
Abstract
Clustered regularly interspaced palindromic repeats (CRISPR) with the associated (Cas) nuclease complexes have democratized genetic engineering through their precision and ease-of-use. We have applied a variation of this technology, known as CRISPR-directed mutagenesis (CDM), to reconstruct genetic profiles within the FLT3 gene of AML patients. We took advantage of the versatility of CDM and built expression vectors that, in combination with a specifically designed donor DNA fragment, recapitulate simple and complex mutations within the FLT3 gene. We generate insertions and point mutations including combinations of these mutations originating from individual patient samples. We then analyze how these complex genetic profiles modulate transformation of Ba/F3 cells. Our results show that FLT3 expression plasmids bearing patient-specific single or multiple mutations recapitulate cellular transformation properties induced by FLT3 ITDs and modify their sensitivity or resistance in response to established AML drugs as a function of these complex mutations.
Keywords:
Introduction
Clustered Regularly Interspersed Palindromic Repeats (CRISPR) and their CRISPR-associated nucleases (Cas) have revolutionized the field of human gene editing Much attention is being devoted appropriately to the potential of CRISPR/Cas as a therapeutic for both diseases caused by inborn errors as well as cancer [Citation1,Citation2]. But, CRISPR has already made a significant impact as a genetic research reagent, new applications for CRISPR/Cas are being presented seemingly on a monthly basis [Citation3–15].
Our laboratory has been studying the mechanism and practical application of CRISPR-directed gene editing in human cells, focusing on the use of CRISPR/Cas in drug discovery protocols. Recently, we discovered a versatile method that enables the creation of expression vectors bearing diverse genetic profiles in a simple reaction that can be used to study more complex genetic diseases. CRISPR-directed mutagenesis (CDM) is a molecular method that allows for the insertion of large fragments of DNA at multiple sites within a plasmid construct to create vectors that can be used to examine the cellular impact of modified protein expression [Citation16]. The versatility of a CRISPR/Cas system in identifying specific sites for fragment insertion within the coding region of eukaryotic genes is an important asset for studying the oncogenic behavior of mutated proteins.
Acute myeloid leukemia (AML) is a disorder of hematopoietic progenitor cells expressed as uncontrolled growth, through mutations in the FLT3, NPM1, CEBPA, RAS, c-KIT genes among others [Citation17–19]. A key gene involved in AML pathogenesis is the fms like tyrosine kinase 3, receptor-type tyrosine-protein kinase (FLT3), located on chromosome 13q12 encoding a class III receptor tyrosine kinase that regulates hematopoiesis. This receptor is activated by the binding of the fms-related tyrosine kinase 3 ligand (FL) to its extracellular domain which induces homodimer formation in the plasma membrane and auto-phosphorylation [Citation20]. The activated receptor kinase phosphorylates multiple cytoplasmic effector molecules in cellular pathways involved in apoptosis, proliferation, and differentiation of hematopoietic cells [Citation20]. Protein kinase activation can be induced by somatic mutations, a common mechanism of tumorigenesis led by the constitutive activation of the receptor resulting in acute myeloid leukemia and acute lymphoblastic leukemia [Citation20–23].
FLT3 is the most common gene mutated in AML [Citation24], most often presenting as internal tandem duplications (ITD) and/or point mutations. FLT3 ITDs range from ten to several hundred bases and are repeated within exon 14, which encodes the juxta-membrane domain (JDM) [Citation25,Citation26]. All duplications are ‘in frame,’ with the number of nucleotides added in multiples of three; thus, the overall reading frame of the protein remains unchanged. Point mutations are found in the tyrosine kinase encoding domain (TKD) in 5–7% of AML patients [Citation27]. FLT3 ITD and TKD mutations induce aberrant activation of FLT3 signaling, leading to proliferation [Citation22].
AML bearing a FLT3 ITD usually presents with high blood blast counts and a normal karyotype but has poor treatment outcomes. It is also penetrant in abnormal karyotype cases. Libura et al. [Citation28] found that 54% of AML, exhibiting either a MLL-PTD or a breakage of the MLL gene at the Topo II (Topoisomerase II) site, carry either an internal tandem (FLT3 ITD) or a variety of point mutations. There is an initial response to treatment, but high relapse rate, short relapse-free survival (RFS) and diminished overall survival often follow [Citation25,Citation29].
Advancement in the treatment of leukemia over the last 40 years has impacted and transformed a uniformly fatal disease into a one that is somewhat manageable. There are, however, several subtypes of pediatric and adult leukemia that evade treatment and continue poor prognosis; many of these involve FLT3 mutations. For example, the FLT3 ITD associated with a single point mutation in the tyrosine kinase domain is known to induce resistance to tyrosine kinase inhibitors (TKI) treatment [Citation30,Citation31].
As with many cancers, the genetic profile of the individual patient impacts the potential of response to a specific drug, including those designed as personalized precision medicine therapy. Because of the versatility and ease of use of CDM, we present here a method for utilizing CRISPR-directed gene editing to construct expression vectors that, in combination with a specifically designed donor DNA fragment, can recapitulate simple and complex mutations residing in FLT3 in a reproducible and robust fashion. We hope to enable the discovery of new therapeutic targets through the screening of anti-leukemic drugs designed to reverse the negative prognosis encountered in infants and adults with leukemia
Methods and materials
CRISPR-directed in vitro gene editing
Cell-free extracts were prepared following the technique outlined by Sansbury et al. [Citation32]. RNP complexes used in in vitro reactions consisted of a purified AsCas12a nuclease (Integrated DNA Technologies, Coralville, IA) and a target-specific crRNA (Integrated DNA Technologies). In vitro DNA cleavage reaction mixtures contained 250 ng of FLT3 plasmid DNA (Gift from Tarlock Lab) and 10 pmol of RNP mixed in a reaction buffer, which was brought to a final volume of 20 µL. Each reaction was incubated for 15 min at 37° C after which DNA was recovered from reaction mixtures and purified using QIAprep Spin Miniprep silica columns (Qiagen, Hilden, Germany). Secondary in vitro recircularization reactions contained DNA recovered from the initial cleavage reaction, 20 µg of cell- free extract supplemented with Quick Ligase (New England Biolabs, Ipswich, MA), and a reaction buffer which was brought to a final volume of 35 µL. Each secondary reaction was incubated for 15 min at 37° C. Double-stranded donor DNA templates (Integrated DNA Technologies), 4.464 µg was added into the secondary reaction mixture. DNA from the secondary in vitro recircularization reactions was recovered from reaction mixtures and purified using silica spin columns. Plasmid DNA recovered from in vitro reactions was transformed into 50 µL of DH5a competent E. coli (Invitrogen, Carlsbad, CA) via heat shock transformation. Competent cells were incubated on ice for 30 min after plasmid introduction, heat shocked for 20 s at 42 °C, placed on ice for 2 min, brought to a final volume of 1 mL in SOC media and incubated for 1 hr at 37 °C, with shaking (225 rpm). Undiluted competent cells were plated on media containing ampicillin antibiotics and incubated overnight at 37 °C. Single ampicillin-resistant colonies were selected, and plasmid DNA was isolated via a QIAprep Spin Mini-prep Kit (Qiagen). Modifications made to the plasmid DNA selected from bacterial colonies were evaluated via DNA sequencing (GeneWiz, South Plainfield, NJ).
Cell lines and transfection
The murine IL-3-dependent pro-B cell line, Ba/F3, was maintained in RPMI 1640 medium (GIBCO, Rockville, MD, USA) supplemented with 10% fetal bovine serum (FBS, Gemini Bio- Products, Calabasas, CA, USA) and 1 ng/ml IL-3. Ba/F3 cells were transfected with plasmid DNA by Nucleofection using the SG Cell Line 4 D-Nucleofector X Kit. BA/F3 cells were transfected with program CM-147 in 20 μl cuvette Strips. Transfected cells were cultured in IL-3-containing medium for 72 hr and then selected in 1 mg/ml Puromycin (GIBCO) for a period of 7 days. The ability of the cells to survive in the absence of IL-3 was determined by trypan blue exclusion.
Cell proliferation of Ba/F3 cells
Cells were seeded at a density of 4 × 104/mL in the presence or absence of IL-3 as indicated. Viable cells were counted at indicated time periods in a standard hemocytometer after staining with trypan blue.
FLT3 inhibitor sensitivity testing
BaF/3 or BaF/3-derived cells were plated in quadruplicate at 1 × 105 cells per well in 96-well plates with or without IL-3 supplement and in the presence of increasing concentrations of Sorafenib (S7397) or Gilteritinib (ASP2215) (purchased from sellecckchem.com) for 72 h. MTS-based assay system (Boehringer Mannheim, Indianapolis, IN, USA) was performed to evaluate the viability of cell in the presence of inhibitors.
Immunofluorescent staining and confocal microscopy imaging
Baf/3 FLT3 expressing cells and un-transfected cells were fixed in 4% formaldehyde for 15 min at room temperature followed by rinsing three times in PBS. For plasma membrane staining 2.0 × 106 cells/mL were incubated with 50 µg/mL of WGA-Alexa 647 (pink) for 10 min at room temperature followed by rinsing two times in PBS. Cells were then permeabilized with 0.3% Triton 100-X for 10 min at room temperature followed by rinsing two times in PBS. Blocking of nonspecific protein-protein interaction was performed in 5% Goat Serum and 0.1% Triton 100-X for 2 hr at room temperature followed by rinsing two times in PBS. To satin the ER cells were then stained in 125 µg/mL of Concanavalin A- Alexa 594 (Red) for 30 min at room temperature followed by rinsing two times in PBS. FLT3 protein was stained by 10 µg/mL of Rabbit anti-FLT3 (ab37847) overnight at 4 °C followed by washing three times in 1% PBS. The secondary antibody was Goat anti-Rabbit IgG(H + L) Alexa Fluor 488 (green) conjugate was used at a 1/1000 dilution for 1 hr. Fixed and stained cells were mounted on to slides for imaging with mountant containing DAPI. The treated cell samples were imaged with ZEISS LSM880 Laser Scanning Confocal Microscope. A 63x oil objective lens and four channel light sources (bright field transmitted light, 405 nm, 488 nm, and 561 nm lasers) were used.
FLT3 expression quantification by flow cytometry
Baf/3 cells expressing FLT3 and un-transfected cells were fixed as described above. Un-permeabilized cells were used to determine the external FLT3 expression. To determine the total FLT3 expression, cells were permeabilized with 0.3% Triton X-100 for 10 min at room temperature followed by two washes with PBS. Cells were then incubated in a blocking solution (5% goat serum, 0.1% Triton x-100 in PBS) for 2 h at room temperature and then washed twice with PBS. Baf/3 cells were stained with rabbit anti-FLT3 primary antibody (abcam ab37847) at a 10 µg/ml concentration overnight at 4 °C and the control cells were stained with the equal concentration of rabbit IgG. Cells were washed three times with PBS and an Alexa Fluor 488 goat anti-rabbit secondary antibody was added to all cells at a 1:1000 dilution for 1 h at room temperature. Cells were washed with PBS and run on the BD FACSAria II flow cytometer to measure FLT3 expression. Samples were analyzed in BD FACSDiva software for the Mean Fluorescence Intensity (MFI). To determine internal FLT3 expression, the MFI from the nonpermeabilized sample was subtracted from permeabilized Baf/3 stained cells.
Results
CDM is initiated with the design of a CRISPR/Cas complex that induces multiple double-strand cleavages within the same or multiple plasmids. The addition of a donor template, either single-stranded or double-stranded with compatible ends, enables large fragments to be inserted at most sites within the expression vector. Importantly, the donor template can be designed and synthesized to contain single or multiple mutations based on genetic profiles where an individual’s genotype has been established. Once the vectors are constructed, the plasmids are transformed into bacteria and isolated as individual colonies, with subsequent genetic and molecular readout through DNA sequencing (). This technique has already been used successfully to a library of exons bearing single polymorphisms into genes such as KRAS, EGFR and PIK3 among others [Citation16]. Subsequent expression of these constructs led to a visualization of the contribution of this mutated protein in the development of tumorigenesis and assisted in the prognosis of the treatment regimen.
Figure 1. In vitro gene editing experimental approach and confirmation. (A) Cas12a RNP is complexed and added to the first in vitro cleavage reaction mixture with plasmid DNA. Plasmid DNA is recovered and added to a second in vitro re-circularizing reaction mixture with cell-free extract. After the reaction is complete, plasmid DNA is recovered from the reaction and transformed into competent E. coli. DNA is then isolated from transformed cells and sequenced to identify modifications made in vitro. Modified from Sansbury et al. [Citation16] (B) FLT3-ITD sequences, designated Patient 4 37 bp ITD (P4) and the Patient 5 76 bp ITD (P5) respectively were previously identified in pediatric AML patients [Citation53] (C) FLT3 expression plasmid template was used in the in vitro gene editing reaction to produce FLT3 mutant plasmids that express FLT3 ITD mutations, ITDs, point mutations or different combination of both. (D) FLT3 mutation molecular description. (E) 1x106 cells that showed IL-3 depletion survival were cultured for 9 days in the presence of puromycin to further confirm the FLT3 cassette stable integration. Top graph was used as control to show the sensitivity of un-transfected Baf/3 cells to puromycin in the presence or absence of IL-3.
![Figure 1. In vitro gene editing experimental approach and confirmation. (A) Cas12a RNP is complexed and added to the first in vitro cleavage reaction mixture with plasmid DNA. Plasmid DNA is recovered and added to a second in vitro re-circularizing reaction mixture with cell-free extract. After the reaction is complete, plasmid DNA is recovered from the reaction and transformed into competent E. coli. DNA is then isolated from transformed cells and sequenced to identify modifications made in vitro. Modified from Sansbury et al. [Citation16] (B) FLT3-ITD sequences, designated Patient 4 37 bp ITD (P4) and the Patient 5 76 bp ITD (P5) respectively were previously identified in pediatric AML patients [Citation53] (C) FLT3 expression plasmid template was used in the in vitro gene editing reaction to produce FLT3 mutant plasmids that express FLT3 ITD mutations, ITDs, point mutations or different combination of both. (D) FLT3 mutation molecular description. (E) 1x106 cells that showed IL-3 depletion survival were cultured for 9 days in the presence of puromycin to further confirm the FLT3 cassette stable integration. Top graph was used as control to show the sensitivity of un-transfected Baf/3 cells to puromycin in the presence or absence of IL-3.](/cms/asset/9c442d72-ea6e-466f-b494-7862ec1cfa65/ilal_a_1805740_f0001_c.jpg)
In this work, CDM was employed on plasmid pMX-WT FLT3 to insert unique FLT3 ITD sequences from two different patient profiles designated as Patient 4 (Ptn4) and Patient 5 (Ptn5). Patient 4 bears a 37-base pair ITD and Patient 5 harbors a 76-base pair ITD () creating two new plasmids known as pMX-Ptn4 and pMX-Ptn5 respectively. The CDM replacement reactions were initiated by double-strand cleavage by CRISPR/Cas12a, a gene editing complex that leaves staggered ends after cutting. The excised 108 base pair fragment in Ptn4 is replaced by a 145 base pair donor DNA fragment which contains the 37-base pair ITD. In the case of Ptn5, a 184 base pair donor DNA fragment containing the 76-base pair ITD was inserted in a similar fashion (, Supplemental Figure 1). A second round of CDM was conducted to insert a fragment from exon 20 that bears a well-established D835Y mutation in the TKD2 domain to create expression vectors pMX-D835Y/Ptn 4 and pMX-D835Y/Ptn5. The ITDs are generated from exon 14 of the human FLT3 gene (). The modified plasmids were transformed into E. coli and individual colonies selected for isolation and confirmation by DNA sequence analysis. Within a 48-hour period, four new plasmid constructs had been created: pMX -Ptn4, pMX-Ptn5, pMX-D835Y/Ptn4 and pMX-D835Y/Ptn5. All variant plasmids were found on the first round of screening of the bacterial readout plates under ampicillin selection and the donor fragment was inserted with a high level of precision and without secondary mutations anywhere within the inserted sequence. Plasmid pMX-WT bearing the WT FLT3 was also propagated under the same conditions.
Each expression vector was transformed into Ba/F3 cells by nucleofection and the cells allowed to grow under Puromycin selection. The cell lines bearing puromycin selection were isolated after day nine and propagated (). After this time, the plasmid has successfully been integrated into the genome of the Ba/F3 cells creating a series of stable cell lines that can be used for further experimentation.
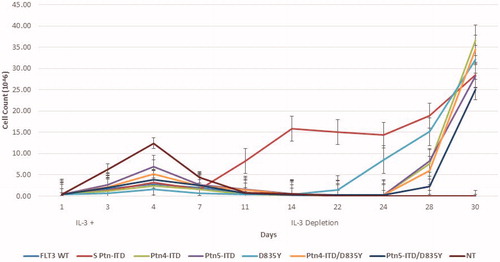
The murine pro-B cell Ba/F3 cell line is dependent on IL-3 for growth and in the absence of IL-3 the cells undergo cell cycle arrest and apoptosis [Citation33]. When transformation of mutated FLT3 expression vectors is achieved, the cells lose their IL-3 dependence [Citation34]; IL3-independent growth is a good marker for cell transformation. All five plasmid constructs described above, and a new plasmid pMX-SPtn, [bearing a 24 base pair ITD, a kind gift form Dr. Katherine G. Turlock (Department of Hematology, Seattle Children’s Hospital)] to serve as a positive control of transformation, were introduced into Ba/F3 cells in the presence of IL-3. After three days of growth, IL-3 was removed and cell growth was measured, after depletion, for a period of 30 days. As predicted, Ba/F3 bearing the WT FLT3 did not survive depletion but all other cell lines, including the positive control, were exhibited robust growth after 28–30 day (). All transformed cell lines continued to grow in a robust fashion even after freeze back and re-initiation of growth in cell culture (data not shown). These data confirm that CDM-generated cell gene expression constructs can be used to create modified cell lines that exhibit a transformation growth phenotype.
Figure 2. Baf/3 cell transformation by FLT3 Expression. 4 × 105 Baf/3 cells were transfected with 2ug of plasmid DNA by Nucleofection using the 4 D-Nucleofector SG Cell Line X Kit and CM-147 program. Cells were incubated for 72 h post transfection in IL-3 supplemented media. At this time 2 x105 cells were plated into a 6-well plate containing IL-3 supplemented media. At 4 days cells were counted and plated into media lacking IL-3 to select for transformed cells. Cell count was performed by trypan blue exclusion.
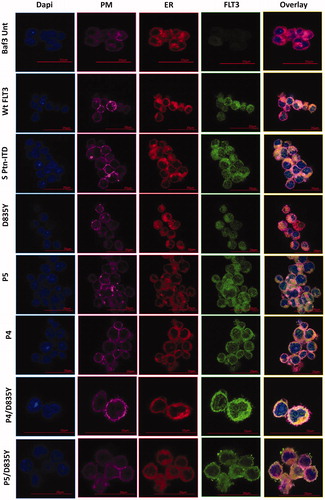
We continued to analyze the nature of the transformed cell by investigating the subcellular localization of the mutated proteins. Subcellular compartmentalization takes place for FLT3 in the plasma membrane and endoplasmic reticulum. To distinguish the cellular compartments, the plasma membrane was identified through staining with WGA-Alexa 647 which is evidenced by a pink like color, while the endoplasmic reticulum was stained by ConA-Alexa 594 which is evidenced by a distinct reddish color. In addition, we utilized anti-FLT3 antibody (FLT3-488, green) to identify the position of this protein (). Untransfected Baf/3 cells were used as controls and as predicted, no FLT3 is expressed in these cells. Baf/3 cells transformed with a plasmid expressing wild type FLT3 reveal its presence in the plasma membrane, reflected most obviously in the overlay column. The signal in Ptn4 and Ptn5 was localized in both the endoplasmic reticulum and the plasma membrane as expected when ITDs are incorporated into the FLT3 gene. Cells transformed with the plasmid bearing the D835Y mutation reveal FLT3 localization to the endoplasmic reticulum and the plasma membrane, while in contrast, cells transformed with a plasmid bearing Ptn4/D835Y are found primarily in the endoplasmic reticulum. FLT3 protein generated from Ptn5/D835Y localizes in both the endoplasmic reticulum and the plasma membrane. The overlay column provides the highest degree of clarity of subcellular localization. Taken together, these results confirm that the expression plasmid library generated by CDM exhibits functional activity faithfully recapitulating the phenotype of FLT3 proteins emanating from FLT3 gene rearrangements [Citation35–39]. The sub-localization of FLT3 was further tested by flow cytometry providing the MFI (Mean Fluorescent Intensity) ratio which quantifies the expression of the different FLT3 variants (Supplemental Figure 3).
Figure 3. Localization of FLT3. Immunofluorescence staining of Baf/3 FLT3 expressing cells and un-transfected cells. Scale bars represent 20 µm and 15 µm.
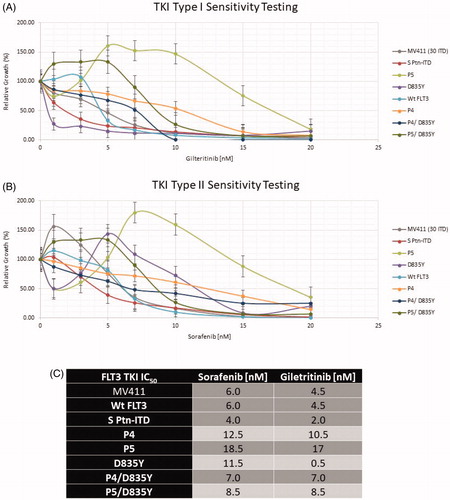
Next, we utilized these newly constructed plasmid expression vectors to determine known drug sensitivity response of transformed cells expressing our individual FLT3 alterations. We utilized first generation and second-generation Tyrosine Kinase inhibitors, Sorafenib and Gilteritinib, which both target FLT3. As shown in , the cells were grown in the presence of the indicated level of each drug (nM). Based on Trypan Blue exclusion, viable cells were seeded in quadruplicate in a 96-well microtiter plate in increasing concentrations of inhibitor for 72 h. Following such treatment, the cells were incubated for four hours in MTS/PMS solution to determine relative cell growth. The results indicate a wide variety of responses but clearly, cells transformed with the FLT3-ITD of patient 5 exhibit an enhanced level of resistance to both drugs. Proliferation of the other cell lines is similar in response to either drug, with the notable exception of the D835Y gene construct which appears to confer significantly greater sensitivity to Gilteritinib. Utilizing data generated from quadruple repeats of the experiment, we can calculate a FLT3 TKI IC 50 and that is presented in . Consistent with the above data, the IC 50 of D835Y reflects an enhanced level of sensitivity to Gilteritinib. These data reflect the known sensitivities of cells bearing FLT3-ITDs in response to these two TKI inhibitors [Citation17,Citation20,Citation40–42].
Figure 4. FLT3 mutation drug sensitivity assay. (A) Gilteritinib and (B) Sorafenib. Relative cell growth of Ba/f3 cells expressing the indicated FLT3 mutants after 72 hr treatment with increasing concentrations of Sorafenib or Gilteritinib. Errors bars represent the Standard Error (n = 4). (C) FLT3 TKI IC50.
Discussion
The complexity of genetic profiles within populations and among individual patients, continues to impact the development of new drugs. While precision and/or personalized medicine have extraordinary potential, it has become clear that the genetic diversity of patients may influence strongly chances for positive response. It is therefore incumbent on researchers to improve methodologies that can quickly assemble testing systems with expandable capabilities to deliver genetically modified genes in a robust and reproducible fashion.
Here, we utilize CDM, a methodology based on an in vitro gene editing system, to generate expression vectors that successfully recapitulate the phenotypic influence of patient based ITDs in the FLT3 gene. The key reaction components of CDM are ribonucleoprotein particles (CRISPR-Cas12a), double-stranded plasmid DNA templates, double-stranded donor DNA fragments and a mammalian cell-free extract. A significant advantage of CDM is that it does not require multiple rounds of PCR which can become problematic as it is sometimes laden with inborn errors or negatively affected by repetitive DNA sequences. This new method improves and expands upon the current strategies for site-directed mutagenesis because it allows for the creation of multiple modified plasmid templates from a single reaction mixture without the need for PCR and with a high degree of precision.
Inhibition of mutated FLT3 kinase activity by pharmacologic agents is an important therapeutic strategy for AML [Citation24]. Most patients with AML and FLT3-internal tandem duplications (ITD) initially show favorable responses to FLT3 inhibitors but the development of resistance emerges over time. One of the most common mechanisms of resistance is based on the acquisition of mutations in the secondary FLT3 tyrosine kinase domain (TKD2) [Citation30]. Therefore, the mutational profile of each patient must be revealed so the most appropriate FLT3 TKI can be selected since the FLT3 point mutations appear to arise frequently during FLT3 TKI therapy. This knowledge is also important during treatment of a patient with a FLT3 ITD mutation because the FLT3 point mutations appear to arise frequently during FLT3 TKI therapy. For example, twelve somatic mutations have been identified at distinct positions within the juxta membrane domain (JMD), the region involved in apoptosis, proliferation and differentiation regulation. These lead to auto phosphorylation of the receptor without the presence of FLT3 ligand (FL) binding [Citation43]. Another parameter influencing FLT3 activation is the length of FLT3 ITD, which causes structural alterations and altered responses to therapy [Citation44]. In the present study, we focused on developing a relevant and novel technology that can enable the discovery of new therapeutic targets through the screening of anti-leukemic drugs designed to reverse the negative prognosis encountered in infants and adults with leukemia. Our results show that by adding CDM to the AML drug discovery toolbox, FLT3 expression plasmids that bear patient-specific single or multiple mutations can be created with greater versatility and a robust fashion. As we expand its capability, CDM may ultimately be used for the assembly and modification of large constructs containing 100 kilobases where individualized amplification reactions of the template backbone are simply not feasible. CDM could be used on artificial chromosomes, including BACs, avoiding the use of the more laborious process of recombineering [Citation45–47]. If this application is reduced to practice, then wider spans of an individual genotype can be programed into drug discovery screens. It is important to note that absolute concentrations indicating drug sensitivity can be affected by the methodology, the workflow, the protocol, and the time at which cell viability is measured [Citation48–50]. In this manuscript, our goal was to provide a foundational system that can be used to establish a CRISPR-based screening system with the discovery of new drugs and not to identify specifically effective concentrations.
We have previously shown that the rate of off-site mutations is significantly lower than standard site directed mutagenesis protocols that rely heavily on multiple rounds of PCR [Citation16,Citation51,Citation52]. Multiple rounds of PCR are often known as the Achilles’ heel of site directed mutagenesis since different degrees of false priming which are usually remain hidden and cannot be seen by the operator prior to the final product being analyzed. This step is an Achilles heel since it can cause various degrees of false priming often unknown to the operator, undesirable mutation insertion and the inability to produce complex DNA mutations. CDM overcomes many of these limitations. For studies of AML, the capacity to catalyze insertion of a synthetic fragment bearing multiple mutations is a significant advantage. Using this approach as a foundation, we will be able to generate deletions, insertions and point mutations within the same reaction including combinations of these mutations, again, important for the construction of the complex genetic profiles often presented by individual patient samples.
GLAL-2020-0862-File005.docx
Download MS Word (13.9 MB)Acknowledgments
We are grateful to members of the Kmiec laboratory for their thoughtful input and comments throughout the course of these experiments. We thank Amanda Hewes for assistance with the CRISPR-directed mutagenesis technique. We thank Dr. Sylvain Le Marchand in the Bio-Imaging Center for assistance with all the confocal microscopy imaging.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826.
- Cho SW, Kim S, Kim JM, et al. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–232.
- Lee J, Bayarsaikhan D, Bayarsaikhan G, et al. Recent advances in genome editing of stem cells for drug discovery and therapeutic application. Pharmacol Ther. 2020;209:107501.
- Shi J, Wang E, Milazzo JP, et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–667.
- Yin H, Xue W, Anderson DG. CRISPR-Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16(5):281–295.
- Quijada-Álamo M, Hernández-Sánchez M, Alonso-Pérez V, et al. CRISPR/Cas9-generated models uncover therapeutic vulnerabilities of del(11q) CLL cells to dual BCR and PARP inhibition. Leukemia. 2020;34(6):1599–1612.
- Jiang C, Meng L, Yang B, et al. Application of CRISPR/Cas9 gene editing technique in the study of cancer treatment. Clin Genet. 2020;97(1):73–88.
- Stewart J, Banerjee S, Pettitt SJ, et al. Modelling the cancer phenotype in the era of CRISPR-Cas9 gene editing. Clin Oncol (R Coll Radiol). 2020;32(2):69–74.
- Ng SR, Rideout WM, Akama-Garren EH, et al. CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proc Natl Acad Sci Usa. 2020;117(1):513–521.
- Mollanoori H, Rahmati Y, Hassani B, et al. Promising therapeutic approaches using CRISPR/Cas9 genome editing technology in the treatment of Duchenne muscular dystrophy. Genes Dis. 2020. DOI:10.1016/j.gendis.2019.12.007
- Dabrowska M, Ciolak A, Kozlowska E, et al. Generation of new isogenic models of Huntington’s disease using CRISPR-Cas9 technology. IJMS. 2020;21(5):1854.
- Komáromy AM. CRISPR-Cas9 disruption of aquaporin 1: an alternative to glaucoma eye drop therapy? Mol Ther. 2020;28(3):706–708.
- Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (80-). 2020;367(6481):eaba7365.
- Couzin-Frankel J. CRISPR takes on cancer. Vol. 367. Washington, DC: American Association for the Advancement of Science; 2020. p. 616.
- Geurts MH, de Poel E, Amatngalim GD, et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell. 2020;26(4):503–510.e7.
- Sansbury BM, Wagner AM, Tarcic G, et al. CRISPR-directed gene editing catalyzes precise gene segment replacement in vitro enabling a novel method for multiplex site-directed mutagenesis. Crispr J. 2019;2(2):121–132.
- Meshinchi S, Arceci RJ. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist. 2007;12(3):341–355.
- Lee BH, Tothova Z, Levine RL, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12(4):367–380.
- Sakamoto KM, Grant S, Saleiro D, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia [Internet]. Vol. 114, Molecular Genetics and Metabolism. 2015. Mol Genet Metab. 2015;114(3):397–402.
- Gary Gilliland D, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542.
- Cauchy P, James SR, Zacarias-Cabeza J, et al. Chronic FLT3-ITD signaling in acute myeloid leukemia is connected to a specific chromatin signature. Cell Rep. 2015;12(5):821–836.
- Leung AYHH, Man C-HH, Kwong Y-LL. FLT3 inhibition: a moving and evolving target in acute myeloid leukaemia. Leukemia. 2013;27:260–268.
- Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3(9):650–665.
- Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300.
- Fathi AT, Chen Y-B. Treatment of FLT3-ITD acute myeloid leukemia. Am J Blood Res. 2011;1(2):175–189.
- Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United King. Blood. 2001;98(6):1752–1759.
- Small D. FLT3 mutations: biology and treatment. Hematology. 2006;2006(1):178–184.
- Libura M, Asnafi V, Tu A, et al. FLT3 and MLL intragenic abnormalities in AML reflect a common category of genotoxic stress. Blood. 2003;102(6):2198–2204.
- de Rooij J, Zwaan C, van den Heuvel-Eibrink M, et al. Pediatric AML: from biology to clinical management. J Clin Med. 2015;4(1):127–149.
- Baker SD, Zimmerman EI, Wang Y-D, et al. Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-positive acute myeloid leukemia. Clin Cancer Res. 2013;19(20):5758–5768.
- Smith CC, Wang Q, Chin C-S, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263.
- Sansbury BM, Wagner AM, Nitzan E, et al. CRISPR-directed in vitro gene editing of plasmid dna catalyzed by Cpf1 (Cas12a) nuclease and a mammalian cell-free extract. Crispr J. 2018;1(2):191–202.
- Warmuth M, Kim S, Gu X, et al. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19(1):55–60.
- Pradhan A, Lambert QT, Reuther GW, et al. Transformation of hematopoietic cells and activation of JAK2-V617F by IL-27R, a component of a heterodimeric type I cytokine receptor. Proc Natl Acad Sci USA. 2007;104(47):18502–18507.
- Chan PM. Differential signaling of Flt3 activating mutations in acute myeloid leukemia: a working model. Protein Cell. 2011;2(2):108–115.
- Reiter K, Polzer H, Krupka C, et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia. 2018;32(2):313–322.
- Moloney JN, Stanicka J, Cotter TG. Subcellular localization of the FLT3-ITD oncogene plays a significant role in the production of NOX- and p22phox-derived reactive oxygen species in acute myeloid leukemia. Leuk Res. 2017;52:34–42.
- Schmidt-Arras D, Böhmer S-A, Koch S, et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood. 2009;113(15):3568–3576.
- Koch S, Jacobi A, Ryser M, et al. Abnormal localization and accumulation of FLT3-ITD, a mutant receptor tyrosine kinase involved in leukemogenesis. Cells Tissues Organs. 2008;188(1-2):225–235.
- Alvarado Y, Kantarjian HM, Ravandi F, et al. FLT3 inhibitor treatment in FLT3-mutated AML is associated with development of secondary FLT3-TKD mutations. Blood. 2011;118(21):1493–1493.
- Schwartz GW, Manning B, Zhou Y, et al. Classes of ITD predict outcomes in AML patients treated with FLT3 inhibitors. Clin Cancer Res. 2019;25(2):572–583.
- Hassanein M, Almahayni MH, Ahmed SO, et al. FLT3 inhibitors for treating acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2016;16(10):543–549.
- Tarlock K, Hansen ME, Hylkema T, et al. Discovery and functional validation of novel pediatric specific flt3 activating mutations in acute myeloid leukemia: results from the COG/NCI target initiative. Blood. 2015;126(23):87–87.
- Meshinchi S, Stirewalt DL, Alonzo TA, et al. Structural and numerical variation of FLT3/ITD in pediatric AML. Blood. 2008;111(10):4930–4933.
- Yamaguchi S, Niwa R, Kazuki Y, et al. Application of a bacterial artificial chromosome modification system for a human artificial chromosome vector. Yonago Acta Med. 2011;54(1):21–31.
- Guo JC, Tang YD, Zhao K, et al. Highly efficient CRISPR/Cas9-mediated homologous recombination promotes the rapid generation of bacterial artificial chromosomes of pseudorabies virus. Front Microbiol. 2016. DOI:10.3389/fmicb.2016.02110
- Narayanan K, Chen Q. Review article bacterial artificial chromosome mutagenesis using recombineering. J Biomed Biotechnol. 2011;2011:971296.
- Kancha RK, Grundler R, Peschel C, et al. Sensitivity toward sorafenib and sunitinib varies between different activating and drug-resistant FLT3-ITD mutations. Exp Hematol. 2007;35(10):1522–1526.
- Auclair D, Miller D, Yatsula V, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia. 2007;21(3):439–445.
- Lierman E, Lahortiga I, Van Miegroet H, et al. The ability of sorafenib to inhibit oncogenic PDGFRβ and FLT3 mutants and overcome resistance to other small molecule inhibitors. Haematologica. 2007;92(1):27–34.
- Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9(1):61.
- Tseng W-CC, Lin J-WW, Wei T-YY, et al. A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal Biochem. 2008;375(2):376–378.
- Crowgey EL, Kolb A, Wu CH. Development of bioinformatics pipeline for analyzing clinical pediatric NGS data. AMIA Jt Summits Transl Sci Proc. 2015;2015:207–211.