
ABSTRACT
The EU Food Supplements Directive (2002/46/EC) mandates the determination of both maximum and minimum permitted levels (MPLs) for micronutrients. In order to determine MPLs which are feasible for particular population groups, a scientific approach should be used in which risk of high intake, risk of inadequacy and benefits are assessed in an integrated way taking all available data and severity and incidence of effect into account. In 2004, Renwick et al. (ILSI Europe) published a scientifically valid, flexible and pragmatic basis for a risk–benefit approach, which has been further developed here to make it a practical and quantifiable approach to be used by risk managers. The applicability of the approach is demonstrated using demo cases on iron and folate. The proposed approach has the capacity to utilize all relevant data available, including data from human studies, bioavailability data showing variability between specific forms of micronutrients and, in the case of animal studies, data on species comparability. The approach is therefore both practical and flexible, making it well suited to risk managers tasked with determining safe intake levels for micronutrients in different forms and for particular population groups.
Abbreviations
| AI | = | Adequate intake |
| AR | = | Average requirement |
| CV | = | Coefficient of variation |
| DRV | = | Dietary reference value |
| EAR | = | Estimated average requirement |
| ED50 | = | 50% effect dose levels |
| EU | = | European Union |
| FSD | = | EU Food Supplements Directive |
| IOM | = | Institute of Medicine |
| ILSI | = | International Life Sciences Institute |
| LOAEL | = | Lowest Observed Adverse Effect Level |
| MPLs | = | Maximum and minimum permitted levels |
| NOAEL | = | No Observed Adverse Effect Level |
| PSI | = | Population Safety Index |
| RDA | = | Recommended daily allowance |
| SCF | = | Scientific Committee on Food |
| ULs | = | Tolerable upper intake levels |
1. Introduction
Article 5 of the European Union (EU) Food Supplements Directive (FSD) (2002/46/EC) mandates the determination of both maximum and minimum permitted levels (MPLs) of vitamins and minerals in food supplements for their subsequent harmonization throughout the EU. In order to determine the maximum amount of vitamins and minerals, the Directive requires that upper safe levels are taken into account, including, where appropriate, varying degrees of sensitivity among different consumer groups. Reference intakes of vitamins and minerals including intakes via other dietary sources should also be considered. Moreover, minimum amounts per daily portion should be set to ensure that nutritionally or physiologically significant amounts of vitamins and minerals are present in food supplements.
In 2006, the European Commission published a discussion paper on the setting of maximum and minimum amounts for vitamins and minerals in foodstuffs (European Commission, Citation2006) in order to obtain stakeholder views. This discussion paper included an overview of existing models for the setting of maximum amounts of vitamins and minerals in foods. After stakeholder consultation, the European Commission prepared an orientation paper on setting maximum and minimum amounts for vitamins and minerals in foodstuffs in July 2007 (European Commission, Citation2007). In this paper, it is concluded that preference should be given to the risk management model developed by European Responsible Nutrition Alliance (ERNA) and the European Federation of Health Products Manufacturers Associations (EHPM) (European Commission, Citation2007). This is a risk management model in which micronutrients are characterized and categorized in three categories based on the calculated Population Safety Index (PSI). For quantitative safety characterization of the vitamin or mineral, the tolerable upper intake levels (ULs) as set by the Scientific Committee for Food (SCF) or European Food Safety Authority (EFSA) are used in the ERNA/EHPM model (European Commission, Citation2007).
The determination of ULs is based on the basic principles of risk assessment: hazard identification, hazard characterization, exposure assessment and risk characterization. Adverse effects of micronutrients are influenced by various factors including differences in requirement according to life-stage, health status, physical activity, and genetic polymorphisms. Accordingly, where necessary, it is appropriate to use separate ULs for specific consumer groups and life-stages. Moreover, bioavailability of a given nutrient (form) strongly determines its absorption, which in turn affects exposure levels which yield both beneficial and adverse effects. It is therefore preferable to specify bioavailability for individual micronutrients when deriving an UL (SCF, 2006). In practice, however, there are often insufficient data to derive specific ULs for particular sub-populations and/or micronutrient forms. In most cases, the ULs established by authorities utilize a precautionary approach that takes into account the level where no adverse effects are reported among sub-populations with the highest intake, or, they are based on the most sensitive adverse effect. Uncertainty factors are used to minimize the risk of any adverse effects at the UL even among these sensitive individuals.
The setting of MPLs typically is based on the Population Reference Intakes (PRI, used by the EFSA (Citation2010a)) or Nutrient Reference Value (NRV) (formerly referred to as the Recommended Daily Allowance, or RDA). This is the average daily dietary intake level that is considered sufficient to meet the nutrient requirements of (almost) all healthy individuals. The PRI or NRV is in turn based on the Average Requirement (AR), the Adequate Intake (AI) or Estimated Average Requirement (EAR), respectively, which is the nutrient intake value that is estimated to meet the requirement of half the healthy individuals in a population group (EFSA, Citation2010a). In the present paper, the abbreviation AR is used equivalent for EAR and AI.
As indicated, in case insufficient data on micronutrient safety and efficacy is present, additional uncertainty factors can be included to cover the uncertainties when deriving an AR and UL. As a consequence, for most micronutrients, the range between the AR and UL is narrow meaning that some sub-populations may readily exceed the UL with intake of a micronutrient. For vitamin A, calcium, copper, fluoride, iodine, iron, manganese and zinc this is known to occur. This poses a dilemma for policy makers. Setting maximum levels that attempt to ensure that the UL is not exceeded by any sub-population, may result in a proportion of the population being at increased risk of an inadequate intake. Moreover, by using the AR and UL as cut-off-points, no information is given about the type and magnitude of the risks when these reference points are surpassed (Bruins et al., Citation2015).
The consequence of this traditional method is that specific (vulnerable) population groups, such as children and pregnant women, or those with higher than average requirements like athletes may risk micronutrient deficiency. This may adversely affect their long-term health status, resilience or function. Moreover, it is known that using a regulatory measure to prevent exposure of sub-populations with greatest sensitivity to a given micronutrient form may also prevent other sub-populations from accessing adequate intake (Verkerk, Citation2010). Given that the FSD also requires determination of MPLs, it follows that the risk of inadequate as well as excess intakes of specific micronutrients should be determined for particular sub-populations. In addition, the nature and severity of effects should be considered in this respect, as the adverse health consequences of intakes below the PRI compared with those above the UL may differ considerably. This requires an approach that would allow the risk manager to weigh the consequences of a change of intake related to the proportion of the population that would have either inadequate or excessive intakes (Renwick, Citation2004).
Considering the above, a scientific approach should be used in which risk of high intake, risk of inadequacy and benefits are assessed in an integrated way forming the basis for risk management decisions that take into account the optimal nutritional status for relevant population groups. In order to make risk management decisions for both risks and benefits, the nature, severity, slope of dose-response curves and incidence of effects should be considered. Furthermore, in case a risk or a benefit applies for a specific sub-population group, this should not be extrapolated for the general population and vice versa. Also the approach should be able to distinguish between chemical forms of micronutrients in terms of differences in bioavailability and effects as a consequence of non-micronutrient parts of the molecule, e.g. methionine in selenomethionine. The main challenge is to develop an approach which takes into account all available data and enables quantification of all relevant aspects (including the severity of the effect and the quality of the data) to determine the optimal dose range for specific population groups. Therefore, the purpose of this paper is to develop a pragmatic and quantitative risk–benefit approach to derive MPLs for specific micronutrients and forms with consideration of previous work in this area. The main aim is to define a methodology or combine methodologies in such a way that for each population group an optimum range of intake of micronutrients can be derived in which the risks of deficiency and excessive intake are taken into account and dose ranges for optimal intakes are established.
2. Current risk–benefit approaches
Several risk–benefit and risk management approaches have been published over the last decade. Although each model has its advantages and disadvantages, some of the starting points used make interpretations, notably the objective assessment of benefits and risks, difficult for a risk manager. Several approaches, such as the Danish budget model (EU, 2006); ERNA/EHPM (2004) updated in 2014 by the International Alliance of Dietary/Food Supplement Associations (Richardson, Citation2014); window of benefit (Palou et al., Citation2009); the EFSA guidance (EFSA, Citation2010b) and others (EU, 2006) use point estimates such as the RDA and UL as starting points for determination of risk of the mironutrient under evaluation. However, herewith variability between micronutrient forms or magnitude and severity of the benefit or risk associated with intakes exceeding these levels among specific population groups is not taken into account. These methods also do not apply any degree of weighing according to the severity of the underlying effects as means of achieving greater proportionality.
Among the most high developed risk–benefit models are those that attempt to develop a common currency between risk and benefit, namely the Disability-adjusted life years (DALY) and Quality-adjusted life years (QALY) approaches, such as the BENERIS, BRAFO and QALIBRA models (Tijhuis et al., Citation2012). Although the DALY/QALY approaches can be very valuable when informing government policy on medical intervention etc., their application to micronutrients and specific forms thereof is unachievable at this time owing to lack of sufficient relevant input data. The European Food Safety Authority (EFSA) has highlighted several additional drawbacks to these approaches, including their applicability to different target populations (e.g. children, pregnant women, adults, elderly), the difficulty of quantifying specific beneficial effects, the incorporation of untested assumptions and the inability to integrate hazard data derived from animal studies (EFSA, Citation2010b).
Moreover, these models directly or indirectly consider diseased populations, while disability states in DALYs do not consider coexisting diseases and the weights are culture/country specific (as are intake and disease occurrence) (Tijhuis et al., Citation2012). QALY values by contrast vary with the actual questionnaire used, often depend on framing of the question, are generally not sufficiently sensitive and revealing differences depending on evaluation by general practitioner, patient, family, or the general public (Tijhuis et al., Citation2012). Thus the data requirements for calculation of a common currency are high and there are numerous challenges in the derivation of suitable inclusion data. Taking the above into account, the DALY/QALY approach was not considered suitable as the basis of a generic approach for use by risk managers for micronutrients.
The approach developed by an ILSI Europe expert group in 2004 (Renwick et al, Citation2004) was found to be the most suitable for risk management decision-making being a scientifically valid, flexible and pragmatic basis for a risk–benefit management of micronutrients in food supplements. The approach is based on two dose related intake-incidence relationships, one for the absence of benefit, the other for toxicity. Each dose response curve is derived from a 50% effect dose (ED50) being the dose that has an effect on 50% of the population, and a coefficient of variation (CV) representing the slope of the effect curve that takes into account the person-to-person variations within the human population. Together, these curves provide a scope for the dose range between benefit and risk for a given sub-population.
Based on the ED50 values for benefit and risk and applying the CV, intake levels can be calculated related to the chance that a specific effect may occur which is represented as the incidence. The ED50 is typically calculated from a dose response curve for which a log-normal distribution is considered to represent biological variability. Moreover, in case insufficient data are available to calculate the dose response curve, default values for the CV may be used instead being a CV of 15% for benefit and 45% for toxicity (Renwick et al, Citation2004). The approach incorporates important aspects for the nature of the hazard, adequacy of the data and the inherent uncertainties related to the available data.
To calculate the intake at predefined incidences based on the ED50s for benefit and risk, Renwick et al. (Citation2004) introduced a methodology. The calculation of the intake at predefined incidences was proposed as follows:
The normal CV is converted onto a log scale by calculating sigma:
sigma = (ln(CV2 +1)0.5;
The log geometric standard deviation (GSD) is calculated:
GSD = esigma
The log normal distribution can be analyzed in Excel by the statistical function NORMSINV of the incidence under consideration
The ratio of doses at median and the incidence (%) is calculated by:
Ratio = 10(logGSD × NORMSINV)
The intake at the predefined incidence considering the ED50 is than calculated:
Intake at incidence = ED50 × ratio
In the widely used Microsoft Excel software package, the formula to calculate intake at a decision incidence is given as follows:
Intake at decision incidence for benefit:
= (ED50*(10ˆ(((LOG(EXP((LN(((CV/100)ˆ2)+1))ˆ0.5)))*(NORMSINV(1-(1/incidence)))))))
Intake at decision incidence for risk:
= (ED50*(10ˆ(((LOG(EXP((LN(((CV/100)ˆ2)+1))ˆ0.5)))*(NORMSINV(1/incidence))))))
The range of intake between the benefit and risk decision incidences is considered to be an intake level which is of benefit for the majority of the population under consideration, without occurrence of excessive adverse effects. This approach is considered a very promising approach for risk management purposes as the weighing of benefit and toxicity can be done using comparable methods and values (ED50 and CV), and conversion from ED50 values to incidences at certain exposure levels can be performed. Furthermore, the severity of the effects underlying the ED50 and uncertainty aspects regarding the data used are included in this methodology, and it can be readily adapted in cases where effects differ for different sub-populations. Moreover, this approach can be applied to different chemical forms of the same micronutrient for the determination of MPLs where differences of bioavailability or risk/benefit profile between the forms are known.
3. Method
While the approach of Renwick et al. (Citation2004) is considered both flexible and appropriate as the basis for the development of a scientific approach for a risk management model for micronutrients, it should be further developed to make it a practical and quantifiable approach to be used by risk managers. Moreover, the model should be adapted to make it suitable for the setting of levels of micronutrients in food supplements and derive, if scientifically needed, different levels for specific micronutrient forms and/or population groups. The purpose of our work was to convert the previously proposed theoretical approach (Renwick et al, Citation2004) to a practical, quantifiable approach for micronutrients in food supplements that may be used directly by risk managers. As a consequence, the approach has been adapted by adding steps and includes a more detailed development of steps, which are crucial making the approach practical.
An overall overview of the design of the proposed risk–benefit approach is presented in section 4.1, after which steps which were further developed compared to the Renwick approach by the present authors are elaborated in section 4.2. Authors insights are given on how to use ED50s and CVs for micronutrients in food supplements and how to relate the severity of the effect considered to incidences reasonably assumed to be acceptable for these effects. Therefore a quantification of the severity of effects is considered, whereas the quality of the data is taken into account using uncertainty factors. Moreover, guidance is offered on how to consider the bioavailability of micronutrients which might lead to different intake levels for different chemical forms of micronutrients evaluated. Examples of relevant sources of food consumption data are given including consumption data on food supplements. Finally, advice is provided on how risk–benefit data might be used by risk managers. In order to evaluate the applicability of the proposed risk–benefit approach for micronutrients in food supplements, two chemical forms of the micronutrients folate and iron were selected for proof of principle (see section 4.3). It should be noted that the proposed approach is intended to be of relevance only for apparently healthy populations or specific subgroups thereof, and therefore it excludes diseased populations or those under any kind of medical supervision.
4. Results
4.1. Overall design of approach for micronutrients in food supplements
The overall risk management approach which is considered suitable for determination of MPLs of micronutrients in food supplements should be flexible to effectively include a broad range of data. The proposed risk management approach involves the following steps:
Identification of relevant (sub-)populations based on the intended use of the micronutrients and/or based on concerns or specific effects known for (vulnerable) sub-populations;
The derivation of ED50 values for benefit and risk from key data for the relevant (sub-) populations;
A severity of effect rating, upon which reasonable benefit and risk incidences can be derived;
An uncertainty assessment, including bioavailability and dataset quality, used for scaling the reasonable incidences. The combined uncertainty factors and reasonable incidences results in decision incidences for benefit and risk. These decision incidences are used for the calculation of optimal intake levels, equivalent to MPLs, at predefined incidences for benefit and risk considering the applicable ED50 and CV;
The decision incidence is calculated by: reasonable incidence × uncertainty factor (e.g. reasonable incidence of 1:100 with an uncertainty factor of 10 results in a decision incidence of 1:1000). This may provide a risk manager with a better understanding of the uncertainties of the choice to make;
Allocation of the intake levels for benefit and risk at the decision incidence;
Intake assessment of micronutrients can be divided in total intake and for food supplement exposure only for the relevant (sub-) populations;
Comparison of the supplement intake levels at the decision incidence with the total intake levels for the relevant sub-populations.
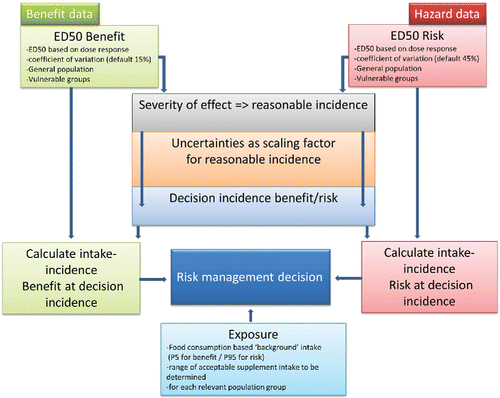
A graphical design of the proposed approach is presented (), based on that of Renwick et al. (Citation2004), including the severity of effect and scaling (uncertainty) factors to determine decision incidences for benefit and risk. A more detailed description of the additional and/ or further developed steps is provided in the subsequent sections of this paper.
Figure 1. Design of the proposed risk–benefit approach for micronutrients for use in risk management decision-making.
4.2. Elaboration of steps which were further developed
4.2.1. Determination ED50's for micronutrients in food supplements
The establishment of the ED50 is considered the starting point of the evaluation. For consideration of benefit for a given micronutrient, an ED50 above which the requirement for the micronutrient is met and below which an adverse effect or lack of benefit as a result of an inadequate intake may be expected should be taken into account. The EFSA derives Dietary Reference Values (DRV), amongst which the AR (EFSA, Citation2010a). The AR can be used as the benefit ED50, as the AR reflects the mean (median) micronutrient requirement adequate for a given population meaning by definition 50% of the population will fall above and 50% will fall below this figure (EFSA, Citation2010a; Fairweather-Taitet al., Citation2011). The AR is therefore the best estimate of requirement that can be obtained from published data as all available literature at the time of determination is evaluated and taken into account.
To determine the risk ED50, the critical dataset to determine the hazard of the micronutrient should be selected first. This dataset should be evaluated for the identification of adverse effects by the micronutrient, usually leading to the derivation of a No Observed Adverse Effect Level (NOAEL) or Lowest Observed Adverse Effect Level (LOAEL). The dataset used to determine the current tolerable upper levels (ULs) for micronutrients (EFSA, Citation2006), is considered a good starting point to determine the ED50 for risk, especially if a dose-related effect is concerned, as all available literature is taken into account and has been checked for its quality and validity. Note that the UL cannot be used as a surrogate for the ED50 as the UL normally is a no effect level.
In case the micronutrient induces adverse effects at all dose levels and a dose response cannot be determined with sufficient certainty, the lowest dose tested can be taken into account as a LOAEL. Given that the LOAEL is actually an effect dose, as is the ED50, the percentage of the population in which the effect is induced at the LOAEL is usually neither indicated nor easy to determine. In case a LOAEL is selected for which based on the data an effect-dose below 50% is likely, the LOAEL can conservatively be used as an ED50. In case a higher effect dose is considered, an uncertainty factor for scaling may be considered (section 4.2.3). If the substance did not induce any adverse effects, not even at the highest dose level, this highest dose level is taken into account as a NOAEL. The use of a NOAEL at the highest dose as a starting point will give a conservative estimation of the ED50. Alternatively, it may be assumed that the intake at this level is already at a dose level which is restricted by the amount normally expected to be consumed. As the NOAEL at the highest dose tested does not indicate any hazard, the severity of the effect is considered absent. In this case the severity at the NOAEL is related to a reasonable incidence of 1:10 (section 4.2.2). Moreover, instead of a default CV of 45%, a CV of 15% may be used instead, which also introduces a small conservative factor into the equation.
For relevant sub-populations, separate ED50s may be determined based on the specific requirements, benefits and/or risk upon consumption of micronutrients. This may be related to age, gender and/or life stages such as pregnancy, for which differences in bioavailability or effect parameter should always be considered. An example of the need for this approach is demonstrated in the case of folate for pregnant women versus that for the elderly (Verkerk, Citation2010). As a result of a specific hazard for a certain sub-population, a risk and/ or benefit ED50 may be elaborated for each relevant sub-population. Moreover, an ED50 may be derived for acute as well as chronic exposure where the available data suggest different hazards. In that case, the derived ED50s should also be compared to short-term and long-term exposure, respectively.
Also, where a very steep dose-response curve is known to occur, the ED50 can be based on the lowest effect observed and/or the most severe effect observed. If the severe effect is of greater health concern than the lowest effect, based on the calculated intake at the decision incidence, either both ED50 values or the most severe effect ED50 value may be chosen as input for the risk–benefit evaluation. This choice should be made according to expert judgment. The ED50 chosen should cover all (relevant) adverse effects for the substance and population concerned. Moreover, the severity and related incidence should be taken into account as well. As a hypothetical example: anemia is found at an ED50 of 1 mg/kg bw/d with a reasonable incidence of 1:100; see ) and histological liver (reversible) damage is observed at a next higher dose level of 10 mg/kg bw/d with a reasonable incidence of 1:100,000 (), the factor of 10 in dose level is much lower than the difference in reasonable incidences being a factor of 1000. In this case the liver damage should be considered as starting point instead of the anemia on which the NOAEL is based.
Table 1. Overview of health effects on which the ED50 is based with their proposed reasonable incidence.
4.2.2. Severity of effects: Quantification
In order to weigh benefit and risk it is noted that the severity of adverse effects for benefit and risk are mostly not comparable. Moreover, the severity of effects should be related to what might be considered an acceptable incidence of the affected population. Therefore, the incidence of effects and the severity of these effects should be taken into account in a risk benefit approach. As acceptable incidences are currently not available for most of the adverse effects, we propose to use the term ‘reasonable incidence' instead. Ultimately, reasonable incidences should be defined for specific effects. These reasonable incidences are, after scaling using the uncertainty factors, called decision incidences which are the incidences considered for risk management decisions.
A list of indicators for severity of adverse effects is given by Renwick et al. (Citation2004). However, these indicators are not further quantified. Quantification of severity of effects is needed in order to take this into account in the risk–benefit assessment. Therefore, for each indicator for severity of effect as described by Renwick, a reasonable incidence is proposed. For carcinogenicity a risk for tumor formation of 1 per 106 is generally accepted. Currently, there is not a generally accepted risk for other effects which can be used for the determination of reasonable incidences. Therefore, we propose reasonable incidences for other effects, which are elaborated relative to the 1 per 106 generally accepted risk for carcinogenicity. We propose also that clinical signs indicative of irreversible organ damage are considered to be of equivalent risk incidence to carcinogenicity (i.e. reasonable incidence of 1 per 106), and that risk incidence is reduced by a factor of 10 incrementally across different categories of declining risk, as shown in . The establishment of reasonable incidences is a risk management rather than a risk assessment responsibility: these incidences are estimations of what society considers to be an acceptable risk in relation to a specified effect. As the proposed reasonable indices are used in the comparison of benefit/risk of deficiency on one side and toxicity on the other, the actual figures used are somewhat arbitrary and primarily reflect the severity of an effect in a numerical sense.
Where different effects are known to occur in different sub-populations, a specific reasonable incidence should be derived for each relevant sub-population.
4.2.3. Uncertainty assessment
4.2.3.1. Quality of data
The dataset used in determination of ED50s might introduce uncertainties which should be taken into account in the risk–benefit assessment. The uncertainty assessment of this approach includes a quantified correction factor for quality of data and for how representative the data is for the human population under consideration (e.g. considering bioavailability aspects). In this respect, the normal correction factors to convert from animal data to the human situation can be considered: a factor 10 as default for interspecies differences and a factor 10 for intra-species differences. Incorporation of these uncertainty factors giving a one-sided precautionary approach could result in significant adverse health effects in the population due to deficiency (Renwick et al., Citation2004). In order to avoid such a situation, it is proposed that an uncertainty factor is introduced as a scaling factor to convert the reasonable incidences for both benefit and risk to a decision incidence. This way the incidence of a less severe risk effect with more uncertainties can be compared more easily with a more severe benefit or risk with fewer uncertainties during risk management.
The allocation of uncertainty factors to a certain dataset can be highly subjective and can vary according to the availability of data, and the consideration of which data are relevant. For an optimal comparison, the uncertainty assessment for benefit and risk should be performed using the same methodology to prevent excessive differences in the uncertainty factors used. The EFSA (Citation2012) has published a guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data (). This document can be used as a background document in order to use a consistent approach. The guidance provides default values for inter- and intra-species extrapolation, extrapolation for the duration of exposure, approaches for handling deficiencies in the data available, and others.
Table 2. Uncertainty factors as published by EFSA (Citation2012).
It should be noted that the CV, as used to determine the intakes at predefined incidences, reflects the dose–response curve for the population underlying the data. Therefore intra-human differences within the population concerned are already taken into account by the CV in case human data are underlying the ED50. Note that one might introduce an uncertainty factor in case information on a specific subpopulation of concern is lacking.
In case the dataset is of relatively low quality or contains data gaps preventing a good estimation of the relevance or the usability of the data, the relevance of using this dataset should be determined before an uncertainty factor is applied: i.e. if the data are questionable with regard to the conclusions drawn, it may be more appropriate to reject the data, rather than applying an uncertainty factor.
As shown in , differences in toxicokinetics and toxicodynamics mainly determine the uncertainty factor to be used. Known differences in bioavailability between the species or population tested, and the population under investigation should therefore be taken into account as well. Note that this may result in a lower as well as higher uncertainty factor to be used. Besides using the bioavailability assessment for the uncertainty assessment of extrapolating animal data to humans, the bioavailability data can be used to determine differences in bioavailability between chemical forms of a micronutrient. In the next section a guidance is provided on how to assess (quality of) bioavailability data of micronutrients.
4.2.3.2. Bioavailability of micronutrients
Bioavailability varies between different forms of micronutrients and so affects systemic exposure and subsequent beneficial or adverse effects. In determining the MPLs of micronutrients potential differences in bioavailability as a consequence of their chemical micronutrient form should therefore be considered. Bioavailability is generally taken into account in the determination of tolerable upper intake levels (EFSA, Citation2006) for which the most bioavailable forms of a given micronutrient is considered to evaluate the risk. However, this may result in deficiency issues when less bioavailable forms of the micronutrient are concerned. Therefore, variations in bioavailability between chemical forms of a given micronutrient should be evaluated. Such variability is known to be important when comparing exposure to polyglutamylfolates from the diet as compared with pteroylmonoglutamic acid taken in supplemental form (Gregory, Citation2001).
4.2.3.2.1. Definition
There is considerable debate about the definition of bioavailability used in assessing bioavailability of the different forms of micronutrients. When comparing the bioavailability between different forms of micronutrients, the absorption of the nutrient moiety is the most important factor determining its systemic availability. In the present work, bioavailability of micronutrients is defined as the proportion of a micronutrient that is absorbed from the diet or supplement and is used in normal metabolic and physiologic processes. The bioavailability factor is expressed as the percentage of intake that is absorbed by the body. This is influenced by a large number of factors: release from dietary matrix, intestinal digestion, binding to and transport over the intestinal mucosa, systemic distribution and deposition, metabolic and functional use, and excretion.
In many cases, however, the term bioavailability is in fact used to actually indicate bio-efficacy, which is one part of bioavailability being the efficiency with which ingested micronutrients are absorbed, thus reflecting only dietary and intestinal components of bioavailability. The latter is also the definition used in the EFSA's report on Tolerable Upper Intake Levels for Vitamins and Minerals (EFSA, Citation2006). Since the risk management approach proposed ideally aims to address beneficial and adverse effects of micronutrient exposure using a systems biology approach taking all bioavailability data into account, the more complete definition of bioavailability is preferred as starting point of the bioavailability assessment. It should be noted, however, that commonly only bio-efficacy data are available.
4.2.3.2.2. Bioavailability data assessment strategy
Many processes and factors are relevant to bioavailability of micronutrients, those deemed among the most important being shown schematically (). Those processes and factors depicted within the red circle highlight the most important considerations.
Figure 2. Schematic depiction of processes and factors relevant to bioavailability of micronutrients. Note: conversion factors and interaction factors can be integrated if known, otherwise an uncertainty factor can be used instead.
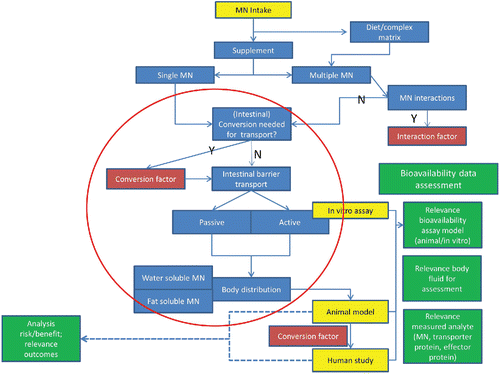
Numerous other factors contributing to the complexity of bioavailability assessment of micronutrients are evident (). However, it will be rare that for a certain micronutrients a full set of bioavailability data is available for both animal and human. Moreover, data will normally be available from different types of studies of varying predicting value whereas data may be present covering different chemical forms of the micronutrient. Therefore, bioavailability should be considered as key information when comparing different chemical forms of a micronutrient, taken into account kinetic data as far as available. As guidance for the bioavailability data assessment, the following considerations are provided.
4.2.3.2.3. Bioavailability data assessment
The amount, nature, and quality of bioavailability data at hand is setting the boundaries to draw conclusions with respect to e.g. comparison of bioavailability between several chemical forms of micronutrients. There are many different types of data in the public domain that are defined as bioavailability data. These include data from in vivo studies in humans and animals, primarily rats, pigs and monkeys, and in vitro studies, covering bio-accessibility and absorption studies. Regarding the human studies bioavailability is assessed in many different study formats, such as acute and long term supplement studies, as well as by assessing different markers for bioavailability, such as isotope-labeled micronutrient studies and analysis of direct or indirect plasma markers. The relevance and validity of the data available is subsequently depending on criteria such as (expected) conversion value of the model used, and relevance of the markers measured for assessment of absorption/bioavailability.
During a Micronutrient Bioavailability Workshop, organized by the EURRECA Network of Excellence, there was general agreement among experts that bioavailability data obtained in human studies incorporating stable isotope methodology is the gold standard (Casgrainet al., Citation2010). Although for a number of micronutrients, including iron and folate, such studies are well-established, for most micronutrients, these data are not available, and assessment of bioavailability mainly relies on animal models and/or in vitro assays. To be able to decide on the value of bioavailability data obtained from studies other than human tracer studies, the following aspects, as schematically presented in , should be taken into consideration:
| – | Translational value of test models | ||||
Figure 3. Schematic depiction of assessment of validity of bioavailability data. Green lines indicate positive answers, red lines negative answers.
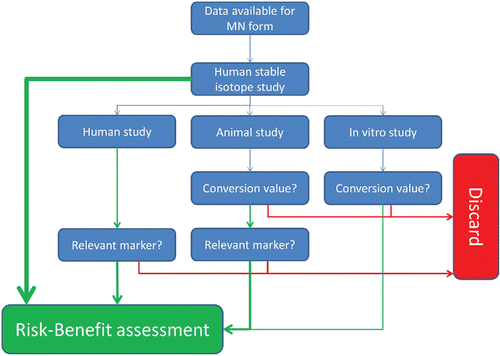
The gold standard for bioavailability studies is a tracer micronutrient human single bolus supplementation study. Where the data originate from human acute or long-term intake-status studies, there is no requirement for a translational or conversion value to be used. In these studies, the relevance of the markers (and observed changes) that are used to determine status as a measure for bioavailability needs to be established (see bullet: ‘Relevance of measured marker to calculate bioavailability' below).
Bioavailability data derived from animal studies, where the bioavailability of reference micronutrient forms have been benchmarked against human tracer studies, should be viewed as of greater value than animal studies in which comparative human studies are not available. The following key factors should be taken into account when considering bioavailability data from animal studies:
Species differences in luminal effects on micronutrient availability. Information on differences in bioavailability due to stability and model species differences in physiology and gut microbiology could be introduced using a conversion factor;
Species differences of uptake with respect to mechanism and kinetics. Depending on the transport or absorption mechanism known, more specifically active receptor-mediated transport or passive absorption, the validity of the bioavailability data available may be affected, for which an uncertainty factor may be introduced;
Species differences in body distribution and deposition. To be able to translate findings in animal models to the human situation, differences in body distribution and deposition need to be taken into account. In case there is information on similarities or differences in this aspect, conversion factors may be available based on the calculated distribution. In other cases, an uncertainty factor may be introduced.
Furthermore, specific for in vitro assays, the correlation between in vitro absorption assays and human bioavailability should be considered. A number of in vitro assays have been used for bioavailability assessment. These include the TNO's dynamic in vitro gastrointestinal TIM model and cell-based assays (Arkbågeet al., Citation2003). Data from these assays contribute to the information on bioavailability of a certain micronutrient. However, they represent only a partial picture of the whole uptake chain. These data can only be used in conjunction with relevant in vivo data, or in case there is proven correlation between valid human bioavailability data and the in vitro results for similar micronutrient forms. If no information on the relevance of these in vitro assays is available these should not be used for quantitative risk–benefit assessment.
| – | Relevance of measured marker to calculate bioavailability | ||||
In many studies, the bioavailability of a micronutrient is assessed by analysis of plasma concentrations of the specific micronutrient or a metabolite thereof. However, the absorption of micronutrients is not necessarily reflected by a fully or partial response in plasma concentrations for a specific micronutrient. Especially in case of minerals, plasma levels of free or bound micronutrients are often tightly regulated and do not (immediately) reflect absorbed material in acute studies. Therefore, special attention has to be given to the validity of plasma markers (in human and animal studies) when used to assess bioavailability. The following aspects need to be considered to assess the validity of a specific plasma (or urine) biomarkers:
Has the biomarker been validated against human tracer studies and as such is there a consistent conversion factor?
Are changes of plasma biomarkers used reflective of intake/bioavailability? This can be a direct or indirect reflection of changes in body stores;
Is the plasma marker used specific for the micronutrient or is it regulated by multiple micronutrients or other mechanisms?
What intrinsic factors affect the plasma levels of biomarker used or the control thereof? For example, inflammation reduces plasma markers of iron status (EFSA, Citation2006).
In conclusion, data from human and animal bioavailability studies using direct or indirect plasma biomarkers of micronutrients status should only be used for risk–benefit assessments if relevant biomarkers truly reflect absorption of the specific micronutrients as assessed. These markers need to have a sound scientific evidence base.
| – | Note on complicating factors | ||||
Micronutrient bioavailability is known to be affected by a wide number of factors. These factors include, but are not limited to, competitive interaction between micronutrients with respect to mucosal transport or systemic transport or storage, and effects of health or nutritional status of individuals on bioavailability regulating processes.
Often micronutrients are provided in multi-micronutrient supplements. Bioavailability of the individual components of these mixtures may strongly differ from the data reported for the individual micronutrients due to matrix interactions or interaction (e.g. competition for absorption). Similar considerations as for single micronutrients need to be taken into account for evaluation of the bioavailability data when a multi-micronutrient exposure is concerned. Some bioavailability data are obtained in studies using complete diets supplemented with tracer-labeled micronutrients, in which case the bioavailability data may be affected by dietary matrix effects.
Another factor often ignored is the influence of health status or micronutrient status on the regulation of absorption of micronutrients. In contrast to bioavailability studies with drugs or chemicals, the starting point in human and animal studies using micronutrients is the biological system, usually heavily loaded with the micronutrient of interest. Since uptake rate and thus bioavailability can be strongly influenced by the nutrient status of the host, this should be taken into account when determining the validity of bioavailability data for healthy and vulnerable populations (Heaney, 2001; Hurrell and Eqli, 2010). In many cases, while the effect of loading has been shown to occur but has found to be highly variable according to micronutrient form and sub-population, the application of uncertainty factors, to be determined by expert judgment, may be relevant.
4.2.4. Intake calculations micronutrients in food supplement
The range of intake between the benefit and risk decision incidences is considered to be an intake level which is of benefit for the majority of the population under consideration without (excessive) adverse effects due to deficiency or toxicity. Using intake data via the diet for the population under evaluation, the daily background exposure can be determined. This background exposure is the starting point for evaluation of desired additional intake levels via food supplements in which both benefit and risk are taken into account. It should be noted, however, that the background exposure is not always taken into account in limit setting. For example in the case of magnesium, the UL is based on the NOAEL of additional Mg exposure, excluding the Mg content in the diet, as this information was not provided in the key study used. The data underlying the UL should therefore be used with care for risk benefit purposes, although not including the amount present in the diet is more conservative.
The importance of reliable exposure data for the (sub)population/geographic area of interest is evident for which some examples containing reliable exposure data are provided. Several European countries have undertaken comprehensive food consumption surveys for which the data are accessible via the website of the EFSA Comprehensive European Food Consumption Database (EFSA, Citation2015a). These surveys generally provide very detailed data which can be used to determine the daily micronutrient consumption for several age groups and (EU) regional differences in intake. The choice of the model may vary with the population of interest. For the United States, the National Health and Nutrition Examination Survey (NHANES) provides detailed information on the health and nutritional status of adults and children in the United States (NHANES). Other detailed intake data are available from Flynn et al. (Citation2009) for Europe, and from the Institute of Medicine (IOM, 2011) for the United States.
For background data on micronutrient exposure from dietary (nonsupplemental) sources, data for different age groups and genders is considered a minimum requirement. These data should include:
5th percentile exposure to compare with benefit decision incidence, as the group of consumers with an intake at or below the 5th percentile are typically at greatest risk of deficiency for the micronutrient in question;
50th percentile (or mean/median) exposure to compare average intake for the given sub-population;
95th percentile exposure to compare with risk decision incidence, as the group of consumers with an intake at or above the 95th percentile might typically be related to the consumers who's intake is above a level which might induce adverse effects as a result of the micronutrient intake.
Flynn et al. (Citation2009) and NHANES also provide intake data including for food supplements and fortified foods. These data allow comparison of intakes from food supplements with intake from conventional and fortified foods as well as with the intake related to the decision incidences calculated respectively for benefit and risk. These are therefore currently considered as the best suitable source of data in respect of the risk–benefit assessment of micronutrients in food supplements.
4.3. Proof of concept via two demo cases
The proposed approach is applied to two forms of folate and iron respectively with the aim of establishing a proof of concept for micronutrients in general. The examples selected cover several issues which are considered informative for risk managers and they provide a demonstration of how other cases, involving specific micronutrients and forms, might be handled within the proposed approach. This can be related to aspects covering specific sub-populations of relevance (folate), different forms of micronutrient (iron) and intake below the decision benefit incidence at the P95 intake (iron for pregnant women). It should be noted that the current manuscript is focused on evaluating the practical applicability of the scientific approach to be used for micronutrient risk management and not intended to provide a full evaluation of the micronutrients considered or the risk management itself.
For the current cases, the benefit, hazard and exposure information used for the proof of concept were taken from publicly available review publications only. The benefit data were obtained from the data underlying the AR values as derived by the EFSA, the hazard data were taken from the data underlying the ULs as derived by the EFSA, and the exposure data were taken from the Dutch Food Consumption Survey (RIVM, Citation2011). The reason for this was that these data are considered to be of good quality and enable assessors and managers to compare outcome with this approach to the current ARs and ULs of folate and iron. Incorporation of new science might change the conclusions of the assessment. If present, the results of some new data in public literature were presented, however, a full evaluation of all data relevant to both folate and iron was considered to be outside of the scope of the current manuscript.
4.3.1. Folate
The oxidized, chemical form of folate, pteroylmonoglutamic (folic) acid, and the bioactive reduced form, 5-methyltetrahydrofolate (5-MTHF), as its calcium salt (calcium methylfolate), are considered for evaluation using the risk–benefit approach as a proof of concept. The concept was considered for the general population (adults), and the sub-populations of interest being pregnant women, lactating women and vitamin B12-deficient persons.
4.3.1.1. Bioavailability
Major discrepancies between results in humans and rats have resulted in the general acceptance that rats cannot be used to quantitatively assess bioavailability of folic acid (Gregory, Citation1995). In vitro assays have clear value in assessing bioavailability of folates as has been described in a recent review (Etcheverry et al., Citation2012). As the data underlying the ED50 are based on human data, these data were not further taken into account.
Bioavailability indices for folates are primarily based on human studies using isotope labeled folic acid supplements or plasma folate assessments following single or long term feeding. Based on these data, the Institute of Medicine (1998) concluded that it is valid to assume that folic acid supplements have a bioavailability of 85% when taken with food and close to 100% when taken with water on an empty stomach.
The gold standard for assessment of bioavailability is measurement of uptake of isotopic labeled micronutrients in well-controlled human studies. For folic acid these studies are readily available. In addition, in the case of folate, there is sufficient evidence for a direct correlation between changes in plasma folate (= 5-MTHF form), or red blood cell (erythrocyte) folate concentration with folic acid intake (Pietrzik et al., 2010). Levels of this active folate form in the circulation is primarily assessed using well standardized microbiological assays on time-course blood samples. Based on these data, it is safe to assume that the bioavailability of the biosynthetic folate forms 5-MTHF-Ca and folic acid are similar (Pietrzik et al., 2010). A number of studies have directly compared the bioavailability of these two supplements in healthy adults (PrinzLangenohl et al. Citation2003 and Pentieva et al. Citation2004; Öhrvik Citation2009). It was concluded that based on the area under the curve (AUC) and Cmax measurements for plasma folate levels there are no significant differences in bioavailability and bioequivalence between either form. This is in line with the EFSA opinion (2004) on the use of 5-MTHF-Ca as a food supplement.
To derive Dietary Folate Equivalents (DFEs) for folate, the following is taken into account: Folic acid taken with food is 85% bioavailable whereas folate naturally present in the food is only about 50% bioavailable. Folic acid taken with food is therefore 85/50 (i.e., 1.7) times more bioavailable (Yang et al. Citation2005). Thus, if a mixture of folic acid plus food folate has been consumed, DFEs are calculated as follows:
1 DFE = 1 μg folate naturally present in the food = 0.6 μg of folic acid from fortified food or as a supplement consumed with food = 0.5 μg of a supplement taken on an empty stomach (EFSA, Citation2014; IOM, Citation2014).
For consideration of bioavailability in the present framework, it is of importance to take into account that polymorphisms in the methylenetetrahydrofolate reductase gene conversion of folic acid to 5-MTHF in intestinal mucosa may result in reduction in systemically available folate from folic acid in comparison to 5-MTHF, which does not require mucosal conversion (Pietzrik et al., Citation2010). In addition, a number of drugs, such as methotrexate and trimethoprim, inhibit dihydrofolatereductase that is also an essential enzyme in conversion of FA into 5-MTHF (Blakley, Citation1984; Pietzrik et al., Citation2010). Therefore, potential interactions may be taken into account.
4.3.1.2. Benefit
Folates play an important role in the transfer of C1-groups (i.e. methyl-, methylene- and formyl-groups), maintaining the methylation balance, such as in the biosynthesis of DNA bases and in amino acid metabolism (Etcheverry et al., Citation2012). The starting point used to derive an ED50 for benefit is the AR or, for pregnant women, AI which are based on the maintenance of normal blood concentrations of folate. AR/AI values of DEFs were obtained from the EFSA (EFSA, Citation2014) and are 250, 600 and 380 µg/day for adults, pregnant women and lactating women, respectively. It is noted that these AR/AI values are in the same order of magnitude to the EAR values as derived by the Institute of Medicine (IOM, Citation2014).
For pregnant women, the AI is based on growth of fetal and maternal tissue and the active transfer of folate to the fetus whereas for lactating women the AR is based on normal folate status, which is higher due to secretion in human milk. Note that for the subpopulation women who want to become pregnant, a specific benefit might be considered for a reduced risk of fetal neural tube defects upon folate intake. Ingestion of 400 µg/day of supplemental folic acid for at least one month before and during the first trimester of pregnancy is commonly advised although the available data on folic acid intake and neural tube disease risk cannot be used for deriving the requirement for folate (EFSA, Citation2014). As only those women who want to become pregnant would benefit from an intervention whereas the critical period for prevention is very short, the AI for women who want to become pregnant is based on the maintenance of normal folate status. The AI for infants aged 7-11 months and AR for children as elaborated by the EFSA were not taken into account in this case as these values are extrapolated from breast-fed infants or from adults (EFSA, Citation2014).
4.3.1.3. Severity of effect
There is indication that folate, in isolation, may reduce the risk of cardiovascular disease, certain types of cancer, and psychiatric or mental disorders. However, this evidence is not yet conclusive. The AR is therefore based on maintenance of normal folate levels in the body (EFSA, Citation2014). Considering that folates play an important role in maintaining the methylation balance, the maintenance of normal folate levels may be related to a ‘biomarker of potential beneficial effects', which indicates an reasonable incidence of 1:100. This incidence also accounts for pregnant and lactating women. As human data are concerned covering a significant amount of data available, a scaling factor of 1 is considered applicable for all populations. The standard CV of 15% is used for the benefit calculations, which is also applied by the EFSA for calculating the PRI from the AR.
4.3.1.4. Risk
The starting point used to derive a decision incidence risk level are the studies underlying the EFSA UL derivation (EFSA, Citation2006). The details of the key study to derive the UL of folate are:
Effect: Masking of hematological signs and the potential of progression of the neurological symptoms in vitamin B12-deficient patients as a result of folic acid supplementation. It should be noted that the EFSA stated that for natural (food) or other reduced folates, no evidence for such an effect is associated with a high intake. This may indicate a different effect or mechanism for the chemical forms at high intake, In this evaluation, it is assumed that it cannot be ruled out that high intake of natural or reduced folates might masks hematological signs of neurological symptoms in vitamin B12-deficient patients as well. A LOAEL of 5 mg/day for folic acid, being 8.3 mg/d folate equivalents, is considered for the derivation of the ED50.
Reversibility: potential neurological consequences are considered as irreversible in nature.
Population: Subjects at risk are those with an (undiagnosed) vitamin B12 deficiency, such as in pernicious anemia (PA) patients and in other conditions associated with cobalamin malabsorption, and groups avoiding animal products, as a result of a marginal intake of vitamin B12, such as vegans and macrobiotics. No data are available to suggest that other populations are at risk. The EFSA considers the UL derived, and as such the data underlying the UL, also applicable for pregnant or lactating women and children (on a bodyweight basis).
Type of study: Case reports (exposure of several days up to 10 years), use of folic acid supplementation.
Based on the data available, an ED50 could not be derived due to lacking dose–response data. Therefore, a LOAEL of 5 mg/day folic acid, being 8.3 mg/day (8,300 µg/day)folate equivalents, is considered as ED50.
It should be noted that the LOAEL is based on studies performed with patients suffering from a vitamin B12 deficiency and may therefore not be representative for the general population. However, considering that the prevalence of pernicious anemia in western Europe is reported to vary between 1.2 to 1.98 per 1000 (mostly in elderly), and considering that vegans and macrobiotics also may risk a marginal intake of vitamin B12, this effect may also be considered of relevance for the general population and is discussed later.
4.3.1.5. Severity of the hazard
General population
For the general population no adverse effects as a result of folic acid or 5-MTHF intake is known, therefore the ED50 may be related to ‘Biochemical changes within or outside the homeostatic range and without known sequelae', which indicates a reasonable incidence of 1:10 with a scaling factor of 2 because of the use of a LOAEL for the ED50. For the general public, including children, a decision risk of 1:20 incidence is therefore taken into account. The standard CV of 45% is used for the risk calculations.
Population with vitamin B12 deficiency
The hazard considered is irreversible in nature for potential neurologic effects, which is related to ‘Clinical signs indicative of irreversible organ damage', which indicates a reasonable incidence of 1:1,000,000. The prevalence of the population of patients suffering from a vitamin B12 deficiency is known and might be used to convert the reasonable incidence towards a decision incidence for the general population correcting for the prevalence of this subgroup. However, as the group of vegans and macrobiotics within the general population is so far not known, it was chosen to use the population with a potential deficiency for vitamin B12 as a separate population.
As the ED50 of 8,300 µg/day is based on a LOAEL with lacking dose-response data and human data are concerned covering a significant amount of data available, an scaling factor of 2 is considered. As the reasonable incidence of 1:1,000,000 already reached the maximum, a decision incidence for risk of 1: 1,000,000 is considered for this population. The standard CV of 45% is used for the risk calculations.
4.3.1.6. Risk-benefit analysis
The intake calculations at predefined benefit/risk incidences is given in for the general population. In an overview of intake values for each of the subpopulations considered is given at the respective decision incidences. Furthermore relevant intake figures via the diet are presented for the present evaluation. In the present case, intake figures for the Dutch diet were used as reported by the RIVM (Citation2011), which may not be representative for other countries/regions. Country specific intake data should be considered including dietary habits of specific regions, for other target populations. The lowest P5 intake figure and the highest P95 figure for adults are considered for the evaluation to evaluate benefit and risk, respectively. As for children the ED50s are equal to that of adults on a bodyweight basis, this subgroup is for the present evaluation not taken into account.
Table 3. Folate intake calculations (in dietary folate equivalents) at predefined incidences for benefit and risk.
Table 4. Overview of ED50s, intake at the decision benefit/risk incidences and intake of folate (in dietary folate equivalents) via the diet.
When considering decision benefit/risk incidences in relation to different intake levels of dietary folate (in folate equivalents [FE]) (), it can be concluded that:
The intake related to a beneficial incidence of 1:100 (at 354 µg/day) for adults in the general population is above the P50 intake via food;
The intake related to a risk incidence of 1:20 (at 4,100 µg/day) for adults in the general population, is well above the P95 intake via food of 492 µg/d;
A supplement intake of 4,100 – 492 (P95 intake) = 3,608 µg folate equivalents/d might be considered beneficial for almost all of the people and especially pregnant and lactating women, in the general public. However, it should be noted that persons in the general public unaware of a vitamin B12 deficiency might receive a folate level which may be related to a significant risk when an intake of maximal 4,100 µg/day is considered, taking into account the intake at the decision risk incidence of 1,100 µg/d for this population;
For the general population, taken into account that persons may be unaware of a vitamin B12 deficiency, therefore a maximum supplement intake of 1,100 – 492 (P95 intake) = 608 µg/d may be considered. This supplement intake covers the intake at the decision benefit of 354 µg/d, as well as the advised additional intake of 400 µg/d for women who want to become pregnant to prevent Neural Tube Disease (EFSA, Citation2014);
For pregnant women, the intake at the decision benefit incidence is 850 µg/day, which covers the growth of fetal and maternal tissue and the active transfer of folate to the fetus in order to maintain normal blood concentrations of folate. Via the diet, a folate intake of 125 (at P5) to 355 (at P95) µg/day is estimated for females in the childbearing age, which intake is below the decision benefit incidence intake of 850 µg/day. A supplement intake of 725 µg folate/day will raise the part of the population at and above the P5 intake via the diet already to an intake at or above 850 µg/day. The part of the population at the P95 intake via the diet (355 µg/day) receiving such a dose will then be exposed to 1,080 µg/day. This intake is well below the intake at the decision risk incidence of 4,100 µg/day and also covers the intake at the decision risk incidence of 1,100 µg/day for the vitamin B12 deficient population. It is noted that the supplement intake of 725 µg/day considered is well above the advised additional intake of 400 µg/d for pregnant women in their first trimester of pregnancy, to prevent Neural Tube Disease (EFSA, Citation2014);
The preferred supplemental intake for lactating women is higher than for the general population because of secretion of folate to human milk. A higher demand for folate, compared to the adult general population, of 186 µg/day is needed compared to the general population to reach the intake at the decision benefit incidence of 540 µg/day for lactating women. Taken into account the maximum supplement intake of 608 µg/day as calculated for the general public, also protecting vitamin B12 deficient persons, a supplement intake of 794 µg/day may be considered;
A risk manager may conclude that based on the intake figures for adults, which were derived from the sub-population of 51 to 69 years of age, a lower supplement folate intake may be advised on the basis of the intake figures for the respective age groups for which a lower intake is reported (data not shown);
In case a higher supplement intake is considered which exceeds the intake at the decision risk index of 1,100 µg/day for vitamin B12 deficient persons, the possible additional risk for the general public unaware of a vitamin B12 deficiency might also be considered taken into account the prevalence of pernicious anemia in western Europe which is reported to vary between 1.2 to 1.98 per 1000. It should be noted that vegetarians and vegans may also risk a marginal intake of vitamin B12, and that this subgroup (of often unknown size) is also of relevance for the younger age groups within the general population.
It should be noted that L-5-MTHF, whether from dietary sources or supplements, has a lower potential for masking vitamin B12-deficiency symptoms (Pietrzik et al. 2010). Furthermore, the cellular uptake of circulating L-5-methyl-THF is subject to tight cellular control, whereas pteroylmonoglutamic (folic) acid, which is not subject to this cellular control, is retained even in folate-replete subjects. For these reasons, Pietzrik et al. (Citation2010) indicate that L-5-MTHF should be considered for use in long-term folate therapies but also for situations close to the decision risk incidence.
4.3.1.7. Inclusion of recent data
Recently, some publications became available which may be used for further fine-tuning the risk–benefit analysis.
The National Toxicology Program (NTP) published a draft monograph on identifying research needs for assessing safe use of high intakes of folic acid (NTP, Citation2015). In this draft monograph four health effect categories were identified being of high priority. These categories were identified based on reported adverse effects of folic acid in studies of intake over 400 µg/day or blood levels above the deficient range, and cover cancer, cognition and vitamin B12, hypersensitivity, and thyroid and diabetes-related disorders. Unfortunately, the draft monograph did not contain the results of the evaluations of these adverse effects. The ED50 taken into account for vitamin-B12 deficient patients in the case above was based on masking of hematological signs and the potential of progression of the neurological symptoms, for which the highest reasonable incidence of 1:1,000,000 already was taken into account. With an ED50 already related to the highest reasonable incidence, only for new adverse effects with an ED50 below the ED50 used for the case above, the intake at the decision risk incidence may be affected and may be taken into account accordingly for risk management purposes.
For the general population a reasonable incidence of 1:20 related to a lack of adverse effects is considered. In cases where serious adverse risks such as cancer, hypersensitivity or thyroid and diabetes-related disorders are related to high intakes by the public in general, the reasonable incidence will raise considerably up to 1:1,000,000. Such effects would therefore significantly affect the calculated intake at the decision incidence. Evaluation of all available relevant evidence is therefore of utmost importance before determinations are finalized. It should be noted that inclusion of vitamin B12 supplementation may be a risk management strategy which is, however, not considered in this case.
It is known that methotrexate (MTX), used as drug for rheumatoid arthritis, depletes intracellular folate, as has been documented in hepatocytes and peripheral blood lymphocytes of MTX treated patients. Folate deficiency may cause side effects such as mouth sores, stomach problems such as nausea or abdominal pain, liver problems or problems with producing blood cells. Folic acid supplementation (0.5 to 2 mg/daily) may ameliorate these side effects of MTX by reducing the frequency and severity of the side effects without lowering the effectiveness of the MTX in the treatment of rheumatoid arthritis (Shea et al. Citation2013). The effect of MTX on folate depletion can be taken into account if more information on the dose–effect relation is known which is currently not the case. As MTX prescription is usually under supervision of a physician, this is out of the scope for supplement risk management purposes.
4.3.2. Iron
The chemical forms iron sulphate and iron bisglycinate are considered for evaluation using the risk–benefit approach as a proof of concept. High-dose iron supplementation is normally restricted to a limited period of time although iron is also commonly found in multivitamins which are intended for extended periods of exposure. For the current case the focus for the hazard was on sub-acute exposure to cover short term high-dose iron supplementation. In case chronic effects are to be taken into account as well, ED50s for sub-acute and chronic exposure can be considered next to each other.
4.3.2.1. Bioavailability
Following oral administration, both iron sulphate and iron bisglycinate adds to the intestinal intraluminal pool of inorganic, non-haem iron. Iron bisglycinate is absorbed intact into the mucosal cells of the intestine, and is subsequently dissociated into its iron and glycine components, the iron part of iron bisglycinate is metabolized like any other source of iron (EFSA, Citation2006). In contrast, the water soluble iron sulphate will be ionized before absorption. Absorption of non-haem iron is known to be influenced as a result of inhibitors, like calcium or polyphenol compounds from beverages, or enhancers, like muscle tissue from several livestock animals or ascorbic acid, in the diet. It might be assumed that the intrinsic bioavailability of iron chelates such as ferrous bisglycinate have a higher bioavailability than iron sulphate in adults based on human studies in iron deficient subjects (Ferrari et al., Citation2012; Milman et al., Citation2014), but for schoolchildren with iron deficiency without anemia there is no indication that there are differences in absorption rates (Duque et al, Citation2014). In the EFSA (Citation2006) opinion on the use of iron bisglycinate in food manufacturing and as a food supplement, it was concluded that no significant differences in bioavailability were observed in infants with normal iron status with bioavailability levels varying with fortified meal type. Therefore, for the case under consideration, it is assumed that the absorption of iron sulphate and iron bisglycinateare rather comparable for infants in the same type of food. For iron deficient adults ferrous bisglycinate has a two to five fold higher absorption when compared to iron sulphate, which should be taken into account by the risk manager when choosing the form of iron in a supplement.
4.3.2.2. Benefit
The Institute of Medicine (IOM, Citation2001) reported that important subclinical and clinical consequences of iron deficiency are impaired physical work performance, developmental delay, cognitive impairment, and adverse pregnancy outcomes. These adverse consequences of iron deficiency are associated with a degree of iron deficiency sufficient to cause measurable anemia, which is the most easily identifiable indicator of functional iron deficiency. The IOM derived an EAR based on the need to maintain a normal, functional iron concentration, but only a minimal store which is related to a serum ferritin concentration of 15 µg/L. However, a more recent evaluation by the EFSA (Citation2015b) considers this serum ferritin level has insufficient taken into account that at this level stores would fall to virtually zero for pregnant women at delivery. Therefore, the EFSA considers a target value of 30 µg/L for serum ferritin, reflecting an adequate level of iron stores for all populations (EFSA, Citation2015b). The starting point used for calculating the ED50 is the AR value for adult men and postmenopausal women as derived by the EFSA (Citation2015b). For the present evaluation an ED50 of 6 mg iron/day is therefore considered for the general (adult) population. The populations of premenopausal women (including pregnant women and lactating women) are not further taken into account separately as the intake via the diet for these sub-populations are unknown and the AR for these populations is slightly higher (7-8 mg iron/day) than the AR for adults being therefore more conservative.
4.3.2.3. Severity of effect
Considering that iron plays an important role in preventing anemia, the maintenance of normal iron levels may be related to a ‘biomarker of potential beneficial effects', which indicates a reasonable incidence of 1:100. As the AR elaborated by the EFSA is based on iron it was chosen not to apply a scaling factor for bioavailability of the chemical forms used for supplement exposure. Instead conversion to the supplement form iron sulphate and iron bisglycinate is made after calculation of the supplement exposure for iron, which is based on the molecular weight (mw) and difference in bioavailability. Therefore, an ED50 of 6 mg iron/day for the general population with a decision incidence of 1:100 is proposed for iron in the present case. The standard CV of 15% is used for the benefit calculations.
4.3.2.4. Risk
Iron bisglycinate
Iron bisglycinate is of low toxicity as in a 13 week study in rats the NOAEL was found to be 500 mg/kg bw/d, being the highest dose tested. Moreover, field trials in developing countries revealed that between 2 and 23 mg/day of supplemental dietary iron can be consumed without any reports of adverse effects. In addition, dietary iron supplementation using iron bisglycinate, at dose levels of approximately 15 to 120 mg iron/day, has been well tolerated by adults and pregnant females with a normal iron status and more in particular by iron-deficient young children. Moreover, there was no evidence of iron overload in iron-replete individuals (EFSA, Citation2006).
Iron sulphate
The EFSA has evaluated several forms of iron including iron sulphate in 2004. In this evaluation, animal toxicity studies were reported for which acute toxicity occurred in mice after oral doses of ferrous compounds in the range 200-650 mg iron/kg body weight, with iron sulphate being the most toxic and iron fumarate the least toxic. Administration of 50 and 100 mg iron/kg body weight/day for 12 weeks decreased growth rates in male rats with a potency in the order iron sulphate> succinate >fumarate>gluconate. Moreover, a daily dose of 50 mg of iron produced a higher incidence of gastrointestinal effects in humans given conventional iron sulphate compared with subjects given the same amount in a wax-matrix, and also in subjects given iron sulphate compared with subjects given the same amount of iron as bis-glycino iron (EFSA, Citation2004).
Based on the data available, an ED50 could not be derived due to lacking dose-response data. However, a lowest effect level can be defined based on the results of studies on gastrointestinal (GI) side effects in human as reported by the EFSA (Citation2004). Side effects of oral iron preparations at therapeutic dose levels of 50-220 mg iron/day include nausea, vomiting, heartburn, epigastric discomfort, diarrhea and intractable constipation. As iron sulphate is indicated as having a higher toxicity than the other forms of iron considered by the EFSA, the lowest effect level mentioned is considered for the ED50. Therefore, an ED50 of 50 mg/day as a low effect level is considered for iron sulphate. Taken into account the uncertainty of this level to be used as an ED50, an uncertainty factor of 2 is considered as scaling factor.
4.3.2.5. Severity of hazard
Considering the difference in the animal toxicity data reported and taken into account that iron sulphate induces gastrointestinal distress at dose levels of 50 mg and higher, whereas for iron bisglycinate these effects were not reported up to 120 mg iron/day, it is concluded that both forms of iron cannot be evaluated using the same ED50.
For the present evaluation, an ED50 of 120 mg iron/day, as iron bisglycinate, is considered. In case no effect is considered at the ED50 taken as starting point, the severity is related to a reasonable incidence of 1:10. Taken into account that a significant amount of human data are underlying this figure no scaling factor is considered. Therefore, an ED50 of 120 mg/day with a decision incidence of 1:10 is proposed for iron bisglycinate.
The GI side effects for iron sulphate in the upper gastrointestinal tract depend on the local iron concentrations and are due to irritation of the mucosa, alteration of gastrointestinal motility and/or rapid transfer of iron into the circulation. The severity of gastrointestinal discomfort is rated as “Clinical symptoms indicative of a minor but reversible change”, for which a reasonable incidence of 1:1000 is considered. Adding the scaling factor of 2 as a result of the use of a LOAEL, a decision incidence of 1:2000 is considered for risk management purposes. Therefore, an ED50 of 50 mg iron/day with a decision incidence of 1:2000 is proposed for iron sulphate.
The standard CV of 45% is used for the risk calculations of both forms.
4.3.2.6. Risk–benefit analysis
The intake calculations at predefined benefit/risk incidences are given in . In an overview of intake values is given at the respective decision incidences. Furthermore, the most relevant intake figures as shown in were taken into account for the present evaluation. As such the lowest P5 intake figure and the highest P95 figure for adults are considered for the evaluation to evaluate benefit and risk, respectively.
Table 5. Iron intake calculations at predefined incidences for benefit and risk.
Table 6. Overview of ED50s, intake at the decision benefit/risk incidences and intake of iron via the diet.
When considering the decision benefit/risk incidences and dietary intakes of iron (), it can be concluded that:
A clear difference is found for the calculated intake at the predefined incidence for iron sulphate and iron bisglycinate based on the respective decision risk incidences concerned. The maximum intake for iron sulphate (13.1 mg iron/day) is about 20% of the maximum intake calculated for iron bisglycinate (69.2 mg iron/day);
The intake of iron at the decision benefit incidence of 1:100, being 8.5 mg/d, is between the P5 (7.2 mg/day) and P50 intake figure (10.7 mg/day) for adults in the general population, a significant amount of individuals therefore have an intake of iron below the intake at the decision benefit incidence;
Supplement use may increase the amount of iron intake, for which an additional 1.3 mg iron/day intake already lift the intake at the P5 intake level (of 7.2 mg iron/day) to the level of the decision benefit incidence of 1:100 for iron. This can be achieved by supplementation of 7 mg iron sulphate (1.3 / 55.8 [mw iron] * 152 [mw iron sulphate] * 2 [bioavailability factor]) or 4.8 mg iron bisglycinate (1.3 / 55.8 [mw iron] * 204 [mw iron bisglycinate]).
For iron sulphate the combined total iron intake via the diet and supplement use is above the intake at the decision risk incidence at the high exposure (P95) intake at which gastrointestinal discomfort may be noticed for 10% of the population exposed to the higher iron intake values.
For iron bisglycinate the combined intake via the diet and supplement intake is well below the intake at the decision risk incidence, even for the high intake consumers, and is therefore not considered of safety concern.
Note that the ED50 for iron sulphate is based on gastrointestinal discomfort. This effect might be related to an intake in the absence of food. In case supplemental intake together with food is considered, these effects are assumed to be limited or even absent and, in the case of supplements, such risks could be adequately managed via labeling. This aspect was not further considered in the present case.
5. Discussion and conclusion
This work presents a feasible, scientifically based methodology that may be used by risk managers to determine MPLs, e.g. given the requirements of Directive 2002/46/EC (Article 5). By taking into account both risk and benefits, the approach allows the determination of an optimum range of supplemental intakes of different forms of micronutrients for each population group of interest, in which not only benefits and risks are taken into account, but in which also the severity of effects are included in decision making. A risk manager must be able to consider the possible residual risk given that, as with conventional foods, it is often not possible to ensure the benefit for the majority of the population, without exposing the high intake part of the population to some type of risk (Verkerk, Citation2010). The risk–benefit approach proposed in this paper is based on the ED50 calculated, the severity of the critical effect and its related ‘acceptable’ incidence for both deficiency and toxicity of a given micronutrient form, and makes comparison on a one-to-one scale possible which will greatly facilitate proportionate risk management decision-making. It should be noted that in risk management evaluations using the risk index, comprised of impact of a risk event × probability of occurrence, is not fundamentally different from evaluating the impact of a risk event according to the severity of a given health effect related to the decision incidence, which is considered acceptable for the health effect observed. The probability of its occurrence can in turn be estimated from data on the incidence of the effect at a given dose level.
5.1. Risk management
Risk managers will require an adequate descriptive narrative about the nature and severity of the beneficial (preventing a deficiency) and adverse toxic health effects used to derive the ED50s, along with the bandwidth of exposure for specific sub-populations. This will allow the risk manager to weigh the acceptability of any incidence against the severity of the effect. For example, a risk manager may be willing to accept an increased incidence of 1: 10 for a change in an enzyme activity at high exposure in favor of a reduced incidence for a deficiency symptom, such as fetal neural tube defects upon folate intake. This might be the case where the enzyme activity is a sensitive indicator of toxicity and is normally related to a reasonable incidence of 1:100. The cases on folic acid and iron as described in section 4.3 provide examples of risk management considerations, implicitly weighing incidence and severity to reach an acceptable additional intake via food supplements. Since there are no guidelines currently available informing risk managers how to manage or deal with different types of risk, good information sharing and cooperation between the risk assessor and risk manager is essential to decide on MPLs to be elaborated.
When assessing the benefit and risk of additional exposure from food supplements above background exposure from conventional diets, it is essential that differences between supplemental intake and those derived from conventional and fortified foods are considered. The outcome of the approach, i.e. the bandwidth of exposure, accounts for the total exposure to a micronutrient. In fact, the risk manager has to decide which part of the total exposure can be taken by the food supplement in relation to the normal intake via the diet. In this process, also sub-populations which might be at risk for a deficiency of a micronutrient via normal dietary exposure should be taken into account. In the iron case described, it is noteworthy that the diet barely leads to an intake at the decision benefit incidence (of 8.5 mg/day) calculated for adults.
It may also be relevant to take into account risks that might arise from the unconscious consumption of micronutrients, notably those in conventional foods (e.g. retinol in liver) vis-à-vis those taken in supplemental form, where the micronutrient source is clearly recognizable as such i.e., pharmaceutical form or appearance, Nutrition Facts statement on label, etc. Moreover, formulation efforts for supplements should also be taken into account using the most recent data available, e.g. possibly affecting population groups or bioavailability for combinations of folate and vitamin B12 or copper with zinc.
The final step in risk management combines the efforts and translates these into (risk) communication(s) most likely through a graded response. This may include clear labeling with precautions directed at any identified at-risk sub-population, detailed usage instructions intended to minimize risk while optimizing benefit, and inclusion of the mandatory statement of amount of micronutrients by comparison with reference intakes. While labeling regulations exist in relation to food supplements, there is as yet no harmonization of warning statements for high intakes that might benefit some sub-populations, while exposing others to some kind of risk. Clear information might also prevent misuse of a specific product by non-intended population groups or overuse by the intended population group. A main point is to create awareness of micronutrient intake by the consumer through all sources. This will empower the consumer/user to make informed decisions.
Risk managers must foster a broad range of responsibilities that include careful consideration of the latest scientific data on both benefit and risk of micronutrients, how this may or may not be related to specific chemical forms of micronutrients, stakeholder interests, social needs and of course consumer safety and choice. It should be noted that the current paper focuses on a scientific approach to be used for micronutrient risk management and not specifically on risk management itself. The value of the outcomes based on using the proposed approach will be affected by the quality of the relevant data available, and the approach in no way makes up for problems associated with inadequate data.
5.2. Advantages risk–benefit approach
Considering this scientific approach to be used for micronutrient risk management, the work by Renwick et al. (Citation2004) is considered a solid platform for further development. With the proposed adaptations and extensions described in this paper, a practical and quantitative risk management approach for micronutrients is available which is able to take into account all relevant data available, e.g. toxicological risk, beneficial effects, bioavailability, differences between chemical forms of nutrients. Moreover, it takes into account quantitatively the nature and severity of effect, incidences which might be acceptable for certain effects and the slope of the dose–response curve, allowing improved quality and sensitivity of risk management decision-making through the balancing of both risks and benefits. With the guidance for assessment of bioavailability data and including harmonized factors allowing conversion of findings from animal studies to humans in cases where human data are lacking, the determination process of an scaling (or uncertainty) factor is as objective as possible.
Apart from application for the general public, the recommended approach can easily be applied to specific population groups in case benefits or risks are specific for a population group. This way optimal supplement intake recommendations for each population group is ensured without prejudicing other groups. Among the factors triggering the need for a sub-population specific approach are bioavailability, and between-species variation. Where relevant biomarkers cannot be identified, omics or systems biology data on functional effects may be used to bridge the comparability of substances used for read-across (Ommen et al., Citation2009 and Citation2010 and Palou et al., Citation2009). Given the rapid emergence of these branches of nutritional science, it is likely that more relevant data of this type will become available in the near future.
In cases where the effects differ for different forms of one micronutrient, it is recommended that the risk management approach be applied separately for each micronutrient form. In cases where only the systemic bioavailability differs for the different micronutrient forms, corrections should be applied to the MPLs derived taking into account these differences in bioavailability.
5.3. Importance of data
Ideally, sufficient data should be available on both risks and benefits to determine dose–response curves. This would require the availability of multiple dose studies on adverse and beneficial effects for each micronutrient (form) for each target population. Unfortunately, this situation is not realistic or feasible given ethical considerations, research costs and available funding. Human study designs mostly include single or limited dose studies assessing nutritional or beneficial effects without considering potential adverse effects. Confounding factors such as dietary habits, nutritional needs among different consumer groups, gender, genetic polymorphisms and geographical variation complicate the possibility to retrieve specific study data further in studying the nutritional and health effects of micronutrients. As a consequence of insufficient data to determine a dose–response curve for many micronutrients the ED50s are point estimates for which standardized CVs can be used for extrapolation purposes simulating a dose–response.
6. Conclusion
The current approach covers the elaboration of micronutrient supplement intake levels considering a minimum exposure for normal and eventual beneficial function of the body (benefit), and maximum exposure related to toxicity (risk) and is therefore considered consistent with the requirements of Article 5 of Directive 2002/46/EC on food supplements. The flexibility and practical applicability has been demonstrated in two cases for each of two forms of folate and of iron. Although we consider briefly how recently published data may affect outcomes, approaches to the evaluation of hazard, risk, benefit and exposure should be further extended by additional literature screening and eventually incorporation of company-confidential scientific information to include all relevant data in relation to micronutrients and their respective forms. In addition, outcomes should ideally be judged against data from observational studies and from clinical experience, in order to determine whether any re-evaluation might be necessary implementing new data, incidence points or scaling/uncertainty factors.
7. Conflict of interest
The current project was sponsored by Alliance for Natural Health International (Dorking, United Kingdom) and Solgar Vitamins (Holland) BV (Haarlem, The Netherlands). Both ANH International and Solgar Vitamins (Holland) BV did not have had any influence on the decisions made or conclusions drawn by the authors related to the project and/or the current manuscript. The authors declare that there are no conflicts of interest.
Acknowledgments
The authors like to thank Geert Houben for his critical review of the manuscript and Suzanne van den Berg for her help in the literature assessment of the selected case studies on folate and iron.
References
- Arkbåge, K., Verwei, M., Havenaar, R. and Witthöft, C. (2003). Bioaccessibility of folic acid and (6S)-5-methyltetrahydrofolate decreases after the addition of folate-binding protein to yogurt as studied in a dynamic in vitro gastrointestinal model. J. Nutr. 133(11):3678–83.
- Blakley, R. L. (1984). Dihydrofolatereductase. In: Folates and Pterins: Chemistry and Biochemistry of Folates (volume 1), pp191–244. Blakley, R. L., Benkovic, S. J., editors. Wiley, New York.
- Bruins, M.J., et al. (2015). Addressing the risk of inadequate and excessive micronutrient intakes: traditional versus new approaches to setting adequate and safe micronutrient levels in foods. Food Nutr. Res. 59.
- Casgrain, A., et al. (2010). Micronutrient bioavailability research priorities. Am. J. Clin. Nutr. 91:1423S–1429S.
- Duque, X., et al. (2014). Effect of supplementation with ferrous sulfate or iron bis-glycinate chelate on ferritin concentration in Mexican schoolchildren: a randomized controlled trial. Nutr. J. 13:71–81.
- EFSA. (2004). Opinion of the scientific panel on dietetic products, nutrition and allergies on a request from the commission related to the tolerable upper intake level of iron. EFSA J. 125:1–34.
- EFSA. (2006). Opinion of the scientific panel on food additives, flavourings, processing aids and materials in contact with food on a request from the commission related to ferrous bisglycinate as a source of iron for use in the manufacturing of foods and in food supplements. EFSA J. 299:1–17.
- EFSA. (2006). Tolerable upper intake levels for vitamins and minerals, scientific committee on food and scientific panel on dietetic products, nutrition and allergies. ISBN: 9291990140; Available at http://www.efsa.europa.eu/en/ndatopics/docs/ndatolerableuil.pdf.
- EFSA. (2010a). Scientific opinion on principles for deriving and applying dietary reference values. EFSA J. 8(3):1458.
- EFSA. (2010b). Guidance on human health risk-benefit assessment of foods. EFSA J. 8(7):1673.
- EFSA. (2012). Guidance on selected default values to be used by the EFSA scientific committee, scientific panels and units in the absence of actual measured data. EFSA J. 10(3):2579.
- EFSA. (2014). Scientific opinion on dietary reference values for folate. EFSA J. 12(11):3893.
- EFSA. (2015a). Available at http://www.efsa.europa.eu/en/datexfoodcdb/datexfooddb.
- EFSA. (2015b). Scientific opinion on dietary reference values for iron. EFSA J. 13(10):4254.
- ERNA/EHPM. (2004). Vitamin and mineral supplements: a risk management model. ISBN 9080920614. Pgs. 1–23.
- Etcheverry, et al. (2012). Application of in vitro bioaccessibility and bioavailability methods for calcium, carotenoids, folate, iron, magnesium, polyphenols, zinc and vitamins B6, B12, D and E. Front. Physiol. 3:1–22.
- European Commission. (2006). Discussion paper on the setting of maximum and minimum amounts for vitamins and minerals in foodstuffs. Available at http://ec.europa.eu/food/food/labellingnutrition/supplements/discus_paper_amount_vitamins.pdf.
- European Commission. (2007). Orientation paper on the setting of maximum and minimum amounts for vitamins and minerals in foodstuffs. Reference no. Sanco/E4/FDA/bs(2007)D/540510. Pgs. 1–23.
- Fairweather-Tait, S. J., et al. (2011). Risk-benefit analysis of mineral intakes: case studies on copper and iron. Proc. Nutr. Soc. 70:1–9.
- Ferrari, P., et al. (2012). Treatment of mild non-chemotherapy-induced iron deficiency anemia in cancer patients: comparison between oral ferrous bisglycinate chelate and ferrous sulfate. Biomed Pharmacother. 66(6):414–418. doi: 10.1016/j.biopha.2012.06.003.
- Flynn, A., et al. (2009). Intake of selected nutrients from foods, from fortification and from supplements in various European countries. Food Nutr.Res. 12:1–51.
- Gregory, J. F. (1995). The bioavailability of folate. In: Folate: Nutritional and Clinical Perspectives (Bailey, L., ed), pp. 195–235. New York: Marcel Dekker.
- Gregory, J. F. (2001). Bioavailability of nutrients and other bioactive components from dietary supplements. case study: Folate bioavailability. J. Nutr. 131:1376S–1382S.
- IOM National Research Council. (1998). Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. The National Academies Press, Washington, DC. Available at http://www.nap.edu/catalog/6015.html.
- IOM National Research Council. (2001). Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; Available at http://www.nap.edu/catalog/10026.html.
- IOM National Research Council. (2014). List of dietary reference intakes. Available at http://iom.edu/Activities/Nutrition/SummaryDRIs/∼/media/Files/Activity%20Files/Nutrition/DRIs/New%20Material/5DRI%20Values%20SummaryTables%2014.pdf.
- Milman, N., et al. (2014). Ferrous bisglycinate 25 mg iron is as effective as ferrous sulfate 50 mg iron in the prophylaxis of iron deficiency and anemia during pregnancy in a randomized trial. J. Perinat. Med. 42(2):197–206. doi: 10.1515/jpm-2013-0153.
- NHANES: National Health and Nutrition Examination Survey of the US Center for Disease Control and Prevention. Available at http://www.cdc.gov/nchs/nhanes.htm.
- NTP. (2015). National toxicology program. Draft NTP monograph for the expert panel: identifying research needs for assessing safe use of high intakes of folic acid. (Available at http://ntp.niehs.nih.gov/ntp/about_ntp/ntpexpanel/2015/draft_monograph_folic_acid_508.pdf; July, 2015).
- Öhrvik, V. (2009). Folate bioavailability in vitro experiments and human trials. Doctoral thesis of the Swedish University of Agricultural Sciences, Uppsala, ISBN 978-91-576-7410-4.
- Palou, A., et al. (2009). Integration of risk and benefit analysis–the window of benefit as a new tool. Cr. Rev. Food Sci.Nutr. 49(7):670–680.
- Pentieva, et al. (2004). The short-term bioavailabilities of (6S)-5-methyltetrahydrofolate and folic acid are equivalent in men. Human Nutr.Metab. 134:580–585.
- Pietzrik, K., et al. (2010). Folic acid and L-5-methyltetrahydrofolate: comparison of clinical pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 49(8):535–548.
- Prinz-Langenohl, R., et al. (2003). Effect of folic acid preload on the bioequivalence of [6S]-5-methyltetrahydrofolate and folic acid in healthy volunteers. J. Inherit. Metab. Dis. 26(Suppl 1):124.
- Renwick, A. G., et al. (2004). Risk-benefit analysis of micronutrients. Food Chem.Toxicol. 42(12):1903–1922.
- Richardson, D. P. (2014). Nutritional risk analysis approaches for establishing maximum levels of vitamins and minerals in food (dietary) supplements. Prepared on behalf of IADSA. Available at http://www.vitaminsinmotion.com/fileadmin/data/pdf/Publications/IADSA_Nutritional_risk_appraoches_for_establishing_max_levels_of_vits___minerals_in_food_supplements.pdf.
- Rossum, C. T. M. van, et al. (2011). Dutch National Food Consumption Survey 2007-2010. RIVM Report 350050006. Available at http://www.rivm.nl/bibliotheek/rapporten/350050006.pdf
- Shea, B., et al. (2013). Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis (Review). Cochrane Database Syst. Rev. 5. DOI: 10.1002/14651858.CD000951.pub2.
- Tijhuis, M. J., et al. (2012). State of the art in benefit-risk analysis: Food and nutrition. Food Chem. Toxicol. 50:5–25.
- van Ommen, B., et al. (2009). Challenging homeostasis to define biomarkers for nutrition related health. Mol. Nutr. Food Res. 53(7):795–804.
- van Ommen, B., et al. (2010). The micronutrient genomics project: A community-driven knowledge base for micronutrient research. Genes. Nutr. 5:285–296.
- Verkerk, R. (2010). The paradox of overlapping micronutrient risks and benefits obligates risk/benefit analysis. Toxicology 278(1):27–38.
- Yang, T. L., et al. (2005). A long-term controlled folate feeding study in young women supports the validity of the 1.7 multiplier in the dietary folate equivalency equation. J. Nutr. 135:1139–1145.