Yamashita Y, Higashi T, et al Supramolecular complex of methyl-β-cyclodextrin with adamantane-grafted hyaluronic acid as a novel antitumor agent. Chem Pharm Bull. 2018;66(3):277–285.
- Upadhyay AK, Singh S, Chhipa RR, et al Methyl-β-cyclodextrin enhances the susceptibility of human breast cancer cells to carboplatin and 5-fluorouracil: involvement of Akt, NF-κB and Bcl-2. Toxicol Appl Pharmacol. 2006;216(2):177–185.
- Sarkar P, Chakraborty H, Chattopadhyay A. Differential membrane dipolar orientation induced by acute and chronic cholesterol depletion. Sci Rep. 2017;7(1):4484.
- Boreddy SR, Pramanik KC, Srivastava SK. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin Cancer Res. 2011;17(7):1784–1795.
- Fahy BN, Schlieman M, Virudachalam S, et al AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003;89(2):391–397.
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18.