
ABSTRACT
Five types of electrostatic complex (macromolecular complexes, core–shell particles, and mixed homogeneous particles) were formed between whey protein (whey protein concentrate [WPC]) and pectin. By controlling the thermal treatment, composition, and order of mixing, it was possible to produce complexes that for the same biopolymer concentration gave differing functional properties. All protein–pectin complexes showed higher foaming ability and stability than native or heated WPC without pectin. Native WPC had higher emulsifying ability than protein–pectin complexes but exhibited the lowest emulsion stability. Ingredients based on such ideas might offer the food manufacturer greater control over food structure, stability, and organoleptic properties.
Introduction
Biopolymer microparticles or complexes have been investigated as functional ingredients in a range of industries, mostly the food industries and more recently the pharmaceutical industries as a drug-delivery system.[Citation1,Citation2] They have also been shown to be useful in the delivery and protection of bioactive compounds in foods.[Citation1–Citation3] They have also been used to replace fats in foods due to their ability to mimic the sensory, optical, and rheaological properties of lipid droplets.[Citation4,Citation5] In view of this, there has been a growing interest in the fabrication of biopolymer microparticles made from proteins alone[Citation6,Citation7] or mixed protein and polysaccharide complexes.[Citation3,Citation8–Citation13] Molecular complexes form via interaction (commonly electrostatic) between two biopolymer molecules. Particle aggregates form through, for example, heat-induced aggregation of biopolymers. Particle aggregates can be homogeneous or heterogeneous. Homogeneous particles are characterised by an even distribution of one or more biopolymer types throughout the particle. Heterogeneous particles may consist of a second phase dispersed in a biopolymer particle or droplets such as lipid emulsion droplets dispersed in the biopolymer particle. Heterogeneous particles may also have a core–shell structure where one biopolymer forms a particle aggregate (core) and a second forms a layer (shell) on the surface. Other particle types that can form are fibrils or non-spherical particles.[Citation3]
Proteins and polysaccharide form electrostatic complexes when they have opposite electrical charge.[Citation3,Citation9,Citation14] For example, the electrical charge on pectin and whey protein molecules is negative at all pH for pectin and at pH above the isoelectric point of the whey protein. At pH below the protein isoelectric point, the net electrical charge changes to positive. Polysaccharide molecules that contain acidic groups are negatively charged over a wide pH range. It is possible to identify a pH range over which the whey protein is positively charged and the pectin negatively charged and thus can form electrostatic complexes. At pH above the protein isoelectric point where the protein is negatively charged, protein and pectin do not interact strongly as a result of electrostatic repulsion between the molecules. When the pH is reduced, complex formation will occur at pHs below the isoelectric point. The complexes are soluble if the charge on the protein is not too high, but if the pH is reduced to far below the isoelectric point, extensive complex formation occurs and this eventually leads to precipitation.[Citation9,Citation15,Citation16] The complex formation also depends on other factors such as temperature, ionic strength, protein:pectin ratio, and protein concentration.[Citation3,Citation17,Citation18]
Due to the gelation and aggregation characteristic of protein/polysaccharide complexes, they can influence the colloidal stability and textural characteristic of food products.[Citation8,Citation19–Citation21] This has found application in a range of new food formulations.[Citation22] Under some conditions, proteins and polysaccharides, including whey protein and pectin, undergo phase separation when mixed due to thermodynamic incompatibility, arising from a positive free energy change on mixing.[Citation19] This usually occurs under conditions where the proteins are uncharged or have the same charge as the polysaccharide.
According to Bouaouina et al.,[Citation23] the functional properties of food proteins in combination with polysaccharides can be categorised into three classes: (a) gelation and aggregation properties, (b) hydration properties (which includes wettability, swelling, dispersibility, viscosity, water-holding capacity, adhesion), and (c) interfacial properties, which includes foaming and emulsification properties. In this study, we used different fabrication techniques to make whey protein concentrate (WPC)–high methoxy pectin complexes under conditions where they interact by electrostatic means. The effect that fabrication method has on particle structure, foaming, and emulsifying properties is studied to determine the potential of these particles as functional food ingredients.
Materials and methods
Materials
WPC (Lacprodan 87) was kindly donated by Arla Foods Ingredients, Arhus, Denmark. The protein concentration as reported by the manufacturer was 87%. Citrus peel pectin (GENU® High Methoxy Pectin ISO USA-SAG type B rapid set 72% DE) was also kindly donated by CP Kelco, Denmark. All sample suspensions were prepared using milli-Q water. Sodium hydroxide (solid pellets) and hydrochloric acid solutions (32% by volume) used for adjusting the pH were purchased from Sigma Aldrich (UK) and Fisher Scientific (UK), respectively.
Biopolymer suspension preparation and fabrication techniques
WPC and pectin complex (WPP) sample suspensions were made by dissolving the appropriate mass of powder in the appropriate mass of water to give a known concentration of WPC or pectin solution (w/w). The solutions were stirred overnight at room temperature using a magnetic stirrer to ensure they were dissolved and hydrated. To remove insoluble material, both protein and pectin suspensions were centrifuged at 3000 rpm for 15 min (Denley BS400 centrifuge, England) and the supernatant removed. The pH of the solutions was adjusted to pH 4 using 1 M HCI solution. pH 4 was chosen because the electrostatic interaction between pectin and whey protein concentration (WPC) is strongest when the pH of WPC is slightly below its isoelectric point (pI ≈ 4.3).[Citation3,Citation9,Citation14] Samples were left standing at room temperature for 20 min with continuous stirring at the desired pH before further use.[Citation3,Citation11,Citation12]
To form molecular complexes or protein–polysaccharide particles, protein and pectin solutions were mixed in the appropriate mass ratio and then heated together or heated individually before mixing with the other (see ). After the samples had been heated in the water bath, they were removed and placed immediately into an ice bath for 3 h to ensure rapid cooling and to bring all aggregation reactions to a halt. The pH of each solution was checked and readjusted to pH 4 if necessary. The protein–pectin complexes were formed by mixing protein and pectin solutions in the appropriate mass ratio. The samples were then kept in a refrigerator (5°C) overnight prior to analysis. A sample where neither WPC nor pectin were heated, but were still mixed in the appropriate mass ratio, was also made and treated in the same way as the heated samples. summarizes the fabrication method for each sample.
Table 1. Sample fabrication techniques.
To determine conditions for the fabrication of WPC pectin complexes, preliminary experiments were carried out to determine the effect of heating temperature, heating time, and protein:pectin ratio on the size of WPP particles. Based on these experiments (Supplementary material Figs. S1–S3), a heating temperature and time combination of 80°C and 25 min and a protein:pectin ratio of 5:1 were chosen for the formation of WPP particles. The temperature of 80°C was chosen as this is known to be close to the temperature at which β-lactoglobulin, the major whey protein, starts to denature. In addition, 80°C was the temperature at which many of the particles have the largest size (Supplementary material Fig. S1). The heating time of 25 min was chosen because all aggregation reactions are complete by this time (Supplementary material Fig. S2). Finally, a protein:pectin ratio of 5:1 was chosen as this gives the biggest differentiation in size between the particles and was stable for the two molecular complexes, which showed very large particle sizes at lower protein:pectin ratios below 4:1 (Supplementary material Fig. S3). Under these conditions and when mixed and adjusted to pH 4, the pectin and WPC proteins form electrostatic complexes.
Five types of whey protein–pectin (WPP) particles were fabricated by the manipulation of heat treatment given to each sample as listed in . It has been shown by other researchers that biopolymer particles can form at a pH just below the isoelectric point (pI) of the protein.[Citation10] Under these conditions, WPC proteins have a net positive charge whilst pectin is negatively charged and complexes are formed through electrostatic attraction. Zetasizer measurements showed that the WPC has a pl ≈ 4.5. Therefore, since our protein and pectin solutions are at pH 4, we would expect the WPC and pectin to form an electrostatic complex when mixed.
Particle size determination
The particle size of the biopolymer particles was measured using dynamic light scattering with a Zetasizer NanoS (model ZEN1600, Malvern Instruments Ltd., UK). The particle size was measured in backscatter mode using a scattering angle of 173° through a 2-mL cuvette of 1.0-cm path length filled with 1 mL of sample dispersions. The samples were characterized by the particle size distribution (diameter, nm). This was calculated by the Zetasizer computer software from the intensity of the scattered light using cumulants analysis of dynamic scattering data. WPP and WPC dispersions were diluted with milli-Q water at a ratio of 1:100 (v/v). A solution of this concentration was found to be sufficiently dilute to remove the effects of multiple scattering. All measurement were performed at 25°C and replicated three times.[Citation3,Citation9,Citation11,Citation14]
Determination of foaming properties
The foaming property of WPP complex suspensions was determined by the method described by Philips et al.[Citation24] Foams were formed by whipping 300 mL of sample dispersion (1.65 wt% WPC and 0.33 wt% pectin) in a 3-L capacity double beater mixer bowl (Breville SHM1, COMET, UK) at room temperature. Each sample was whipped for 5 min whilst the rotational speed was set at 5 (maximum). The foaming ability and foam stability were then measured by the methods described below.
Foaming ability
The foaming ability was estimated by measuring the volume (mL) of foam formed by each sample immediately after whipping was stopped. The method was simple and suitable for the purpose, and it was designed in our laboratory to serve the purpose. To enable estimation of foam volume, a standard curve of volume of sample versus measured height of sample in the bowl was plotted. This was carried out by filling up the whipping bowl with a varied volume of sample suspension and then measuring the height of fluid. The height of suspension and the corresponding volume were then used to plot a standard curve (data not shown) which was fitted to a linear equation (Eq. 1). Foam volume was then calculated by measuring the height of foam in the bowl and using Eq. (1) to calculate the volume.
Foam stability
The foam stability was determined at room temperature by the drainage method described by Phillips et al.[Citation24] To ensure continuous measurement of liquid drainage from foams, a 0.6-cm hole was drilled 5.0 cm from the centre of the bottom of the whipping bowl. The hole was sealed during whipping with a rubber stopper. After 5 min of whipping, the rubber stopper was removed and the bowl was seated on a glass funnel placed in a ring stand at a 30° angle above a measuring cylinder so that the drainage hole was at the lowest point. The drained liquid was collected into the measuring cylinder and the cumulative increase in liquid volume was continuously measured and recorded. The time taken for the foam to drain 50% (i.e., 150 mL) of its initial volume (i.e., before whipping) was used as a measure of foam stability (i.e., the half-life of the foam).
Preparation of emulsions
Oil-in-water emulsions containing 30% sunflower oil (w/w) and 6% (w/w) WPP (ratio 5% WPC: 1% pectin) were prepared. Sunflower oil was purchased from Tesco Supermarkets (Edinburgh, United Kingdom). The emulsions were made by premixing the oil and WPP solution using an Ultra Turrex high shear mixer, followed by high pressure homogenisation with an APV systems homogeniser (Model APV 1000, Albertslund, Denmark) using a single stage homogenisation at 200 bar pressure with continuous recirculation for 15 min.[Citation14,Citation20]
Emulsifying ability
The emulsifying ability was determined by measuring the oil droplet size in an emulsion made with the WPC and WPP samples. Oil droplet size (d4,3) and the droplet size distribution were measured using a Malvern Mastersizer 2000 (Malvern Instruments, Worcestershire, UK) within 1 min of making the emulsion. The particle refractive index, solvent (water) refractive index, and the assumed absorbance were 1.47, 1.33, and 0, respectively. The d4,3 was chosen to express the average particle size because this is more sensitive to any small populations of large droplets that may be formed during homogenisation.[Citation14,Citation20]
Emulsion stability
The emulsion stability of each sample was observed over a 24-week (6 months) period by following changes in the particle size distribution and serum separation due to creaming. Sodium azide (Sigma Aldrich, UK) was added to each emulsion sample to prevent microbial growth or spoilage during the storage period.
For particle size analysis, the emulsions were left to stand in transparent plastic bottles at room temperature (25°C) for 24 weeks. The droplet size (d4,3) and particle size distribution were measured using a Malvern Mastersizer at time = zero (the day samples were made and within 30 min of manufacture) and after 1, 2, 3, 5, 10, 18, and 24 weeks of storage. For measurement of creaming stability, the emulsion samples were stored in 15 mL plastic tubes (with added sodium azide) at 25°C for 24 weeks and the serum fraction that separated over the storage period was measured (in mm) at various times over this period. The change in d4,3 and cream height were found to be approximately linear over time and the slope of the plots of d4,3 or cream height versus time was used as an indicator of the rate of emulsion instability.
Statistical analysis
SPSS (version 22.0) was used to do a one-way ANOVA and Duncan multiple range test to establish significant differences (p < 0.05) between results. All tests were replicated three times.
Results and discussion
Particle size
Based on the methods used to make the WPP particles and the descriptions of electrostatic complex particle structure by Jones and McClements,[Citation3] we can hypothesize about the structure of the WPP particles in this study. Heating of WPC leads to denaturation and aggregation[Citation25] whilst prolonged heating of pectin is known to lead to depolymerisation.[Citation26–Citation28] Since in WPP01 the protein and pectin are mixed and heated together, we expect these to gel and to form a homogeneous spherical particle. We envisage that the WPP01 particle has a composite mixed structure of aggregated WPC intermixed with degraded pectin. Matalanis et al.[Citation29] have described such a structure as a “heterogeneous continuous” biopolymer complex and are similar to the “type 2” particles described in the work of Jones et al.[Citation12] For WPP02 and WPP03, the protein is heated first and then mixed with pectin after heating. The pectin will form a layer on the surface of the protein particles through electrostatic interaction to form heterogeneous core–shell particles with a core of denatured WPC and an outer shell of pectin. In WPP03, only the protein is heated and the WPC aggregates become coated with an extensive layer of oppositely charged pectin molecules giving particles larger than that formed by WPC alone (. 1 and ). In WPP02, WPC and high methoxy pectin are unstable under heating conditions. When heated, the individual proteins in WPC denature and aggregate to form protein particle, and this becomes coated with degraded pectin molecules that are smaller than the unheated pectin and so WPP02 particles are smaller than those of WPP03 ( and ). Various authors[Citation3,Citation29,Citation30] have described a similar structure as a “heterogeneous core–shell” biopolymer complex that is the same as the “type 1” particles described by Jones et al.[Citation12] With WPP04 and WPP05, the protein is unheated and will form a macromolecular complex with the unheated (WPP04) or heated pectin (WPP05). For WPP04 and WPP05, protein molecules (and pectin [WPP04]) were not heated, so extensive aggregations of protein will not occur. However, we hypothesize that the aggregation mechanism in these samples can be explained in terms of complex coacervation. Various researchers[Citation9,Citation16,Citation31,Citation32] describe complex coacervation as a spontaneous separation of a biopolymer system in which one phase is rich in the two biopolymers and the other is depleted of the two biopolymers. It is likely that the aggregation mechanism of the two samples was similar except for the fact that in WPP05, the pectin was heated and this would cause structural change in the pectin, whilst WPP04 was unheated. In both WPP04 and WPP05, we would expect the oppositely charged protein and pectin to associate through electrostatic interactions, with smaller individual protein molecules attached to the larger pectin chains. WPP04 and WPP05 particles are of a similar size which may indicate that they are formed through interaction of several protein and pectin molecules, which may be held together through the WPC molecules acting as a “bridge” between the pectin molecules. The heated WPC (WPC-H) will also form spherical particles, but these will be homogeneous as they are only composed of WPC. The WPC and WPP particles form a range of particle sizes which can be described by a particles size distribution. The distribution of particle size for both native and WPC-H is shown in and and indicates a broad range of particles present naturally or formed during heating. For the native unheated WPC (), these aggregates formed as a consequence of the heat applied during processing of the powder.[Citation33] The particle size distributions for WPP01–WPP03 (–) reveal that the particles formed under these conditions have a lower degree of polydispersity than the other particles. It also reveals that some WPP01 () particles were larger than in WPP02 () and WPP03 (), but the average was reduced by a population of smaller particles.
Figure 1. Particle size distribution of whey protein–pectin complexes (WPP) used in the foaming experiments measured by the Malvern Zetasizer. Whey protein–pectin samples have a protein concentration of 0.33% (w/w) pectin and 1.65% (w/w) WPC (protein–pectin ratio of 5:1). The concentration of WPC-only samples is 1.65 wt% and pectin is 0.33 wt%. WPP samples are prepared according to the methods given in .
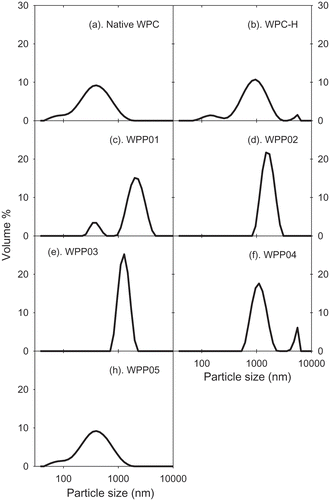
Figure 2. Z-average particle size (µm) of WPP particles used in foaming experiments as measured with the Malvern Zetasizer. Whey protein–pectin samples have a protein concentration of 0.33% (w/w) pectin and 1.65% (w/w) WPC (protein–pectin ratio of 5:1). The concentration of WPC-only samples is 1.65 wt%. WPP samples are prepared according to the methods given in .
Foaming properties
Foaming ability
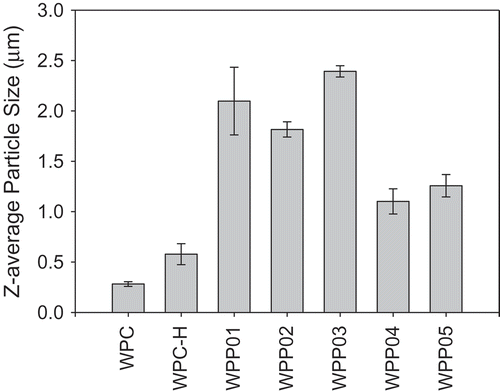
The ability of WPP particles, native and WPC-H to form and stabilise foams was assessed. All WPP samples showed a highly significant increase in foaming ability over the native and WPC-H samples. Within the set of WPP samples, all samples showed significant difference (p < 0.05) in foaming ability except for WPP01 and WPP02 (). shows that sample WPP04 had the highest foaming ability (2470 mL), followed by sample WPP05 (2307 mL), while the WPC-H had the lowest foaming ability (1160 mL).
Table 2. The foaming ability and foam stability of whey protein and protein–pectin complexes.
Based on the results above, there was no apparent correlation between the WPP particle size and foaming ability. This result was in contrast with the relationship observed between whey protein aggregate size and foaming ability by other researchers.[Citation34–Citation36] For example, Rullier et al.[Citation36] report that foams made with β-lactoglobulin aggregates had a lower foaming ability and foam stability than those made with the native protein. Furthermore, foaming ability and foam stability decreased with increasing aggregates size in contrast with our own results. However, when native protein was also present in the foaming solution as well as protein aggregates, more stable foams were formed than for the native protein alone. To explain this, Rullier et al.[Citation36] believe that the lower surface activity of the protein aggregates means they are not able to form fine air bubbles in foams, and the larger bubbles are less stable. However, if sufficient native protein is present, this can form fine foam bubbles which are stabilised more efficiently by the large protein aggregates. Various researchers[Citation37–Citation40] explain that foaming ability is guided by the surface tension and the rate of diffusion of particles onto the air–water interface. Particles that diffuse rapidly to the air–water interface and are able to reduce surface tension rapidly will stabilise the air bubbles in foam more quickly and will give rise to smaller bubbles and a greater foam volume. So, a possible explanation for the correlation between increasing particle size and decreasing foaming ability is that the surface tension at the air–water interface is lower when smaller particles are adsorbed than for bigger particles (which also diffuse more slowly to the interface). The results of Rullier et al.[Citation36] confirm this as they have measured surface tension for β-lactoglobulin aggregates and have found that the larger the aggregates, the slower the rate of decrease of surface tension and the higher the final equilibrium surface tension. The differences between our foaming ability results for protein–pectin particles and those observed by others for protein-only aggregates suggest that the composition and structure of the particles plays a more important role in WPP foaming properties than does particle size. The differences observed between complex particles (WPP) and whey protein-only samples (WPC) are related to the presence or absence of pectin, and it is likely that the increased viscosity caused by pectin is a major contributor to foaming ability. The increased viscosity of the aqueous phase in pectin containing foams helps to trap air bubbles and reduce bubble coalescence, thus leading to a smaller average bubble size. However, clearly it is not only the presence or absence of pectin that is important, otherwise all pectin containing samples would have the same foaming properties. The state of the pectin and how it interacts with the protein in the WPP aggregates is also important.
Foam stability
The foam stability is a measure of the time it takes for the foam bubbles to burst or rupture. The time taken for this to occur depends on the nature of the stabilising particles at the air–water interface. shows significant differences in foam stability among samples (p < 0.05). WPP samples produced significantly more stable foams than the native and WPC-H. Samples WPP04 and WPP05 formed the most stable foams with half-lives of ≈17 and ≈14 min, respectively (), whilst WPC-H has the lowest foam stability with a half-life of 20 s. The foam stability also did not show any correlation or dependence on the particle size. The latter is probably a result of the reduced surface activity of the aggregated proteins mentioned in the discussion of foaming ability. Although the aggregated WPP samples (WPP01, WPP02, and WPP03) are likely to have a reduced ability to adsorb at the air–water interface, their larger size will allow them to form thicker more dense adsorbed layers at the air bubble interface. The adsorbed layer in these systems provides a greater stability to the bubbles against coalescence.
The different types of WPP particle exhibited differing foaming ability and foam stability. The key observations in this respect are as follows: (1) the presence of pectin improves foaming ability and foam stability; (2) the molecular complexes (WPP04 and WPP05) showed a statistically significant higher foaming ability and foam stability than other WPP particles; and (3) the homogeneous spherical WPP01 particles had a substantially lower foaming ability than core–shell particles of WPP02, indicating that where the pectin is located within the aggregate structure is highly important to functionality.
It is not unsurprising that the presence of pectin improves foaming properties as this will increase the aqueous phase viscosity, a factor known to increase foaming ability and foam stability.[Citation41] However, pectin was present in all WPP particles, so this cannot alone explain the differences between the foaming properties of the WPP aggregates. Simply adding pectin (heated or unheated) to native unheated WPC gave macromolecular complexes with the highest foaming ability and foam stability. In these systems, it is possible that the complex formed acted in a similar way to some naturally occurring protein-containing polysaccharides such as gum arabic.[Citation42,Citation43] That is the protein inferred some hydrophobic character on the pectin molecule and allowed it to adsorb to the air–water interface to allow formation of foam bubbles. At the same time, the pectin part of the complex located in the aqueous phase and increased the viscosity in the foam plateau borders which reduced the rate of foam drainage and increased foam stability.
We can also compare the state of the pectin in WPP04 and WPP05 to the pectin found in WPP01 and WPP02. In these, the pectin was either incorporated into the aggregate with the protein (WPP01) or sat on the surface of the protein aggregate (WPP02). In both cases, the pectin was likely to be in a state where it has a reduced interaction with the water phase and consequently a reduced effect on the viscosity. Thus, we might explain the reduced foaming properties of WPP01 and WPP02 compared to WPP04 and WPP05 as being due to a reduced effect of the pectin on aqueous phase drainage in the foam. If this interpretation is correct, then clearly the state of the pectin in WPP01 is such that its effect on aqueous phase viscosity is less than that of pectin in WPP02 as the foam stability of WPP01 particles is much less than that of WPP02.
Emulsifying ability
The mean particle size for native WPC, WPC-H, and WPP particles and mean particle size (d4,3) for emulsions made with these are shown in and . The largest protein–pectin particles were formed for WPP03, the heterogeneous core–shell particle, where we hypothesize that the particle was made up of a core of aggregated protein, with a layer of pectin electrostatically bound to the surface. The smallest particles were found in the unheated WPC solution (). The order of increasing WPP particle size was WPC < WPC-H < WPP05 < WPP04 < WPP02 < WPP01 < WPP03. If we compare the relative protein–pectin particle sizes for the emulsion experiments with those used in the foaming experiments, we observe a strong linear correlation (Supplementary material Fig. S4). This gives us confidence that the mechanism of formation and the structure of the two sets of particles are the same, albeit with larger particle sizes at the higher protein + pectin concentrations used to make emulsions.
Figure 3. Z-average particle size (µm) of WPP particles used in emulsifying experiments as measured with the Malvern Zetasizer. Whey protein–pectin samples have a concentration of 1.0% (w/w) pectin and 5.0% (w/w) WPC (protein–pectin ratio of 5:1). The concentration of WPC only samples is 5.0 wt%. WPP samples are prepared according to the methods given in .
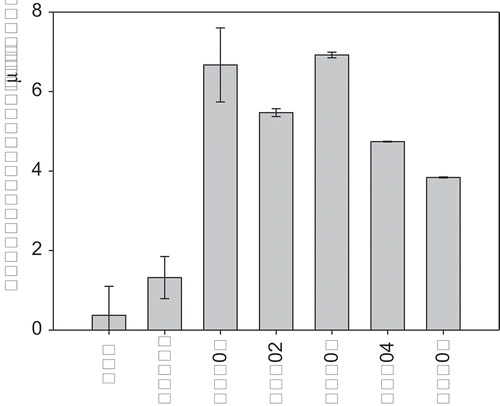
Figure 4. Mean particle size d4,3 (µm) of emulsion droplets made using WPP particles.
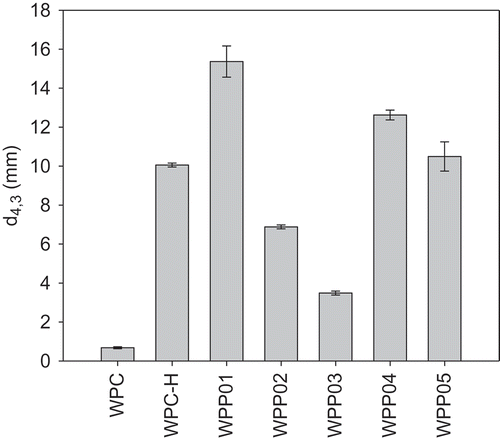
In , the emulsifying ability of the WPC and WPP particles is presented expressed as the d4,3 of the emulsion droplets, where a smaller particle size indicates a better emulsifier. The order of increasing emulsifying ability is WPP01 < WPP04 < WPP05 < WPC-H < WPP02 < WPP03 < WPC. If the emulsifying ability is plotted against the size of the WPC–pectin particles, no correlation is observed between the two (data not shown). This suggests that the emulsifying ability is independent of the particle size of the emulsifiers, a finding in agreement with those of Ghosh and Bandyopadhyay.[Citation22] However, it is clear that the conformation of the protein molecules within the complex does have an impact on the emulsifying ability of all the samples. The WPC was the best emulsifier, which is perhaps not unexpected. The proteins in WPC powder are largely un-aggregated, will adsorb readily to the oil droplet surface, and are accepted as being good at stabilising the interface. A similar response has been observed in dissociated caseins which were significantly better emulsifiers than large aggregates of proteins, such as micelle fragments of milk caseins found in skim milk powder and milk protein concentrate.[Citation44,Citation45] This was because the aggregated proteins do not spread as easily at the oil–water interface and are thus less efficient at stabilising oil droplets. The adsorption of protein at an oil–water interface is followed by the unfolding of the protein, and this unfolding helps in promoting the interactions and reduction of surface tension.[Citation46] The surface denaturation process is not as efficient in aggregated proteins, as their structure is held together by intra-molecular interactions that oppose surface unfolding. This explains why WPC is the best emulsifying sample, but WPC-H which has been heated and aggregated has reduced emulsifying ability. When pectin is present in the WPP particles, however, the emulsifying ability is modified depending on how the pectin interacts with the WPC.
The poorest emulsifier of the WPP particles was WPP01 where the protein and pectin were heated together and we believe form a homogeneous spherical particle where the protein and pectin are dispersed evenly through the particle. Here, the surface of the particle is likely to be a mixture of protein and pectin, and clearly the presence of the pectin at the surface interferes with the ability of the particles to adsorb and stabilize the droplet surface. WPP02 and WPP03, where the protein was heated separately, and the pectin was added after (either heated pectin, WPP02, or unheated pectin, WPP03), form a different structure where the pectin forms a layer on the surface of aggregated protein particles through electrostatic interaction. These WPP particles were considerably more efficient as emulsifiers than WPP01 and WPC-H, but not as good as WPC. There was also an effect of pectin treatment observed in WPP02 and WPP03. The WPP03 particle, which contained unheated pectin on the surface of the particles, was a significantly better emulsifying agent than WPP02. Heating of the pectin is believed to lead to degradation via either a β-elimination reaction where atoms or groups are lost from adjacent atoms joined by a single (σ) bond, leading to formation of a double (π) bond,[Citation28] or through acid hydrolysis if the pH is low. Thus, it is conceivable that WPP02 had smaller pectin fragments at the surface of the aggregated protein core than were found for the WPP03 complexes. It is possible that this affected the hydrophobicity of the surface of WPP particle, possibly through greater coverage of the surface by the smaller pectin fragments, which made the WPP particle less hydrophobic.
An interesting observation was made when comparing the size of the WPP complexes with the size of the emulsions made from them. For all emulsions, with the exception of WPP02 and WPP03, the emulsion droplets were significantly larger than the WPP particles. The average particle size of samples WPP02 and WPP03 was 6.7 and 5.5 μm, respectively, whilst the average emulsion droplet sizes were 6.9 and 3.5 µm, respectively. This suggests that the WPP02 and WPP03 aggregates cannot be the primary emulsifiers/stabilizers for the emulsion, since they would be too large to fit on the droplet interface. A possible explanation for this effect could be that the WPP02 and WPP03 particles, which were made from WPC-H and unheated pectin, were unstable under the high shear conditions of the homogenizer and broke up into smaller particles. The presence of smaller WPP aggregate particles would also explain the relatively small droplet size of the emulsions compared to other WPP samples since smaller aggregates might be expected to be better emulsifiers. An alternative explanation could be that the relatively large protein particles in WPP02 and WPP03 contributed to the scattering of light when the particle size was measured and that the average particle size measured for the emulsion droplets made with WPP02 and WPP03 contained a significant contribution from the protein particles themselves.
Emulsion stability
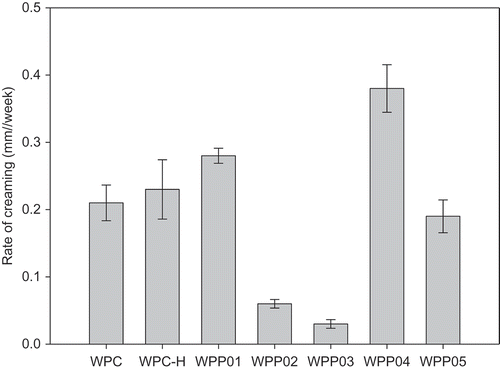
The long-term stability of the emulsions made with WPC and WPP was studied over a period of 24 weeks at room temperature (25°C) by following the change in mean particle size (d4,3) and height of cream layer formed. These were measured at weekly intervals and the rate of change of d4,3 and rate of creaming determined from the slope of plots of these as a function of time. Plots of these two emulsion stability measures are presented in and . The most stable emulsions were formed by the WPP02 and WPP03 complexes, for both change in d4,3 with time () and creaming stability (). These are the two heterogeneous core–shell particles (). These two WPP particles performed well in terms of foam stability as well () but did not give the highest foam stability. If we look at the least stable emulsions, then WPC had the highest rate of change of d4,3 followed by WPP04, whilst for creaming, WPP04 was the least stable with WPC more stable to creaming. WPC was anomalous when comparing rate of change of d4,3 with creaming rate. For all other WPP particles and WPC-H, there was a linear correlation between the two measures of emulsion stability, except for WPC emulsions. A correlation plot is shown in the Supplementary material (Fig. S5). In this plot, the emulsions formed from native WPC appear as an outlier point not close to the best-fit line. The order of increasing stability for the emulsions made with WPP particles (excluding WPC) was WPP04 > WPP01 > WPC-H > WPP05 > WPP02 > WPP03 for both creaming and change in d4,3.
Figure 5. Stability of emulsions made with WPP particles measured as the rate of change of droplet size (d4,3 in µm/week).
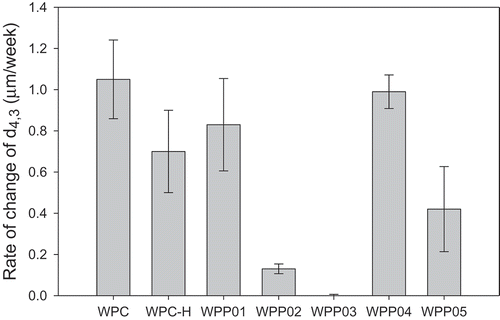
Figure 6. Stability of emulsions made with WPP particles measured as the rate of change of cream layer height (mm/week).
To explain the differences in emulsifying ability and emulsion stability, a number of factors must be considered. The different WPP structures and sizes will play a role, as might the presence of pectin. We have noted that aggregated proteins are known to be poorer emulsifiers than non-aggregated proteins.[Citation44] Euston and Hirst[Citation44] studied the emulsifying properties of aggregated proteins products (milk protein concentrate and skim milk powder) and compared them to native WPC and sodium caseinate. The aggregated protein products produced emulsions with significantly larger emulsion droplet than the non-aggregated proteins. At low protein emulsifier concentrations, the aggregated protein emulsions were also significantly less stable than those made with the non-aggregated proteins. However, as the protein concentration in the emulsions was increased, a point was reached where the stability of the aggregated protein emulsions increased rapidly and became greater than that of the non-aggregated proteins. The poor emulsifying properties of the aggregated proteins can be explained by their rigid, compact structure. The aggregated proteins are unable to unfold and spread at the oil droplet surface and therefore are unable to stabilize the droplets when they are small in the homogenizer. However, the aggregated proteins pack more densely at the oil–water interface, and once the adsorbed layer reaches a certain thickness, Euston and Hirst[Citation44] speculated that the effective density of the emulsion droplets (oil + protein layer) becomes great enough so that the density of the droplets becomes closer to that of the aqueous phase, and creaming reduces. We might expect a similar effect to be observed for our WPP complex particles, since they contained aggregated protein and polysaccharide. The particle size of the aggregates increased in the order WPC > WPP05 > WPP04 > WPP02 > WPP01 > WPP03 and we might expect creaming stability to decrease in this order based on the observations of Euston and Hirst.[Citation44] However, plotting creaming stability (and change in d4,3 with time) (Supplementary Figs. S6 and S7) against WPP particle size, we find that the emulsion stability characteristics of WPP particles were more complex. There was some indication of a correlation between particle size and emulsion stability, with in general larger WPP particle size leading to more stable emulsions. However, there were two exceptions to this, WPP01 and WPP04, where in both cases the emulsions were less stable than other emulsions containing WPP particles of a similar size. The reason for this lower stability was unclear. WPP01 was a homogeneous particle made when both WPC and pectin were heated together and were both incorporated into the particle. WPP04 on the other hand was a macromolecular complex formed when unheated WPC and pectin were mixed. With WPP04, it is possible that depletion flocculation was occurring due to the presence of the native pectin molecules. Depletion flocculation is a phenomenon during which the large pectin molecules are eliminated or excluded from the gap between two approaching droplets.[Citation47] This leads to an osmotic imbalance and a net force pushing the emulsion droplets together. Depletion flocculation in emulsions has been observed in the presence of un-adsorbed aggregated caseins[Citation44,Citation48] and with polysaccharides. This might explain why WPP05 emulsions were more stable than those made with WPP04, since the pectin in those complexes had been heated and was likely to be degraded, and thus smaller in molecular weight/size.
For WWP03, where unheated pectin was also present, we did not see a lower than anticipated emulsion stability because we expect the pectin not to be free in solution, but to be adsorbed to the surface of the WPC-H particles to form a core–shell WPP particle. An argument against the depletion flocculation explanation for the low stability of WPP04 emulsions is that depletion flocculation is reversible, and flocs would be expected to dissociate into individual droplets when dispersed in water during particle size analysis with the Mastersizer. However, clearly the Mastersizer detected large droplets in the emulsions. In addition, a depletion flocculation explanation for the low stability of WPP01 emulsions is also not compelling, as there is no clear reason why WPP01 particle would cause this and other particles of a similar size would not. An alternative explanation for the lower than expected stability of WPP01 emulsions is that we could be seeing the effects of a bridging type of flocculation. In emulsions where the protein emulsifier is present in too low a concentration to fully saturate the surface of the oil droplets, the protein can be shared between separate droplets to form a bridge that leads to a more permanent form of flocculation. These flocs are less stable to creaming and to coalescence than individual droplets. The fact that this occurred with WPP01 aggregates and no other aggregates may be because the surface of WPP01, particles might reasonably be expected to be more hydrophobic because it was not fully covered by a layer of hydrophilic pectin as would be expected in a core–shell particle (WPP02 and WPP03).
Conclusion
WPC–pectin electrostatic complexes can be made to adopt, spherical homogeneous particles, core–shell particles, or macromolecular complexes depending on how the protein and pectin are heated and mixed. This study provides evidence that the structure of WPC–pectin electrostatic complexes can be manipulated to alter foaming and emulsifying properties of their solutions and suggests that the structure of these particles can be tuned to give optimal foaming and emulsifying properties in food system applications. It is also possible that similar effects will be observed with thickening and gelation properties and work continues in this area. Our own work on controlling structure in WPC aggregates[Citation6,Citation7] has shown that control over solution viscosity can be achieved through controlled thermal aggregation so we are hopeful that similar effects can be achieved in the more complex binary aggregates of WPC and pectin. If this is achieved, it will open up the possibility of functional ingredients that can be more closely tailored to the functional requirements of a particular product or manufacturer.
LJFP_A_1396478_supplementary_figures.docx
Download MS Word (90.8 KB)Supplemental data
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Emerich, D. F.; Thanos, C. G. Target Nanoparticle-Based Drug Delivery and Diagnosis. J. Drug Target. 2007, 15, 163–183.
- Goldberg, M.; Langer, R.; Jia, X. Q. Nanostructured Materials for Applications in Drug Delivery and Tissue Engineering. J. Biomaterial Sci. 2007, 18, 241–268.
- Jones, O. G.; McClements, D. J. Functional Biopolymer Particles: Design, Fabrication, and Applications. Compr. Rev. Food Sci. Food Saf. 2010b, 9, 374–397.
- Janhoj, T.; Ipsen, R. Effect of Pre-Heat Treatment on the Functionality of Microparticulated Whey Protein in Acid Milk Gels. Milchwissenschaft 2006, 61, 131–134.
- Janhoj, T.; Peterson, C. B.; Frost, M. B.; Ipsen, R. Sensory and Rheological Characterization of Low-Fat Stirred Yogurt. J. Texture Stud. 2006, 37, 276–299.
- Zhang, Z.; Arrighi, V.; Campbell, L.; Lonchamp, J.; Euston, S. R. Properties of Partially Denatured Whey Protein Products: Formation and Characterisation of Structure. Food Hydrocolloid 2016a, 52, 95–105.
- Zhang, Z.; Arrighi, V.; Campbell, L.; Lonchamp, J.; Euston, S. R. Properties of Partially Denatured Whey Protein Products 2: Solution Flow Properties. Food Hydrocolloid 2016b, 56, 218–226.
- McClements, D. J.;. Non-Covalent Interactions between Proteins and Polysaccharides. Biotechnol. Adv. 2006, 24, 621–625.
- Turgeon, S. L.; Schmitt, C.; Sanchez, C. Protein-Polysaccharide Complexes and Coacervates. Curr. Opin. Colloid Interface Sci. 2007, 12, 166–178.
- Jones, O. G.; McClements, D. J. Biopolymer Nanoparticles from Heat-Treated Electrostatic Protein–Polysaccharide Complexes: Factors Affecting Particle Characteristics. J. Food Sci. 2010a, 75, 36–43.
- Jones, O. G.; McClements, D. J. Formation of Biopolymer Particles by Thermal Treatment of Beta-Lactoglobulin-Pectin Complexes. Food Hydrocolloid 2009, 23, 1312–1321.
- Jones, O. G.; Decker, E. A.; McClements, D. J. Comparison of Protein-Polysaccharide Nanoparticle Fabrication Methods: Impact of Biopolymer Complexation before and after Particle Formation. J. Colloid Interface Sci. 2010a, 344, 21–29.
- Jones, O. G.; Decker, E. A.; McClements, D. J. Thermal Analysis of β-lactoglobulin Complexes with Pectins or Carrageenan for Production of Stable Biopolymer Particles. Food Hydrocolloid 2010b, 24, 239–248.
- Tolstoguzov, V. B.;. Microstructural Elements and Their interactions-Ingredient Interactions: Aggregation and Phase Separation. In McClements, D. J., ed. Understanding and Controlling the Microstructure of Complex Foods; Cambridge: Woodhead, 2006; 185–206.
- De Kruif, C. G.; Tuinier, R. Polysaccharide Protein Interactions. Food Hydrocolloid 2001, 15, 555–563.
- Turgeon, S. L.; Beaulieu, M.; Schmitt, C.; Sanchez, C. Protein-Polysaccharide Interactions: Phase-Ordering Kinetics, Thermodynamic and Structural Aspects. Curr. Opin. Colloid Interface Sci. 2003, 8, 401–414.
- Hattori, T.; Hallberg, R.; Dubin, P. L. Roles of Electrostatic Interaction and Polymer Structure in the Binding of β-lactoglobulin to Anionic Polyelectrolytes: Measurement of Binding Constants by Frontal Analysis Continuous Capillary Electrophoresis. Langmuir 2000, 16, 9738–9743.
- Wang, X.; Lee, J.; Wang, Y. W.; Huang, Q. Composition and Rheological Properties of β-lactoglobulin/pectin Coacervates: Effects of Salt Concentration and Initial Protein/Polysaccharide Ratio. Biomacromolecules 2007, 8, 992–997.
- Benichou, A.; Aserin, A.; Garti, N. Protein-Polysaccharide Interactions for Stabilization of Food Emulsions. J. Dispers. Sci. Technol. 2002, 23, 93–123.
- McClements, D. J.;. Food Emulsions: Principles, Practice and Techniques, 2nd edn.; Boca Raton, Florida: CRC Press, 2005.
- McClements, D. J.;. Understanding and Controlling the Microstructure of Complex Foods; Woodhead: Abington, 2007.
- Ghosh, A. T.; Bandyopadhyay, P. Polysaccharide-Protein Interactions and Their Relevance in Food Colloids. In Karunaratne, D. N., ed. The Complex World of Polysaccharides; Croatia: InTech, 2012; 395–408.
- Bouaouina, H.; Desrumaux, A.; Loisel, C.; Legrand, J. Functional Properties of Whey Proteins as Affected by Dynamic High-Pressure Treatment. Int. Dairy J. 2006, 16, 275–284.
- Philips, L. G.; Haque, Z.; Kinsella, J. E. A Method for the Measurement of Foam Formation and Stability. J. Food Sci. 1987, 52, 1074–1077.
- De Wit, J. N.; Swinkels, G. A. A Differential Scanning Calorimetric Study of the Thermal Denaturation of Bovine Beta-Lactoglobulin. Thermal Behaviour at Temperatures up to 100°C. Biochimica Et Biophysica Acta 1980, 624, 40–50.
- Kelly, R.; Gudo, E. S.; Mitchell, J. R.; Harding, S. E. Some Observations on the Nature of Heated Mixtures of Bovine Serum with an Alginate and a Pectin. Carbohydr. Polymer. 1994, 23, 115–120.
- Morris, G. A.; Foster, T. J.; Harding, S. E. A Hydrodynamic Study of the Depolymerisation of a High Methoxyl Pectin at Elevated Temperatures. Carbohydr. Polymer. 2002, 48, 361–367.
- Diaz, J. V.; Anthon, G. E.; Barrett, D. M. Nonenzymatic Degradation of Citrus Pectin and Pectate during Prolonged Heating: Effects of pH, Temperature, and Degree of Methyl Esterification. J. Agric. Food Chem. 2007, 55, 5131−5136.
- Matalanis, A.; Jones, O. G.; McClements, D. J. Structured Biopolymer-Based Delivery Systems for Encapsulation, Protection, and Release of Lipophilic Compounds. Food Hydrocolloid 2011, 25, 1865–1880
- Abd el-Salam, M. H.; El-Shibiny, S. Formation and Potential Uses of Milk Proteins as Nano Delivery Vehicles for Nutraceuticals: A Review. Int. J. Dairy Technol. 2012, 65, 13–21.
- Bungenberg De Jong, H. G.;. Crystallisation-Coacervation-Flocculation. In Kruyt, H. R., ed. Colloid Science. Amsterdam: Elsevier, 1949; Vol. 2, 232.
- De Kruif, C. G.; Weinbreck, F.; De Vries, R. Complex Coacervation of Proteins and Anionic Polysaccharides. Curr. Opin. Colloid Interface Sci. 2004, 9, 340–349.
- Ryan, K. N.; Zhong, Q.; Foegeding, E. A. Use of Whey Protein Soluble Aggregates for Thermal Stability—A Hypothesis Paper. J. Food Sci. 2013, 78, R1105–R1115.
- Schmitt, C.; Bovay, C.; Rouvet, M.; Shojaei-Rami, S.; Kolodziejczyk, E. Whey Protein Soluble Aggregates from Heating with NaCl: Physicochemical, Interfacial, and Foaming Properties. Langmuir 2007, 23, 4155–4166.
- Unterhaslberger, G.; Schmitt, C.; Shojaei-Rami, S.; Sanchez, C. β-Lactoglobulin Aggregates from Heating with Charged Cosolutes: Formation, Characterization and Foaming. In Dickinson, E., Leser, M. E., eds. Food Colloids: Self Assembly and Material Science; Cambridge: Royal Society of Chemistry, 2007; 175–192.
- Rullier, B.; Novales, B.; Axelos, M. A. V. Effect of Protein Aggregates on Foaming Properties of β–Lactoglobulin. Colloid Surface A 2008, 330, 96–102.
- Borcherding, K.; Lorenzen, P. C.; Hoffman, W.; Schrader, K. Effect of Foaming Temperature and Varying Time/Temperature-Conditions of Pre-Heating on the Foaming Properties of Skimmed Milk. Int. Dairy J. 2008, 18, 349–358.
- Kamath, S.; Huppertz, T.; Houlihan, A. V.; Deeth, H. The Influence of Temperature on the Foaming of Milk. Int. Dairy J. 2008, 18, 994–1002.
- Huppertz, T.;. Foaming Properties of Milk: A Review of Influence of Composition and Processing. Int. J. Dairy Technol. 2010, 63, 477–488.
- Medrano, A.; Abirached, C.; Araujo, A. C.; Panizzolo, L. A.; Moyna, P.; Añón, M. C. Correlation of Average Hydrophobicity, Water/Air Interface Surface Rheological Properties and Foaming Properties of Proteins. Food Sci. Technol. Int. 2012, 18, 187–193.
- Von Klitzing, R.; Muller, H. J. Film Stability Control. Curr. Opin. Colloid Interface Sci. 2002, 7, 42–49.
- Dickinson, E.; Galazka, V. B.; Anderson, D. M. W. Emulsifying Behaviour of Gum Arabic. Part 1: Effect of the Nature of the Oil Phase on the Emulsion Droplet-Size Distribution. Carbohydr. Polymer. 1991a, 14, 373–383.
- Dickinson, E.; Galazka, V. B.; Anderson, D. M. W. Emulsifying Behaviour of Gum Arabic. Part 2: Effect of the Gum Molecular Weight on the Emulsion Droplet-Size Distribution. Carbohydr. Polymer. 1991b, 14, 385–392.
- Euston, S. R.; Hirst, R. L. Comparison of the Concentration-Dependent Emulsifying Properties of Protein Products Containing Aggregated and Non-Aggregated Milk Protein. Int. Dairy J. 2000, 9, 693–701.
- Euston, S. R.; Hirst, R. L. The Emulsifying Properties of Commercial Milk Protein Products in Simple Oil-in-Water Emulsions and in a Model Food System. J. Food Sci. 2000, 65, 934–940.
- Dickinson, E.; McClements, D. J. Advances in Food Colloids; Blackie: Glasgow; 1995, 81.
- Euston, S. R.; Finnigan, S. R.; Hirst, R. L. Aggregation Kinetics in Heated Whey Protein Stabilized Emulsions iii. Effect of Polysaccharide Stabilizers. Food Hydrocolloid 2002, 16, 499–505.
- Dickinson, E.; Golding, M.; Povey, M. J. W. Creaming and Flocculation of Oil-in-Water Emulsions Containing Sodium Caseinate. J. Colloid Interface Sci. 1997, 185, 515–529.