
Abstract
Nanoparticle-based drug delivery system offers a promising platform for combination cancer therapy. However, the inefficient drug release in cells reduces the therapeutic efficacy of cancer nanomedicines. Herein, a PEGylated poly(α-lipoic acid) copolymer (mPEG-PαLA) was prepared and used as a reduction/pH dual responsive nanocarrier to simultaneously deliver paclitaxel (PTX) and doxorubicin (DOX) for osteosarcoma therapy. The amphiphilic mPEG-PαLA could efficiently encapsulate both PTX and DOX during its self-assembly into micelles in aqueous solution to generate PTX and DOX co-loaded nanoparticles (NP-PTX-DOX). The as-prepared NP-PTX-DOX showed enhanced PTX and DOX release in response to reductive and acidic stimuli. Moreover, the dual-drug loaded nanoparticles were efficiently internalized by K7 osteosarcoma cells and released drugs intracellularly, as confirmed by flow cytometry analysis and confocal laser scanning microscopy. Consequently, NP-PTX-DOX exhibited synergistic therapeutic effects and induced enhanced cell apoptosis in K7 cells. Furthermore, NP-PTX-DOX presented improved biodistribution and higher tumor growth inhibition efficacy compared to the control groups in a murine osteosarcoma model. Altogether, the results of this work indicate that the proposed strategy is promising for osteosarcoma therapy using mPEG-PαLA copolymer as a dual-responsive nanocarrier to co-deliver anticancer drugs.
1. Introduction
Osteosarcoma is one of the most common malignant bone cancers in adolescents with a very high mortality rate (Ji et al., Citation2015; Anderson, Citation2016; Heymann et al., Citation2016; Jackson et al., Citation2016; Harrison et al., Citation2018). The standard treatment methods for osteosarcoma include preoperative chemotherapy, surgery, and postoperative adjuvant chemotherapy. The most widely used chemotherapy approach for osteosarcoma treatment involves using a single drug. Although single-drug chemotherapy has a favorable therapeutic effect and improved survival rate, it also has the limitations of poor targeting ability, limited anticancer effects, severe systemic toxicity, and possible development of drug resistance (Giantonio et al., Citation2007; Leader et al., Citation2008; Park et al., Citation2012). The combination of two or more anticancer drugs with different antitumor mechanisms can enhance the therapeutic effects by the synergy between the different drugs, reduce the adverse effects, and prevent the occurrence of drug resistance. Therefore, it is necessary to develop novel combination therapies to improve clinical osteosarcoma treatment (Fu et al., Citation2014; Jiang et al., Citation2014; He et al., Citation2016; Chiper et al., Citation2018).
Recently, nanoparticle-based drug delivery systems are widely used to improve the selectivity, reduce the side effects, and enhance the anticancer effects of chemotherapeutic drugs by enhanced permeability and retention (EPR) effect (Landesman-Milo et al., Citation2015; Kamaly et al., Citation2016; Lu et al., Citation2018; Samanta et al., Citation2019; Wang et al., Citation2019). Importantly, nanoparticles can concurrently encapsulate two or more kinds of chemotherapeutics for combination cancer therapy (Torchilin, Citation2011; Eldar-Boock et al., Citation2013; Ramasamy et al., Citation2015; Fan et al., Citation2017; Mo et al., Citation2017). However, inefficient drug release may significantly reduce the anticancer efficiency of the drug-loaded nanoparticles after being internalized by cancer cells (Zhang et al., Citation2014; Li et al., Citation2018; Yang et al., Citation2018; Xiaoya et al., Citation2019). Therefore, there is an urgent need to develop smart intracellular stimuli-responsive nanocarriers. The acidic endolysosomes (pH 5.0–6.0) and the reductive cytosol (containing 2–10 mM GSH) are ideal intracellular stimuli for the development of stimuli-responsive nanomedicines due to their significant differences from the extracellular environment (pH 7.4 and 2–20 μM GSH; Hoffman, Citation2013; Huang et al., Citation2013; Yamawaki et al., Citation2016; Hu et al., Citation2017). Therefore, intracellular reduction/pH-responsive nanocarriers can protect the loaded drugs from leaking in the extracellular environment and release the drugs only in the cells (Chakravarthi & Bulleid, Citation2004; Cheng et al., Citation2011; Wu et al., Citation2014; Chen et al., Citation2015; Xu et al., Citation2015; Chen et al., Citation2016; Gao et al., Citation2017; Hao et al., Citation2018).
DOX is a cationic small-molecule drug that intercalates into nuclear DNA and thus induces apoptosis. PTX induces cell apoptosis by interfering with the normal functions of microtubules during cell mitosis (Wu et al., Citation2017; Zhao et al., Citation2017; Feng et al., Citation2019; Yang et al., Citation2019). The synergistic effects of DOX and PTX have been investigated in the treatment of breast cancer, lung cancer, melanoma, and hepatic carcinoma, however, their synergistic therapeutic efficacy against osteosarcoma has been poorly understood (Jin et al., Citation2010; Wang et al., Citation2011, Citation2016; Zhao et al., Citation2011, Citation2017; Liu et al., Citation2014; Baabur-Cohen et al., Citation2017; Qiu et al., Citation2017; Jiang et al., Citation2020).
In this study, a PEGylated poly(α-lipoic acid) copolymer (mPEG-PαLA) was prepared and applied as a reduction/pH dual responsive nanocarrier to concurrently deliver doxorubicin (DOX) and paclitaxel (PTX) for osteosarcoma therapy (Scheme 1). mPEG-PαLA could simultaneously encapsulate cationic DOX and PTX by electrostatic and hydrophobic interactions to generate PTX and DOX co-loaded nanoparticles (NP-PTX-DOX). After cellular internalization, the protonation of the carboxyl groups in mPEG-PαLA and primary amine in DOX in acidic intracellular microenvironment reduced the electrostatic and hydrophobic interactions between mPEG-PαLA and the loaded drugs. Moreover, the degradation of disulfide-containing PαLA backbone in the reductive cytosolic environment resulted in the dissociation of nanoparticles. Both these effects mediated the reduction/pH dual responsive release of DOX and PTX in the cells (Scheme 1). The results demonstrated that NP-PTX-DOX exhibited synergistic anticancer effects in vitro and presented improved biodistribution and increased antitumor efficacy in a murine K7 osteosarcoma model.
Scheme 1. Schematic illustration of the synthesis of mPEG-PαLA, the preparation and in vivo delivery of NP-PTX-DOX nanoparticles and the reduction/pH dual responsive intracellular release of PTX and DOX for combination cancer therapy.
2. Materials and methods
2.1. Materials
α-Lipoic acid (αLA) and methoxy poly(ethylene glycol) (mPEG, Mn = 2000 g mol−1) were purchased from Changchun Third Party Pharmaceutical Technology Co., Ltd. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), 4-dimethylaminopyridine (DMAP), 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and dihydrochloride (DAPI) were purchased from Sigma-Aldrich LLC. (Shanghai, China). Doxorubicin hydrochloride (DOX·HCl) and PTX were obtained from Dalian Meilun Biological Technology Co., Ltd. Dulbecco’s modified Eagle’s medium (DMEM, Gibco) and fetal bovine serum (FBS, Gibco) were obtained from Thermo Fisher Scientific (Shanghai, China).
2.2. Characterization
1H and 13 C NMR spectra were recorded on a Bruker AV-300 NMR spectrometer. Dynamic light scattering (DLS) measurements were performed on a Wyatt QELS instrument with a vertically polarized He–Ne laser (Wyatt Technology Co., Santa Barbara, CA, DAWN EOS). Transmission electron microscopy (TEM) measurements were performed on a JEOL JEM-1011 transmission electron microscope (Tokyo, Japan) with an accelerating voltage of 100 kV. Fluorescence intensity was measured on a Fluorescence Master System (Photon Technology International, Birmingham, NJ).
2.3. Synthesis of PαLA
α-Lipoic acid (2.0632 g) was added to a flask under a nitrogen atmosphere and then stirred at 98 °C for 2 h. The crude product was dissolved in 100 mL of THF and then was slowly poured into a beaker containing 500 mL of cold diethyl ether to obtain a viscous solid. PαLA was then obtained after filtration and vacuum drying (yield: 76.4%).
2.4. Synthesis of mPEG-PαLA
mPEG (2 g) dissolved in a mixed solution of THF (100 mL) and DMSO (100 mL) was added to a solution of PαLA (0.824 g, 1 mmol) in 100 mL of THF. Then, EDC·HCl (381 mg) and DMAP (62.03 mg) were added to the solution and the mixture was stirred at room temperature (RT) for 24 h. Subsequently, EDC·HCl (381 mg) and DMAP (62.03 mg) were added again and the mixture was stirred at RT for another 24 h. Then, the mixture was dialyzed against deionized (DI) water for 4 d (MWCO = 3500 Da). After lyophilization, mPEG-PαLA was obtained as a white powder (Mn = 51.3 kDa, PDI = 1.45, determined by GPC, yield: 64.3%).
2.5. Preparation of mPEG-PαLA nanoparticles (NP) and drug-loaded nanoparticles
To prepare mPEG-PαLA nanoparticles (NP), 100 mg of mPEG-PαLA was dissolved in 6 mL of DMF and was added dropwise to 60 mL of rapidly stirred water. After 90 min, the solution was transferred to a dialysis bag (MWCO = 3500 Da) and dialyzed against DI water for 12 h to remove DMF. NP was obtained after lyophilization.
Drug-loaded nanoparticles were prepared by a similar method. Briefly, 100 mg of mPEG-PαLA and corresponding drugs (20 mg of DOX·HCl was used for NP-DOX; 10 mg of PTX was used for NP-PTX; 20 mg of DOX·HCl and 10 mg of PTX were used for NP-PTX-DOX) were dissolved in 6 mL of DMF and added dropwise to 60 mL of rapidly stirred DI water in the dark. After 90 min, the solution was transferred to a dialysis bag (MWCO = 3500 Da) and dialyzed against DI water for 12 h. NP-DOX, NP-PTX or NP-PTX-DOX was obtained after lyophilization. The drug loading content (DLC) and drug loading efficiency (DLE) of DOX and PTX were determined by fluorescence spectrophotometry and calculated as follows: DLC (%) = (weight of loaded drug/weight of drug-loaded nanoparticles) × 100%; DLE (%) = (weight of loaded drug/weight of feeding drug) ×100%.
2.6. In vitro drug release
NP-PTX-DOX solution (2 mL) was added to a dialysis bag (MWCO = 3500 Da) and incubated with 80 mL of release media under continuous shaking at 37 °C in dark. The release media for DOX were (i) phosphate-buffered saline (PBS) at pH 7.4, (ii) PBS at pH 5.5, and (iii) PBS at pH 7.4 containing 5 mM GSH. The release media for PTX were (i) PBS at pH 7.4 containing 1% Tween 80, (ii) PBS at pH 5.5 containing 1% Tween 80, and (iii) PBS at pH 7.4 containing 1% Tween 80 and 5 mM GSH. Release medium (2 mL) was taken out and replaced with fresh medium at preselected time points. The release of DOX and PTX was measured by fluorescence measurements (480 nm excitation, 592 nm emission) and HPLC, respectively.
2.7. Cellular internalization
K7 murine osteosarcoma cells were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China). For flow cytometry analysis, K7 cells were seeded in a 6-well plate at a density of 3.0 × 105 cells per well in 1.8 mL of DMEM containing 10% FBS and cultured for 24 h at 37 °C in 5% CO2 atmosphere. Then, free DOX, NP-DOX, and NP-PTX-DOX (final concentration of DOX: 5.0 μg/mL) dissolved in 200 μL of PBS were added and incubated for another 2 or 6 h. Subsequently, the original medium was removed and the cells were washed with PBS for 3 min four times. The cells were then detached with trypsin and collected. Finally, the cells were resuspended in 0.5 mL of PBS and were measured by a Guava EasyCyteTM 12 Flow Cytometer.
For confocal laser scanning microscopy (CLSM) experiment, K7 cells were seeded on coverslips in a 6-well plate at a density of 1 × 105 cells per well in 1.8 mL of DMEM containing 10% FBS at 37 °C in 5% CO2 atmosphere. After 24 h, free DOX, NP-DOX, and NP-PTX-DOX (final concentration of DOX: 5.0 μg mL−1) dissolved in 200 μL of PBS were added and the cells were incubated for another 3 or 6 h. Then, the original medium was removed and the cells were washed with PBS for 3 min four times. Subsequently, the cells were fixed with 4% formaldehyde for 20 minutes. The nuclei of the cells were stained with DAPI, and the cells were observed under a confocal microscope (Carl Zeiss LSM 700).
2.8. In vitro cytotoxicity analysis
K7 cells were seeded into 96-well plates at a density of 7500 cells per well in 180 μL of DMEM containing 10% FBS and cultured for 24 h at 37 °C in 5% CO2 atmosphere. Then, free DOX, free PTX, NP-DOX, NP-PTX, and NP-PTX-DOX dissolved in 20 μL of PBS or DMSO were added and the cells were incubated for another 48 or 72 h. The cells were then subjected to MTT assay. The optical absorbance at 490 nm was determined by a microplate reader (Tecan Spark). The cell viability (%) was calculated as follows: (Asample/Acontrol) × 100%. The synergistic effect of PTX and DOX was evaluated by the Chou-Talalay method (Chou, Citation2006): CI = D1/Dm1 + D2/Dm2, where Dm1 and Dm2 represent the IC50 values of drug 1 alone and drug 2 alone, respectively; D1 and D2 represent the concentrations of drug 1 and drug 2 in the combination system at the IC50 value. CI >1 represents antagonism, CI = 1 represents additive and CI < 1 represents synergism.
2.9. Biodistribution
Female Balb/C mice (6–8 weeks) were purchased from the Laboratory Animal Center of Jilin University. All animal experiments were reviewed and approved by the Animal Care and Use Committee of Jilin University. For the biodistribution study, the mice were subcutaneously injected with 1 × 106 K7 cells in the right flank. When the tumor volume reached 100 mm3, the mice were divided into two groups (n = 3). Each group received an intravenous injection of free DOX or NP-PTX-DOX at an equivalent dosage of DOX (2 mg kg−1). After 6 h, the mice were sacrificed. Tumors and major organs including heart, liver, spleen, lung, and kidney were collected and washed with PBS, then subjected to fluorescence imaging using a Maestro In Vivo Imaging System (Cambridge Research & Instrumentation, Inc., Cambridge, MA).
2.10. In vivo anti-tumor analysis
Female Balb/C mice were subcutaneously injected with 1 × 106 K7 cells in the right flank. When the tumor volume reached about 90 mm3, the mice were randomly divided into six groups (n = 5). Each group received an intravenous injection of PBS, free DOX, free PTX, NP-DOX, NP-PTX, and NP-PTX-DOX, respectively, with an equivalent dosage of DOX (2 mg kg−1) or PTX (4 mg kg−1), on day 0, 3, 6, and 9. Tumor volumes and body weights were measured every other day. Tumor volume (V) was calculated by the following equation: V = length × width2/2.
2.11. Histological analysis
K7 tumor-bearing mice received the same treatment as described above. On day 16, the mice were sacrificed. Tumors and main organs were collected, fixed in 4% paraformaldehyde, and embedded in paraffin. The sliced tumors and organs were then analyzed by hematoxylin and eosin (H&E) staining and TUNEL assay using One Step TUNEL Apoptosis Assay Kit (Meilunbio) according to the manual.
2.12. Statistical analysis
The data were shown as mean ± standard deviation (SD). The statistical significance of experiments was analyzed by one-way ANOVA, and p < .05 was considered statistically significant in all analyses (95% confidence interval).
3. Results and discussion
3.1. Preparation and characterization of NP-PTX-DOX
To prepare NP-PTX-DOX, a PEGylated-PαLA copolymer mPEG-PαLA was designed and synthesized (Scheme 1). The use of mPEG can reduce nonspecific protein adsorption and prevent immune recognition to prolong the circulation time of the nanoparticles in blood. The disulfide-bond-containing PαLA backbone can be degraded by intracellular GSH to induce a reduction-responsive drug release. The hydrophobic PαLA backbone with carboxyl groups could also be used to load PTX and cationic DOX by hydrophobic and electrostatic interactions and release the drugs in response to intracellular acidic pH. The synthetic route of mPEG-PαLA is shown in Scheme 1. First, PαLA was synthesized by ring-opening polymerization of αLA at 98 °C for 2 h. The successful synthesis of PαLA was confirmed by the right shift of proton peaks a and c of αLA (Figure S1) to a1 and c1 (Figure S2) after its ring-opening polymerization. The successful synthesis of PαLA was also confirmed by 13 C NMR (Figure S4 and S5). Then, mPEG-PαLA was synthesized by conjugating mPEG to the side chains of the obtained PαLA via esterification reactions. The successful conjugation of mPEG molecules to the PαLA backbone was confirmed by the appearance of proton peaks h, i, and j, as shown in Figure S3. Moreover, the integration ratio of peaks h to g (1:5) revealed that about 20% of carboxyl groups on PαLA were modified by mPEG. The structure of mPEG-PαLA was further confirmed by 13 C NMR analysis (Figure S6). The presence of peak i2 indicated the formation of ester bonds between mPEG molecules and the PαLA backbone. The appearance of peak h2 showed that some free carboxyl groups were unmodified and could be used for drug loading (Figure S6).
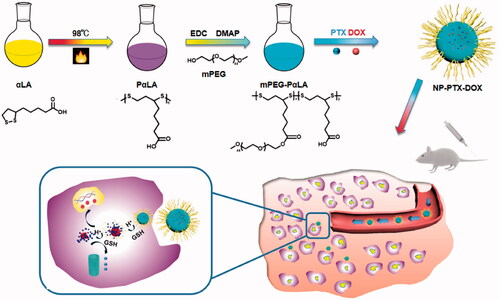
Figure 1. Size distribution and morphology of NPs (A), NP-PTX (B), NP-DOX (C) and NP-PTX-DOX (D) measured by DLS and TEM, respectively. Scale bar = 100 nm. In vitro release of DOX (E) and PTX (F) from NP-PTX-DOX in PBS (pH 7.4), PBS (pH 5.5), and PBS (pH 7.4, 5 mM GSH). Data are shown as mean ± SD (n = 3).
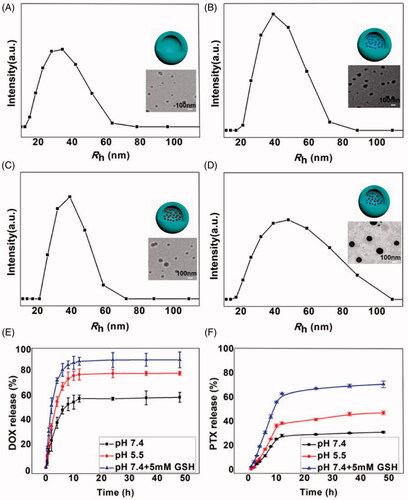
Figure 2. Cellular internalization of DOX and NP-PTX-DOX incubated with K7 cells for 2 h (A) or 6 h (C) at 37 °C determined by flow cytometry. Mean fluorescence intensity of K7 cells treated with DOX and NP-PTX-DOX for 2 h (B) or 6 h (D) determined by flow cytometry. ***p < .001. Data are shown as mean ± SD (n = 3).
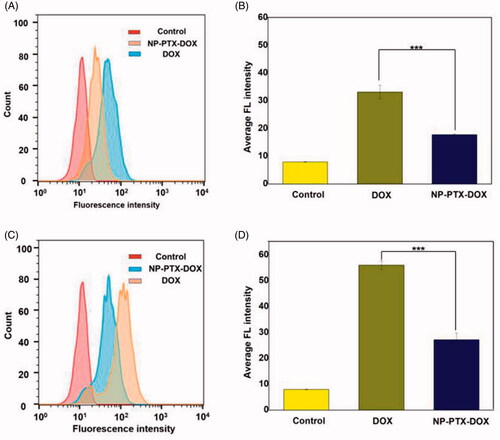
Figure 3. CLSM images of K7 cells treated with DOX or NP-PTX-DOX for 3 h (A) or 6 h (B) at 37 °C. The cell nuclei were stained with DAPI. Scale bar = 20 μm.
Then, the synthesized amphiphilic mPEG-PαLA was used to encapsulate both DOX and PTX by electrostatic and hydrophobic interactions to generate NP-PTX-DOX. As shown in , NP-PTX-DOX had a hydrodynamic radius (Rh) of ∼48 nm as measured by DLS, which was slightly larger than that of empty mPEG-PαLA nanoparticles (NP, ∼33 nm), single-DOX loaded nanoparticles (NP-DOX, ∼39 nm) and single-PTX loaded nanoparticles (NP-PTX, ∼39 nm). Transmission electron microscopy (TEM) images of NP, NP-DOX, NP-PTX and NP-PTX-DOX nanoparticles exhibited a homogeneous distribution of spherical nanoparticles. Furthermore, the drug loading efficiency (DLE) of DOX and PTX in NP-PTX-DOX was 21.2 and 80.1%, respectively, while the drug loading content (DLC) of DOX and PTX in NP-PTX-DOX was 4.3 and 8.2%, respectively. The above results revealed that mPEG-PαLA could be used as a novel nanocarrier to simultaneously load PTX and DOX with high efficiency.
3.2. In vitro drug release
To investigate the reduction/pH dependence of drug release, the time-dependent release of DOX and PTX from NP-PTX-DOX was performed in PBS at pH 7.4, PBS at pH 5.5 and PBS at pH 7.4 containing 5 mM GSH to mimic the normal extracellular environment, acidic endolysosomal circumstance, and the reductive cytosolic environment, respectively. As shown in , the release of DOX at pH 5.5 was faster than that at pH 7.4. This could be attributed to the enhanced hydrophilicity of DOX and the weakened electrostatic interactions between carboxyl groups in mPEG-PαLA and the amine group in DOX under acidic conditions. Furthermore, NP-PTX-DOX exhibited the maximum release of DOX at pH 7.4 containing 5 mM GSH than at pH 7.4 or pH 5.5. This could be explained by the cleavage of disulfide bonds in the PαLA backbone and the resultant disintegration of NP-PTX-DOX-induced by reducing GSH. This result also demonstrated that NP-PTX-DOX was more sensitive to reduction than acidic pH. Similarly, the release of PTX at pH 5.5 was higher than that at a pH of 7.4 in the absence of GSH. The PTX release from NP-PTX-DOX increased in the presence of GSH and was higher compared to the control groups (). In sum, these results indicated that NP-PTX-DOX effectively prevented drug release in the extracellular environment but mediated reduction/pH dual responsive release of PTX and DOX in cancer cells.
3.3. Cellular internalization of NP-PTX-DOX
The cellular internalization of NP-PTX-DOX was investigated by detecting the fluorescence of DOX using flow cytometry analysis (FCA) and confocal laser scanning microscopy (CLSM). K7 osteosarcoma cells were incubated with DOX and NP-PTX-DOX for 2 or 6 h before being analyzed by FCA and CLSM. As shown in , NP-PTX-DOX presented higher fluorescence intensity than the PBS treated control after 2 h incubation, indicating that NP-PTX-DOX was efficiently internalized by K7 cells. Furthermore, the DOX treated group presented higher fluorescence intensity than the NP-PTX-DOX treated group, possibly because DOX could freely diffuse into the cancer cells, whereas NP-PTX-DOX entered the cells by the slower process of cell endocytosis. Moreover, prolonging the incubation time from 2 to 6 h significantly increased the fluorescence intensity of DOX and NP-PTX-DOX treated groups (), confirming the continuous time-dependent cellular internalization of both formulations.
The cellular uptake of NP-PTX-DOX was further investigated by CLSM to detect the red fluorescence of DOX. The nuclei of the cells were stained blue by DAPI. As shown in , the red fluorescence from DOX overlapped with the blue fluorescence of cell nuclei in DOX and NP-PTX-DOX treated groups, indicating that NP-PTX-DOX was efficiently internalized by K7 cells and released DOX into the nuclei after intracellular stimuli-mediated DOX release. In agreement with the FCA results, the fluorescence intensity of NP-PTX-DOX treated cells increased with the increase in incubation time from 2 to 6 h. Altogether, these results indicated that NP-PTX-DOX was efficiently internalized by cancer cells and subsequently released the loaded drugs.
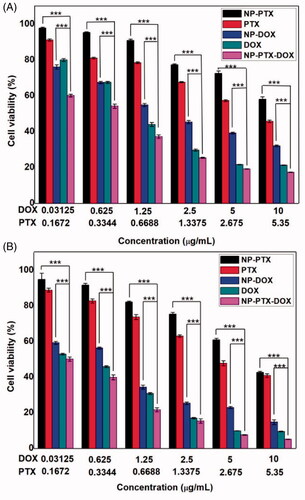
3.4. In vitro cytotoxicity
To investigate the in vitro synergistic anticancer effect of NP-PTX-DOX, K7 cells were incubated with free PTX, free DOX, NP, NP-PTX, NP-DOX, and NP-PTX-DOX for 48 or 72 h. The cell viability was then determined by the MTT assay. NP treated cells manifested negligible reduction in the cell viability (Figure S7). In contrast, as shown in , all the drug-containing formulations presented concentration-dependent cytotoxicity against K7 cells. Free PTX and DOX displayed higher tumor cell killing than NP-PTX and NP-DOX, respectively. This could be attributed to the fact that free PTX and DOX could diffuse into cancer cells. However, NP-PTX and NP-DOX entered the cells by endocytic pathways. Thus, the release of drugs from the nanoparticles needed more time. Moreover, DOX-containing formulations presented higher cytotoxicity than the PTX-containing formulations. NP-PTX-DOX treated group presented more reduction in the cell viability than NP-PTX and NP-DOX treated groups. This may be explained by the synergy between the released DOX and PTX from NP-PTX-DOX. To verify this assumption, the CI value (CI < 1 represents synergism) was calculated according to the cell viability results in . The CI value was calculated to be 0.84 (<1) after 72 h incubation, demonstrating the excellent synergistic anticancer effect of DOX and PTX released from the nanoparticles.
Figure 4. (A) Viability of K7 cells incubated with free DOX, free PTX, NP-PTX, NP-DOX or NP-PTX-DOX for 48 h at 37 °C. (B) Viability of K7 cells incubated with free DOX, free PTX, NP-PTX, NP-DOX or NP-PTX-DOX for 72 h at 37 °C. ***p < .001. Data are shown as mean ± SD (n = 3).
3.5. Biodistribution
NP-PTX-DOX is anticipated to improve the biodistribution of loaded drugs by EPR effect. To verify this hypothesis, the in vivo biodistribution of free DOX and NP-PTX-DOX was investigated after intravenous injection into K7 osteosarcoma tumor-bearing mice for 6 h. As shown in Figure S8, NP-PTX-DOX treated group showed stronger fluorescence than the free DOX-treated group, demonstrating that NP-PTX-DOX can effectively prolong the in vivo circulation time of DOX. Furthermore, the tumor of the NP-PTX-DOX treated group presented stronger fluorescence than that of the free DOX treated group. This was confirmed by quantitative analysis of the region-of-interest. The fluorescence intensity of the tumor in NP-PTX-DOX treated group was about 1.4-fold that of the tumor in the free DOX treated group. Therefore, these results indicated that NP-PTX-DOX could efficiently improve the tumor accumulation and retention of loaded drugs to enhance the anticancer effect.
3.6. In vivo antitumor efficacy
Finally, the in vivo synergistic antitumor efficacy of NP-PTX-DOX was evaluated by intravenously injecting free PTX, free DOX, NP-PTX, NP-DOX and NP-PTX-DOX (2 mg kg−1 DOX, 4 mg kg−1 PTX) into K7 osteosarcoma tumor-bearing mice every three days. As shown in and , free PTX, free DOX, NP-PTX, NP-DOX, and NP-PTX-DOX significantly delayed the tumor growth compared to PBS. NP-PTX and NP-DOX presented higher tumor growth inhibition effect than free PTX and DOX, respectively (), which is in contrast with the in vitro results (). This may be caused by the enhanced tumor accumulation of NP-PTX and NP-DOX through the EPR effect.
Figure 5. Individual tumor growth curves of the PBS (A), PTX (B), NP-PTX (C), DOX (D), NP-DOX (E), and NP-PTX-DOX (F) treated groups.
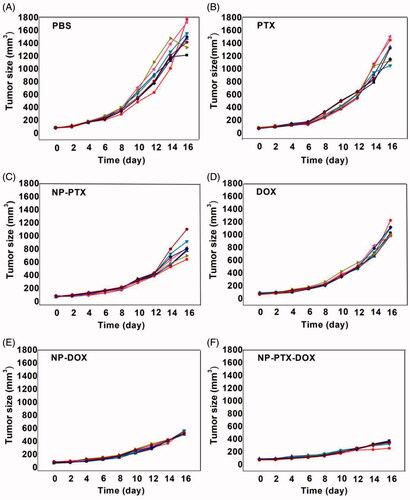
Figure 6. (A) Mean tumor volumes of K7 tumor-bearing mice after intravenous injection of PBS, free PTX, free DOX, NP-PTX, NP-DOX and NP-PTX-DOX on day 0, 3, 6 and 9 (2 mg kg−1 DOX, 4 mg kg−1 PTX). Data are shown as mean ± SD (n = 5). (B) Images of the excised tumors from the mice treated as described in (A) on day 16. (C) Tumor inhibition ratios of the treated groups on day 16. (D) Change in body weights of K7 tumor-bearing mice during treatment. (E) H&E staining and TUNEL analyses of K7 tumors from the mice treated as described in (A) on day 16. ***p < .001. Scale bar = 100 μm.
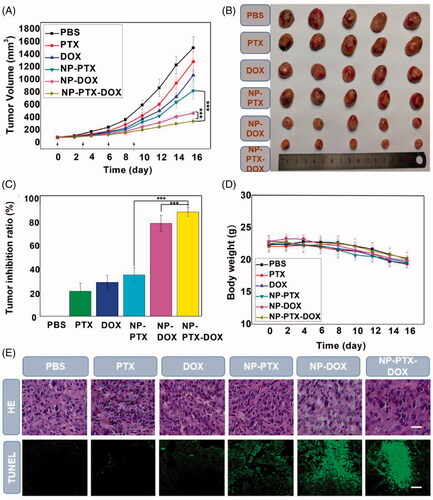
Furthermore, NP-PTX-DOX presented better antitumor effect than NP-PTX and NP-DOX (), which could be ascribed to the synergy between intracellularly released DOX and PTX as demonstrated above (). In agreement with the tumor growth inhibition results, hematoxylin and eosin (H&E) staining results revealed that NP-PTX-DOX treated group presented the highest extent of cancer cell death compared to the control groups. The TUNEL images indicated that tumors from NP-PTX-DOX treated group had the most tumor cell apoptosis (). The in vivo toxicity of free PTX, free DOX, NP-PTX, NP-DOX, and NP-PTX-DOX at the tested dose was investigated by monitoring the body weight changes of the treated mice. As shown in , none of the tested formulations presented any significant systemic toxicity because no obvious reduction in body weight was observed in the treatment groups compared to the PBS treated group. The reduction in body weight in all the groups may be due to the tumor burden of the mice, which would reduce the appetite and increase the energy consumption of the ill mice. H&E staining of the major organs from the treated mice was also carried out to further evaluate the systemic toxicity of the tested formulations. As shown in Figure S9, the organs, including the heart, liver, spleen, lung, and kidneys, from all the treatment groups showed negligible pathological changes compared to the PBS treated group. In summary, NP-PTX-DOX can efficiently deliver DOX and PTX into cancer cells and induce synergistic therapeutic effect against osteosarcoma with high biocompatibility
4. Conclusions
Combination therapy has the advantages of enhanced anticancer efficacy and reduced side effects. Nanoparticle-based DDS offers a promising route for combination cancer therapy by concurrently loading two or more kinds of chemotherapeutics. However, inefficient drug release from the nanomedicines in cancer cells may reduce the antitumor effects. Herein, a reduction/pH dual responsive nanocarrier mPEG-PαLA was developed which could self-assemble into micelles in aqueous media and concurrently encapsulate PTX and DOX. This dual-drug loaded micellar nanoparticle exhibited efficient drug release based on reduction/pH dual response and could be internalized by K7 osteosarcoma cells. Furthermore, the dual-drug loaded mPEG-PαLA micelles exerted effective synergistic anticancer effects both in vitro and in vivo in a murine K7 osteosarcoma model because of the synergy between PTX and DOX and the improved biodistribution of the nanoparticles. In conclusion, this work demonstrates the excellent potential of mPEG-PαLA copolymer as a reduction/pH dual responsive nanocarrier to co-deliver anticancer drugs for osteosarcoma therapy.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Anderson ME. (2016). Update on survival in osteosarcoma. Orthop Clin North Am 47:283–92.
- Baabur-Cohen H, Vossen LI, Kruger HR, et al. (2017). In vivo comparative study of distinct polymeric architectures bearing a combination of paclitaxel and doxorubicin at a synergistic ratio. J Control Release 257:118–31.
- Chakravarthi S, Bulleid NJ. (2004). Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J Biol Chem 279:39872–9.
- Chen H, Tham HP, Ang CY, et al. (2016). Responsive prodrug self-assembled vesicles for targeted chemotherapy in combination with intracellular imaging. ACS Appl Mater Interf 8:24319–24.
- Chen W, Achazi K, Schade B, Haag R. (2015). Charge-conversional and reduction-sensitive poly(vinyl alcohol) nanogels for enhanced cell uptake and efficient intracellular doxorubicin release. J Control Release 205:15–24.
- Cheng R, Feng F, Meng F, et al. (2011). Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release 152:2–12.
- Chiper M, Niederreither K, Zuber G. (2018). Transduction methods for cytosolic delivery of proteins and bioconjugates into living cells. Adv Healthc Mater 7:e1701040.
- Chou T-C. (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–81.
- Eldar-Boock A, Polyak D, Scomparin A, Satchi-Fainaro R. (2013). Nano-sized polymers and liposomes designed to deliver combination therapy for cancer. Curr Opin Biotechnol 24:682–9.
- Fan W, Yung B, Huang P, Chen X. (2017). Nanotechnology for multimodal synergistic cancer therapy. Chem Rev 117:13566–638.
- Feng C, Zhang H, Chen J, et al. (2019). Ratiometric co-encapsulation and co-delivery of doxorubicin and paclitaxel by tumor-targeted lipodisks for combination therapy of breast cancer. Int J Pharm 560:191–204.
- Fu A, Tang R, Hardie J, et al. (2014). Promises and pitfalls of intracellular delivery of proteins. Bioconjug Chem 25:1602–8.
- Gao L, Wang T, Jia K, et al. (2017). Glucose-responsive supramolecular vesicles based on water-soluble pillar[5]arene and pyridylboronic acid derivatives for controlled insulin delivery. Chemistry 23:6605–14.
- Giantonio BJ, Catalano PJ, Meropol NJ, et al. (2007). Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the eastern cooperative oncology group study E3200. J Clin Oncol 25:1539–44.
- Hao Q, Chen Y, Huang Z, et al. (2018). Supramolecular chemotherapy: carboxylated pillar[6]arene for decreasing cytotoxicity of oxaliplatin to normal cells and improving its anticancer bioactivity against colorectal cancer. ACS Appl Mater Interf 10:5365–72.
- Harrison DJ, Geller DS, Gill JD, et al. (2018). Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther 18:39–50.
- He C, Tang Z, Tian H, Chen X. (2016). Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev 98:64–76.
- Heymann MF, Brown HK, Heymann D. (2016). Drugs in early clinical development for the treatment of osteosarcoma. Expert Opin Investig Drugs 25:1265–80.
- Hoffman AS. (2013). Stimuli-responsive polymers: biomedical applications and challenges for clinical translation. Adv Drug Deliv Rev 65:10–6.
- Hu X, Zhang Y, Xie Z, et al. (2017). Stimuli-responsive polymersomes for biomedical applications. Biomacromolecules 18:649–73.
- Huang H, Zhang X, Yu J, et al. (2013). Fabrication and reduction-sensitive behavior of polyion complex nano-micelles based on PEG-conjugated polymer containing disulfide bonds as a potential carrier of anti-tumor paclitaxel. Coll Surf B Biointerf 110:59–65.
- Jackson TM, Bittman M, Granowetter L. (2016). Pediatric malignant bone tumors: a review and update on current challenges, and emerging drug targets. Curr Probl Pediatr Adolesc Health Care 46:213–28.
- Ji GR, Yu NC, Xue X, Li ZG. (2015). PERK-mediated autophagy in osteosarcoma cells resists ER stress-induced cell apoptosis. Int J Biol Sci 11:803–12.
- Jiang T, Mo R, Bellotti A, et al. (2014). Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater 24:2295–304.
- Jiang Y, Zhou Y, Zhang CY, Fang T. (2020). Co-delivery of paclitaxel and doxorubicin by pH-responsive prodrug micelles for cancer therapy. Int J Nanomed 15:3319–31.
- Jin C, Li H, He Y, et al. (2010). Combination chemotherapy of doxorubicin and paclitaxel for hepatocellular carcinoma in vitro and in vivo. J Cancer Res Clin Oncol 136:267–74.
- Kamaly N, Yameen B, Wu J, Farokhzad OC. (2016). Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev 116:2602–63.
- Landesman-Milo D, Ramishetti S, Peer D. (2015). Nanomedicine as an emerging platform for metastatic lung cancer therapy. Cancer Metastasis Rev 34:291–301.
- Leader B, Baca QJ, Golan DE. (2008). Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 7:21–39.
- Li Y, Yang H, Yao J, et al. (2018). Glutathione-triggered dual release of doxorubicin and camptothecin for highly efficient synergistic anticancer therapy. Coll Surf B Biointerf 169:273–9.
- Liu Y, Fang J, Kim Y-J, et al. (2014). Codelivery of doxorubicin and paclitaxel by cross-linked multilamellar liposome enables synergistic antitumor activity. Mol Pharm 11:1651–61.
- Lu S, Zhao F, Zhang Q, Chen P. (2018). Therapeutic peptide amphiphile as a drug carrier with ATP-triggered release for synergistic effect, improved therapeutic index, and penetration of 3D cancer cell spheroids. Int J Mol Sci 19:2773.
- Mo J, Wang L, Huang X, et al. (2017). Multifunctional nanoparticles for co-delivery of paclitaxel and carboplatin against ovarian cancer by inactivating the JMJD3-HER2 axis. Nanoscale 9:13142–52.
- Park J, Wrzesinski SH, Stern E, et al. (2012). Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater 11:895–905.
- Qiu Y, Wu C, Jiang J, et al. (2017). Lipid-coated hollow mesoporous silica nanospheres for co-delivery of doxorubicin and paclitaxel: preparation, sustained release, cellular uptake and pharmacokinetics. Mater Sci Eng C Mater Biol Appl 71:835–43.
- Ramasamy T, Ruttala HB, Choi JY, et al. (2015). Engineering of a lipid-polymer nanoarchitectural platform for highly effective combination therapy of doxorubicin and irinotecan. Chem Commun 51:5758–61.
- Samanta K, Setua S, Kumari S, et al. (2019). Gemcitabine combination nano therapies for pancreatic cancer. Pharmaceutics 11:574.
- Torchilin V. (2011). Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev 63:131–5.
- Wang H, Wu J, Williams GR, et al. (2019). Platelet-membrane-biomimetic nanoparticles for targeted antitumor drug delivery. J Nanobiotechnol 17:60.
- Wang H, Zhao Y, Wu Y, et al. (2011). Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials 32:8281–90.
- Wang Y, Zhang H, Hao J, et al. (2016). Lung cancer combination therapy: co-delivery of paclitaxel and doxorubicin by nanostructured lipid carriers for synergistic effect. Drug Deliv 23:1398–403.
- Wu C, Xu J, Hao Y, et al. (2017). Application of a lipid-coated hollow calcium phosphate nanoparticle in synergistic co-delivery of doxorubicin and paclitaxel for the treatment of human lung cancer A549 cells. Int J Nanomed 12:7979–92.
- Wu X, Sun X, Guo Z, et al. (2014). In vivo and in situ tracking cancer chemotherapy by highly photostable NIR fluorescent theranostic prodrug. J Am Chem Soc 136:3579–88.
- Xiaoya D, Yu W, Gao L, et al. (2019). Iminoboronate ester cross-linked hydrogels with injectable, self-healing and multi-responsive properties. Acta Polymerica Sinica 50:505–15.
- Xu XD, Zhao L, Qu Q, et al. (2015). Imaging-guided drug release from glutathione-responsive supramolecular porphysome nanovesicles. ACS Appl Mater Interf 7:17371–80.
- Yamawaki K, Asoh TA, Kikuchi A. (2016). Redox-responsive minimized fragmentation of three-armed oligo(ethylene glycol) gels for protein release. Coll Surf B Biointerf 146:343–51.
- Yang H, Shen W, Liu W, et al. (2018). PEGylated poly(α-lipoic acid) loaded with doxorubicin as a pH and reduction dual responsive nanomedicine for breast cancer therapy. Biomacromolecules 19:4492–503.
- Yang M, Ding H, Zhu Y, et al. (2019). Co-delivery of paclitaxel and doxorubicin using mixed micelles based on the redox sensitive prodrugs. Coll Surf B Biointerf 175:126–35.
- Zhang Y, Xiao CS, Li MQ, et al. (2014). Core-cross-linked micellar nanoparticles from a linear-dendritic prodrug for dual-responsive drug delivery. Polym Chem 5:2801–8.
- Zhao D, Wu J, Li C, et al. (2017). Precise ratiometric loading of PTX and DOX based on redox-sensitive mixed micelles for cancer therapy. Coll Surf B Biointerf 155:51–60.
- Zhao L, Zhu L, Liu F, et al. (2011). pH triggered injectable amphiphilic hydrogel containing doxorubicin and paclitaxel. Int J Pharma 410:83–91.