Abstract
The single drop microextraction method was developed for the determination of ten organochlorine pesticides in tea brews using gas chromatograph-electron capture detector. The optimized parameters for effective extraction required a 2 μL drop of n-hexane at the tip of a microsyringe immersed in 5 mL of a diluted tea brew sample for 25 min and stirred at 600 rpm. The limit of detection in the range of 0.01 to 0.025 μg/L, with relative standard deviation of repeatability and reproducibility, was in the range of 1–10 and 8–24%. The relationship of the measured signal was a linear function (>0.9863) of concentration and the recovery method ranged from 92–116%. The single drop microextraction method showed the advantage of being easy to operate, low consumption of organic solvent, and high extraction efficiency.
INTRODUCTION
Recently increased concern on food safety issues has emphasized the need of detection of organic contaminants at very low levels for their monitoring and better management in the food supply chain. Under the paradigm of risk analysis approach for achieving food safety, regulatory laws in many countries forbid the consumption and trade of food items contaminated with the pesticides even below the level of part per billion or declares zero tolerance level particularly for genotoxic compounds.Citation[1] Thus, in the regimes of recent safety mechanisms and analytical controls of food, it is highly desirable to develop simple analytical methods to measure the lowest possible concentration of pesticides with acceptable accuracy and precision in the desired matrix. The innovative sample preparation strategies, particularly those involving enrichment of analytes, may substantially contribute to the decrease in limit of detection (LOD) with acceptable method performance characteristics.Citation[2] Some of the shortcomings, such as use of hazardous solvents, costly reagents, and adsorbents associated with available sample preparation techniques[Citation3–8] fostered the need of exploring improved preconcentration methods. In this regard, solid-phase microextraction (SPME) developed by Arthur and PawliszynCitation[9] is one of the most promising preconcentration methods that is relatively simple, solvent free, and may also help in reducing the LOD through enrichment of analytes on the fiber.Citation[10,Citation11] SPME technique has been applied extensively to determine a wide range of analytes,Citation[10,Citation12] including acaricides, organochlorine pesticides (OCPs), nitrogen containing pesticides, and organophosphate pesticides in a range of different foodsCitation[2] and water.Citation[13,Citation14] Although the use of SPME is increasingly gaining popularity, the technique suffers from drawbacks, such as low operating temperature, instability and swelling with organic solvents, breakage of the fiber, stripping of coatings, expensive coated fibers, and for some applications, limited lifetime. Due to the cost, analytes specificity and fragility of the SPME fiber may limit its wider applications.Citation[15]
Single drop microextraction (SDME), developed in 1996 by Jeannot and Cantwell,Citation[16] provides an alternative technique, which integrates sampling, extraction, concentrating, and sample introduction in a single step.Citation[15,Citation17] It provides target analytes in a single drop of solvent and directly injected into an injector port of gas chromatograph (GC). The enrichment of the analytes on the single dropCitation[18] may reduce the LOD manifold. Thus, SDME may further overcome the drawbacks associated with other sample preparation strategies including SPME. SDME has been successfully applied for the determination of alcohols,Citation[19] nitro aromatic explosives,Citation[20] chlorobenzenes,Citation[21] drugsCitation[22,Citation23] for the screening of pesticides,Citation[24,Citation25] and volatile organic compounds in water.Citation[26]
Tea is the most popular beverage throughout the world. The widespread use of pesticides in the tea crop has led to the presence of pesticide residues in tea, which gets transferred into the brew, and is offered commercially for public consumption where they contribute to the total dietary intake.Citation[27] Various international authorities (Codex Alimentarious Commission, US EPA, and EU) have been involved in regulating the pesticides by fixing a very low level of maximum residues limits (MRLs) for trade and consumption of tea. Thus, it is of great importance to develop a simple, fast, and eco-friendly analytical method, which could work at a very low level with a desired degree of accuracy and precision. Although the funnel SDME applied for the determination of OCPs in tea brew,Citation[28] it further needs a skilled manpower for making such type of small device.
Keeping in view the above fact, the aim of the present work was to develop a simple multi-residue method using SDME-GC-electron capture detector (ECD) for determination of OCP residues in tea brew. For the purpose of the present studies, different parameters affecting the extraction process were studied and optimized followed by its validation. The optimized parameters were applied to real samples of tea collected from market to evaluate the application of this method to real samples.
MATERIALS AND METHODS
Reagents and Chemicals
Standards of α-hexachlorocyclohexane, β-hexachlorocyclohexane, γ-hexachlorocyclohexane, α-endosulfan, dieldrin, endrin, and β-endosulfan were obtained from Dr. Ehrenstorfer Laboratory (Augsburg, Germany). Alachlor, aldrin, and heptachlor were obtained from Sigma Aldrich (Mumbai, India). Purity level of all the pesticide standards was >97%. The solvents (n-hexane, cyclohexane, and acetone) used in the experiments were procured from Merck India (Mumbai) and the toluene used was from sd. Fine Chemicals Ltd. (Mumbai, India). The water was purified using a Milli-Q water purification system (Millipore, Bedford, MA, USA). The micro syringes were obtained from Hewlett and Packard (Switzerland). The magnetic stirrer was obtained from Genetix Asia Pacific Pvt Ltd. (Mumbai, India).
Sampling
All brands of the tea samples were collected from a local market. All the tea samples were Indian brand made in India and stored at 4°C until analysis.
Stock Standard Solution
Stock standard solution (10 mg/L) of individual pesticide or pesticides mixture was prepared by dissolving 1 mg of analyte into 100 mL of acetone. Standard solution of different concentration range of the analytes was prepared by diluting the stock solution in acetone (0.005 to 10 μg/L). All solutions were stored at 4°C.
SDME Procedure
The tea brew was prepared from the made tea (procured from our Institute's tea manufacturing unit) through an infusion process wherein 2 g of made tea was infused in 200 mL of boiling water. After 3 min of brewing, the water extract was filtered through filter paper (0.4 μm), cooled and used in the study as liquid matrix test model. An aliquot of 5 mL of tea brew sample was placed in a 15 mL glass vial equipped with a polytetrafluoroethylene (PTFE)-coated magnetic stir bar and screw capped with a PTFE-faced silicone septum. The n-hexane was drawn into a 10-μL syringe. The microsyringe needle was inserted through the septum and directly immersed into the tea brew sample. The n-hexane drop was made by a depressed microsyringe plunger and the drop (2.0 μL) was exposed to the sample. SDME was performed for a range of time intervals (5–30 min) and at different stirring rates (200–1000 rev/min) at room temperature (24 ± 1°C) with different solvents (n-hexane, toluene, and cyclohexane). After microextraction, the organic drop was retracted into the syringe and transferred immediately into the injector port of the gas chromatograph for analysis. For optimization of extraction parameters, SDME was performed in a spiked tea brew by varying a single parameter at a time with respect to that of the other. The optimized conditions were used for development of method and its validation.
Apparatus and Operating Conditions
A Shimadzu 2010 series gas chromatograph (Kyoto, Japan) equipped with an electron capture detector was used for the analysis of organochloine pesticides. The chromatographic separation was performed using a Restek column (30 m × 0.25 mm I.D. × 0.25 μm) in split mode. The gas chromatograph (GC) oven was programmed as follows: initial temperature of 220°C held for 2 min, ramped at 5°C/min to 260°C held for 4 min. The injector and detector temperatures were kept at 250 and 300°C, respectively. The nitrogen gas (99.99%) was used as the carrier gas at a flow rate of 1 mL/min.
RESULT AND DISCUSSION
Optimization of the SDME
In order to obtain the optimized extraction conditions, the peak area of analyte as the gas chromatograph-electron capture detector (GC-ECD) response was used to evaluate the extraction efficiency and influenced several factors at different conditions.
Selection of Solvent
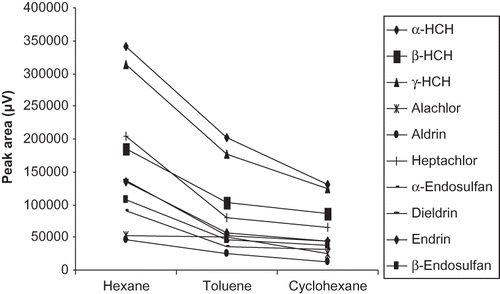
The organic solvents (n-hexane, cyclohexane, and toluene) were tried out. The peak response of pesticides for the individual extraction solvent was recorded. Average peak areas obtained for each organochlorine pesticide (OC) with three different solvents are shown in . The result revealed that the best extraction efficiency was achieved for all organochlorine pesticides with n-hexane. This is due to low polarity (polarity index, 0.9) of n-hexane favored towards non-polar compounds and its low solubility in water (1.4 × 10−4 g/100 mL), which results in low dissolution of solvent and enhances the extraction of analyte.Citation[18] Thus, n-hexane was selected for the succeeding experiments.
Figure 1 Optimization of solvent for extraction of organochlorine pesticides.
Extraction Time
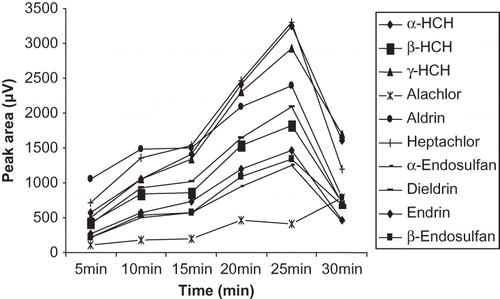
The time of contact should be sufficient to allow the solvent drop to absorb the maximum amount of analyte. The extraction efficiency was checked at varied time intervals ranging 5 to 30 min until the equilibrium between the spiked tea brew and n-hexane drop was attained for all the analytes. The result revealed () that the extraction efficiency increased with an increase in extraction time up to 25 min. Peak response of the analytes reached steady state on increasing the extraction time beyond 25 min. Moreover, the phenomenon of drop dissolution was also observed as reported in some earlier studies.Citation[29] Thus, in order to gain short enrichment time, 25 min was considered to be the reasonable selection for the rest of the studies.
Figure 2 Optimization of extraction time for extraction of organochlorine pesticides.
Exposure Volume of Extractant Phase
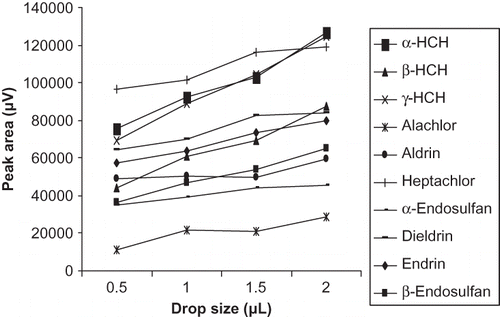
As described elsewhere, the amount of the analyte extracted into an organic drop can be increased with the larger organic solvent volume.Citation[15] To optimize the drop size, experiments were performed by increasing the drop size of n-hexane from 0.5–2.5 μL. The peak response showed a linear relationship with the drop sizes in the range of 0.5–2 μL (). When drop size was increased beyond 2 μL, the n-hexane drop slided upward along the needle beyond the point of retraction. The same phenomenon was observed in earlier reported literature.Citation[15] This explains why, in the present study, a drop size beyond 2 μL became unstable at the microsyringe needlepoint and linear relationship between the solvent size and analyte extraction was limited to a solvent volume of 2 μL; thus, 2 μL drop volume was selected for further experiments.
Figure 3 Optimization of solvent drop size for extraction of organochlorine pesticide.
Agitation Rate
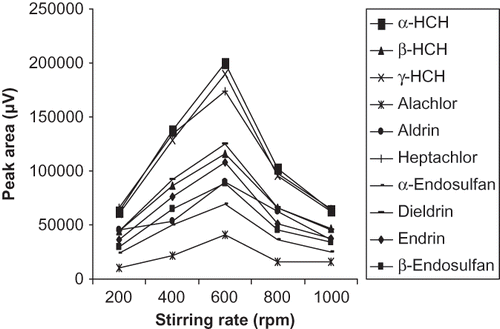
The effect of stirring was studied using 2 μL of n-hexane drop for 25 min at different stirring rates (200, 400, 600, 800, and 1000 rpm). The results shown in indicated that the stirring speed enhanced the extraction efficiency significantly and the maximum peak response of the analytes was obtained at 600 rpm under the given extraction time of 25 min. The results are in agreement with the explanation based on film theory of convective-diffusive mass transfer. The higher stirring speed may accelerate the analyte diffusion into the solvent microdrop by decreasing the thickness of the Nernst diffusion film in the aqueous phase and thus shortening the time to achieve the equilibrium concentration.Citation[18,Citation28] However, too vigorous agitation leads to generation of mechanical forces in the sample solution that disrupt the stability of the drop.Citation[18,Citation28,Citation30] In the present study, stirring speed above 600 rpm also caused instability in solvent drop due to formation of bubbles into the solvent and it was difficult to maintain the integrity of the solvent drop. Therefore, a stirring speed of 600 rpm was selected for the subsequent experiments.
Figure 4 Optimization of stirring rate for extraction of organochlorine pesticides.
Matrix Effect
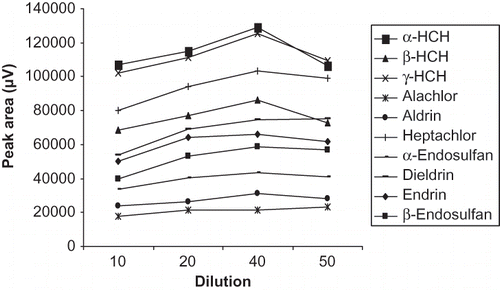
A tea sample matrix consists of a variety of interfering components, which may interfere with the extraction of analytes through SDME.Citation[14] Dilution of the sample matrix may enhance the extraction efficiency as observed in earlier studies.Citation[14,Citation31] Our results also supported this explanation and showed that the recovery of the analytes linearly improved in the dilution range 10 to 40 times as shown in . No significant change was observed on further dilution. Thus, dilution of the sample matrix in the ratio of 1:40 was considered optimum in the given range of analytical limits.
Figure 5 Optimization of dilution for extraction of organochlorine pesticides.
Effect of Temperature and Salt
The effect of temperature was studied by performing SDME under the optimized conditions at a temperature range of 24 to 50°C. The result indicated that the extraction efficiency improved on increasing the temperature. However, as reported earlier, we also observed the damage and dissolution of drop at a higher temperature.Citation[14] Thus, in the present work, SDME was performed at ambient temperature (∼25°C). The effects of salt on extraction of analytes were tested by adding different concentration (50–200 mg/5 mL) of sodium chloride (NaCl) salt to the sample matrix. The results revealed that there was no significant increase on the detector response on adding the salt. Similar observations have been mentioned in the earlier reports.Citation[14,Citation29] Hence, for convenience no salt was added in the subsequent experiments
Method Validation
In order to consider the method for practical applications, it is necessary to validate the method, which involves optimization of the characteristic parameters for method: linearity, precision, accuracy, LOD, and enrichment factor. The developed SDME method was evaluated for validation characteristics under the optimized extraction conditions in spiked tea brew. As shown in , under optimal conditions, the linearity was observed over the ranged from 0.6 to 10 μg/L for all analytes. The coefficient of determination (r2) ranged from 0.986 to 0.995. The LODs, based on a signal-to-noise (S/N) of 3, ranged from 0.01 to 0.025 μg/L. While determining LOD, rinsing of the syringe with n-hexane was performed to avoid sample carryover. The present method demonstrated superiority over the earlier reported methods in terms of lowered LOD.Citation[15,Citation18,Citation32] The repeatability and reproducibility of the proposed method presented and acceptable precision with a RSD <20% (except β- hexachlorocyclohexane (HCH) and alachlor). The International Conference of Harmonization (ICH) guidelineCitation[33] has recommended that the RSD was less than 20% acceptable for a concentration of <0.1%.
Table 1 Validation parameter for SDME followed by GC-ECD, for determination of organochlorine pesticides from tea brew
The enrichment factor of α-HCH, γ-HCH, heptachlor α-endosulfan, and dieldrin was >50 fold except for β-HCH (40 fold), γ-HCH (38 fold), alachlor (24 fold), and both endrine and β-endosulfan (28 fold); some analytes could be preconcentrated nearly or greater than 100 fold. In the present method, accuracy was determined by comparing detector response of fortified tea brew and water sample (Milli-Q) for the analytes at the target concentration. The relative recovery for all the analytes was in the ranged from 92–116%, which is acceptable as described in the ICH guideline.
Application of the Method in Real Samples
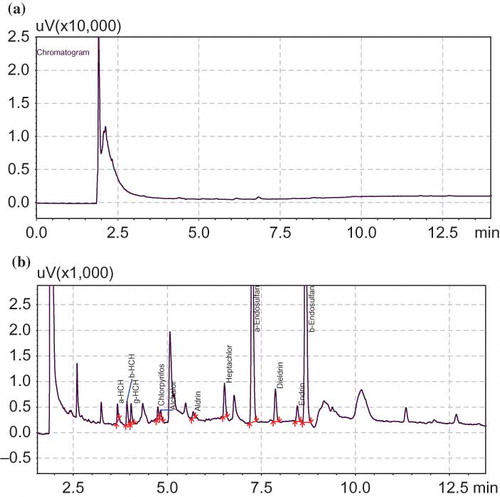
The ten tea samples collected from a local market were extracted using the developed SDME method and analyzed by GC-ECD. The result for tea brew showed that most of the samples were free from OCP contamination except two (β-HCH–9.0 μg/L, γ-HCH–1.0 to 9.0 μg/L) and they were confirmed by spiking the proposed pesticides in the tea brew. shows the chromatogram obtained for real sample and shows the chromatogram of tea brew spiked samples.
Figure 6 (a) Chromatogram obtained for tea brew unspiked sample. (b) Chromatogram obtained for tea brew spiked sample (α-HCH, β-HCH, γ-HCH, alachlor, aldrine, heptachlor, α-endosulfan, dieldrin, endrin, and β- endosulfan). (Color figure available online.)
CONCLUSION
It is evident that the single-drop microextraction sample preparation technique has advantages of detecting very low levels of analytes in tea brew. In the present work, it was found that SDME technique followed by GC-ECD considerably lowered the LOD for the organochlorine pesticides investigated. Real sample analysis results indicated practical application of the developed method. In view of the increasingly stringent regulatory limits for health standards of many pesticides below the detection limit of the existing instrument, the research focused on SDME use may provide a cost effective solution for analytical inspection in various liquid matrices at a very low level to address various food safety management issues concerning the presence of pesticide residues in the food supply chain.
ACKNOWLEDGMENT
The authors are grateful to Dr. P. S. Ahuja for providing valuable suggestions throughout the experimental work.
Notes
This article has been republished with minor changes. These changes do not impact the academic content of the article.
REFERENCES
- Heberer, T., Wiederholt, M.L., Schafft, H., Abrahamc, K., Pzyrembel, H., Henning, K.J., Schauzu, M., Braeunig, J., Goetz, M., Niemanni, L., Remy, U.G., Luchk, A., Appel, B., Banasiak, U., Bol, G.F., Lampen, A., Wittkowski, R., and Hensel, A., 2007. Zero tolerances in food and animal feed-Are there any scientific alternatives?: A European point of view on an international controversy, Toxicology Letters 175 (2007), pp. 118–135.
- Lambropoulou, D.A., and Albanis, T.A., 2007. Methods of sample preparation for determination of pesticide residues in food matrices by chromatography-mass spectrometry-based techniques: A review, Analytical and Bioanalytical Chemistry 389 (2007), pp. 1663–1683.
- Fatoki, O.S., and Awofolu, R.O., 2003. Methods for selective determination of persistent organochlorine pesticide residues in water and sediments by capillary gas chromatography and electron-capture detection, Journal of Chromatography A 983 (2003), pp. 225–236.
- Anastassiades, M., Lehotay, S.J., Stajnbaher, D., and Schenk, F.J., 2003. Fast and easy multiresidue method employing acetonitrile extraction partitioning and “Dispersive solid-phase Extraction”, Journal of AOAC International 86 (2003), pp. 412–431.
- Chen, Y., Xie, M.Y., and Gong, X.F., 2007. Microwave-assisted extraction used for the isolation of total triterpenoid saponins from Ganoderma atrum, Journal of Food Engineering 81 (2007), pp. 162–170.
- Obana, H., Akutsu, K., Okihashi, M., Kakimoto, S., and Hori, S., 1999. Multiresidue analysis of pesticides in vegetables and fruits using a high capacity absorbent polymer for water, The Analyst 124 (1999), pp. 1159–1165.
- Valverde-Garcia, A., Fernandez-Alba, A.R., Contreras, M., and Agüera, A., 1996. Supercritical fluid extraction of pesticides from vegetables using anhydrous magnesium sulfate for sample preparation, Journal of Agriculural and Food Chemistry 44 (1996), pp. 1780–1784.
- Sohrabvandi, S., Mousavi, S.M., Razavi, S.H., and Mortazavian, A.M., 2010. Application of advanced instrumental techniques for analysis of physical and physicochemical properties of beer: A review, International Journal of Food Properties 4 (2010), pp. 744–759.
- Arthur, C.L., and Pawliszyn, J., 1990. Solid phase microextraction with thermal desorption using fused silica optical fibers, Analytical Chemistry 62 (1990), pp. 2145–2148.
- Kumar, A., Singh, B., Malik, A.K., and Tewary, D.K., 2007. Determination of some aldehydes by using solid-phase microextraction and high-performance liquid chromatography with UV detection, Journal of AOAC International 90 (2007), pp. 1689–1694.
- Kumar, A., Malik, A.K., and Tewary, D.K., 2009. A new method for determination of myricetin and quercetin using solid phase microextraction-high performance liquid chromatography-ultra violet/visible system in grapes, vegetables and red wine samples, Analytica Chimica Acta 631 (2009), pp. 177–181.
- Aghlara, A., Mustafa, S., Yazid, A.M., and Mohamad, R., 2009. characterization of headspace volatile flavor compounds formed during kefir production: application of solid phase microextraction, International Journal of Food Properties 12 (2009), pp. 808–818.
- Magdic, S., and Pawliszyn, J., 1996. Analysis of organochlorine pesticides using solid-phase microextraction, Journal of Chromatography A 723 (1996), pp. 111–122.
- Lopez-Blanco, M.C., Balanco-Cid, S., Cancaho-Grande, B., and Simal-Gandara, J., 2003. Application of single-drop microextraction and comparison with solid-phase microextraction and solid-phase extraction for the determination of α- and β-endosulfan in water samples by gas chromatography–electron-capture detection, Journal of Chromatography A 984 (2003), pp. 245–252.
- Zhang, M., Huang, J., Wei, C., Yu, B., Yang, X., and Chen, X., 2008. Mixed liquids for single-drop microextraction of organochlorine pesticides in vegetables, Talanta 74 (2008), pp. 599–604, .
- Jeannot, M.A., and Cantwell, F.F., 1996. Solvent microextraction into a single drop, Analytical Chemistry 68 (1996), pp. 2236–2240.
- Cardoso, A.A., and Dasgupta, P.K., 1995. Analytical chemistry in a liquid film/droplet, Analytical Chemistry 67 (1995), pp. 2562–2566.
- Zhao, L., and Lee, H.K., 2001. Application of static liquid-phase microextraction to the analysis of organochlorine pesticides in water, Journal of Chromatography A 919 (2001), pp. 381–388.
- Tankeviciute, A., Kazlauskas, R., and Vickackaite, V., 2001. Headspace extraction of alcohols into a single drop, Analyst 126 (2001), pp. 1674–1677.
- Psillakis, E., and Kalogerakis, N., 2001. Application of solvent microextraction to the analysis of nitroaromatic explosives in water samples, Journal of Chromatography A 907 (2001), pp. 211–219.
- Wang, Y., Kwok, Y.C., He, Y., and Lee, H.K., 1998. Application of dynamic liquid-phase microextraction to the analysis of chlorobenzenes in water by using a conventional microsyringe, Analytical Chemistry 70 (1998), pp. 4610–4614.
- Rasmussen, K.E., Pedersen-Bjergaard, S., Krogh, M., Ugland, H.G., and Gronhaug, T., 2000. Development of a simple in-vial liquid-phase microextraction device for drug analysis compatible with capillary gas chromatography, capillary electrophoresis and high-performance liquid chromatography, Journal of Chromatography A 873 (2000), pp. 3–11.
- de Jager, L.S., and Andrews, A.R.J., 2001. Development of a screening method for cocaine and cocaine metabolites in urine using solvent microextraction in conjunction with gas chromatography, Journal of Chromatography A 911 (2001), pp. 97–105.
- de Jager, L.S., and Andrews, A.R.J., 1999. Solvent microextraction of chlorinated pesticides, Chromatographia 50 (1999), pp. 733–738.
- de Jager, L.S., and Andrews, A.R.J., 2000. Development of a rapid screening technique for organochlorine pesticides using solvent microextraction (SME) and fast gas chromatography (GC), Analyst 125 (2000), pp. 1943–1948.
- Buszewski, B., and Ligor, T., 2002. Single-drop extraction versus solid-phase microextraction, LC GC Europe 2 (2002), pp. 92–97.
- Tewary, D.K., Kumar, V., Ravindranath, S.D., and Shanker, A., 2005. Dissipation behavior of bifenthrin residues in tea and its brew, Food Control 16 (2005), pp. 231–237.
- Qian, L., and He, Y., 2006. Funnelform single-drop microextraction for gas chromatography–electron-capture detection, Journal of Chromatography A 1134 (2006), pp. 32–37.
- Liang, P., Guo, L., Liu, Y., Liu, S., and Zhang, T.Z., 2005. Application of liquid-phase microextraction for the determination of phoxim in water samples by high performance liquid chromatography with diode array detector, Microchemical Journal 80 (2005), pp. 19–23.
- Ye, C., Zhou, Q., and Wang, X., 2007. Improved single-drop microextraction for high sensitive analysis, Journal of Chromatography A 1139 (2007), pp. 7–13.
- Simplicio, A.L., and Boas, L.V., 1999. Validation of a solid-phase microextraction method for the determination of organophosphorus pesticides in fruits and fruit juice, Journal of Chromatography A 833 (1999), pp. 35–42.
- López, F.J., Pitarch, E., Egea, S., Beltran, J., and Hernández, F., 2001. Gas chromatographic determination of organochlorine and organophosphorus pesticides in human fluids using solid phase microextraction, Analytica Chimica Acta 433 (2001), pp. 217–226.
- 1994. International Conference of Harmonization. Validation of analytical procedure text and methodology Q2 (R1), International Conference on Harmonization Guideline (1994), pp. 1–13.