
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The most rampant and complex form of dementia is Alzheimer’s disease (AD) which is characterized by various cognitive deficits and personality abnormalities. AD is a neurodegenerative condition marked by the buildup of beta-amyloid peptide fragments and Tau protein in the form of tangles in brain neurons. The presence of β-amyloid peptide, Tau protein, oxidative stress, and an aggravated neuro-inflammatory response are all part of its pathophysiological pathway. Quite a number of invertebrates have been genetically modified such that they express human proteins that play a role or two in the pathogenesis of AD. Also, in order to create an animal model of AD, quite a number of substances have been investigated for their potential to cause cognitive dysfunctions. The following keywords were used to search and retrieve articles from reputable databases, including Scopus, Science Direct, Google Scholar and PubMed: “Alzheimer’s Disease,” “Tau protein,” “beta-secretase,” “Cognitive Impairments,” “Amyloid beta,” “phytochemicals and AD,” “neurofibrillary tangles” and “Neurotoxins that induce AD,” The aim of this review is to advance a better understanding of the functions and roles of Tau and amyloid precursor proteins in the pathogenesis of AD; and to offer updated and recent information on the use of plant chemicals in the treatment of AD. It is expedient to state that not all biochemical, cognitive, behavioral and histological disorders are recapitulable. Nevertheless, research into the etiology of this severely debilitating disease is being aided by experimental models of AD created by chemicals and bioengineered model organisms.
Introduction
Alzheimer’s disease (AD), the most common kind of age-related dementia, is marked by gradual fall of cells allied with the central nervous system, which ultimately causes reduction in cognitive function and leads to dementia. Alzheimer’s disease (AD) is recognized as a complex disorder influenced by multiple factors, encompassing both environmental and genetic components that contribute to its development and progression.[Citation1] Research has shown that oxidative stress plays a significant part in neurodegenerative illnesses like AD through the peroxidation of lipids of the neurons’ cell membranes.[Citation2] Due to brains’ low antioxidant capacity, consumption of about 20% of oxygen required for metabolic processes, the incapability of neurons to manufacture reduced glutathione, and higher concentration of polyunsaturated fatty acids; the brain and the entire central nervous system are understood to be principally susceptible to oxidative onslaught.[Citation3,Citation4] Alzheimer’s disease is distinguished by malfunctioning cholinergic systems, cognitive decline, memory deterioration, neuronal loss, and disturbances in behavior. The development of AD involves intricate mechanisms that disrupt the neurological processes associated with memory function. While some cases of AD can manifest in individuals younger than 65, the vast majority, accounting for over 90% of diagnoses, occur in individuals aged 65 and above.[Citation5] Extensive research is underway to investigate the neuropathological aspects of Alzheimer’s disease (AD), such as neuronal loss and dysfunction, neurofibrillary tangles and senile plaques. Despite these efforts, a complete understanding of the underlying pathogenic process remains elusive. Additionally, existing treatments have not demonstrated significant success in practical application.[Citation6] The primary hypotheses presented for the development of Alzheimer’s disease involve amyloid deposition, disruption of intracellular signaling, tau phosphorylation oxidative stress, inflammation and dysregulation of metal ions.[Citation7]
Metals like iron (Fe) and aluminum (Al), as well as mercury (Hg) are the substances attracting the most interest in AD.[Citation8] Due to its loosely bonded electron and capacity to exist in multiple valence states, iron is believed to confer the most significant pathologic role among these metals; as a catalyst for the formation of free radicals.[Citation9] The Fenton reaction, which leads to the liberation of hydroxyl radical (OH*) when iron reacts with hydrogen peroxide (H2O2), is also believed to be the mechanism by which iron brings about its harmful effect, but the superoxide reaction, which produces iron (III), can restore the divalent iron that took part in the Fenton reaction.[Citation10] Excessive production of ROS can cause lipid peroxidation by directly attacking the cell membrane’s polyunsaturated fatty acids.
Around 46.8 million individuals worldwide had dementia as of 2015; AD is the main underlying origin of dementia.[Citation11] If there are no interventions, this value is anticipated to rise rapidly to no less than one hundred and thirty-one million by 2050.[Citation11,Citation12] Women have reportedly a longer life expectancy than males, in terms of prevalence between genders.[Citation13] As a result, women make up around two thirds of the elderly AD patients. According to studies, AD management cost no less than eight hundred million USD in 2016, and is estimated to increase to 1 trillion USD in 2018. AD does, in fact, have a direct economic burden on the world as a whole.[Citation14,Citation15]
Review methodology
The following databases PubMed, Scopus, Science Direct, and Google Scholar were searched for appropriate studies published on the phytochemicals with neuroprotective properties and their mechanisms of action; and the role of tau and amyloid proteins in Alzheimer’s disease. The search terms used were “Alzheimer’s disease/phytochemicals,” “tau protein/Alzheimer’s disease,” “Plant Chemicals,” “beta-secretase,” “amyloid-beta,” “Cognitive Impairments,” “Neurofibrillary Tangles,” “Neurotoxins/Alzheimer’s disease,” “Dietary Supplements,” “Neurophytotherapy.”
Preclinical research on AD and cell lines models in lab animals, phytochemical therapies (bioactive substances and essential oils), outcomes supported by histopathology and behavioral evidence, and English-language articles were the inclusion criteria. Summary reports of incomplete research and conference presentations, duplicate articles, trials involving homeopathic remedies, and experiments without a control group were all excluded.
Both the chemical formula and the botanical names of the plants were verified using online resources WorldFlora and ChemSpider, respectively (http://www.worldfloraonline.org/; http://www.chemspider.com/). The most pertinent information regarding the neuroprotective effects of phytochemicals and the role of tau and amyloid proteins in AD has been condensed into set of tables and images.
Causes of AD
Although the precise causes of AD are yet unknown but there are many theories about its pathogenesis. Some theories contend that proteins with flaws or less acetylcholine cause AD.[Citation16] There are three major theories that try to explain how the disease is triggered: First, the cholinergic hypothesis,[Citation17] which opined that decreased synthesis of the neurochemical acetylcholine causes AD, is the oldest and the foundation for the majority of currently utilized pharmacological therapy.[Citation17] The lack of effectiveness of treatments for acetylcholine insufficiency has made cholinergic hypothesis’ lacks popular acceptance.[Citation18] Several genes have been recognized as potential menace factors for AD, despite the fact that the disease does not normally appear to be inherited. Regardless of the precise origin, Alzheimer’s leads to the loss of neurons in the brain’s memory centers (often the temporal and parietal lobes), which culminates in memory loss. This neuronal loss is followed by an upsurge in beta-amyloid plaque and tangles in the brains of Alzheimer’s patients.[Citation19]
Oxidation stress is a major influence in the progression of the disorder.[Citation20] Due to their low antioxidant capacity, the brain and the entire CNS are principally susceptible to oxidative menace. The brain is prone to lipid peroxidation caused by free radicals because it has a high concentration of polyunsaturated fatty acids. Additionally, neurons rely on nearby astrocyte cells for the production of usable glutathione precursors because they are unable to produce glutathione, an essential constituent of aerobic cell antioxidant system.[Citation4,Citation21]
Stages of ad development
The four stages of the disease’s progression are marked by escalating patterns of cognitive and functional deficits. These include pre-dementia, early, moderate, and late stages of dementia.
Pre-dementia stage of AD
The initial symptoms are frequently misdiagnosed as stress or aging-related.[Citation22] A thorough neuropsychological assessment can detect mild cognitive anomalies up to 8 years before an individual reaches the clinical conditions for the diagnosis of AD.[Citation23] These early signs can interfere with even the most difficult daily duties.[Citation23] The most conspicuous impairment is memory forfeiture, which shows itself as issues recalling recently taught information and a lack of capacity to learn new material.[Citation24] The initial stages of this disease can also be characterized by subtle difficulties with devotion, scheduling, and abstract thinking, as well as impairments in the memory that has to do with “meanings” and “concept relationships;” which is otherwise known as semantic memory.[Citation24] At this stage, apathy can be seen, and it will continue to be the disease’s most enduring neuropsychiatric symptom.[Citation25] Slight intellectual impairment has also been used to describe the disease’s preclinical stage,[Citation26] but it is debatable whether this word refers to a new diagnostic stage or the early stages of AD.[Citation27]
Early phase of AD
The progressive loss of cognition and deterioration of memory in persons living with AD eventually results in a conclusive diagnosis. Memory issues are less common than challenges with linguistic, executive processes, awareness (otherwise called agnosia), or movement accomplishment (otherwise called apraxia) among a tiny percentage of people.[Citation28] Not all memory functions are adversely affected by AD. Episodic memory, semiotic memory, and implicit memory – the body’s recall of how to perform actions, like using a fork to eat – are all less influenced by the change than new information or memories.[Citation29] The fundamental characteristics of language impairment are a decreasing lexis eloquence and word articulacy, which result in an overall depletion of spoken and philological written ability.[Citation30] At this stage, the Alzheimer’s patient typically has adequate basic communication skills. Some movement synchronization and planning issues may be present while executing fine motor tasks like writing, sketching, or dressing, although they are typically not observed.[Citation30] People with AD can frequently still complete many chores on their own as the condition develops, although they may require aid or monitoring and evaluation with the most cognitively demanding tasks.[Citation30]
Moderate phase in AD
Progressive decline eventually makes it difficult for subjects to maintain their independence, leaving them unable to carry out the majority of basic daily tasks.[Citation30] Speech difficulties arise from a lack of vocabulary recall, which frequently results in incorrect word substitutions (paraphasias). Additionally, reading and writing abilities deteriorate over time.[Citation30,Citation31] As time goes on and AD worsens, complex motor processes become less coherent, which raises the chance of falling.[Citation30] Memory issues deteriorate during this stage, and the person might not be able to identify close family members. Long-term memory that was previously uncompromised becomes impaired. More people have behavioral and neuropsychiatric anomalies. Wandering, irritation, and labile affect are common symptoms that can cause sobbing, irrational anger, or reluctance to caregiving.[Citation30] The percentage of patients who have illusionary misidentifications and other delusional symptoms is around 30%.[Citation32] Additionally, subjects lose awareness of their limitations and disease process (anosognosia).[Citation32] Patients can get urinary incontinence.[Citation32] Moving the patient to long-term facilities where he would be given adequate care, might ease the burden these symptoms cause for family members and caregivers.[Citation32]
Advanced phase in AD
In the final stage of the illness, the patient is totally dependent on the carers.[Citation33] Simple sentences or even single words are used more frequently, eventually resulting in speech loss.[Citation34] Despite their inability to speak, patients may frequently understand and respond to emotional cues.[Citation34] Despite the possibility of aggression, extreme indifference and fatigue are considerably more frequent outcomes. Patients eventually will not be able to complete even the most straightforward tasks without help.[Citation34] They become bedbound and do not have the capacity to self-feed as their muscle mass and movement decline. As a fatal illness, AD usually results in an external cause of death, such as pneumonia or an infection of pressure ulcers.[Citation35]
Tau protein in AD
Deposits of proteins (that are not soluble) in cells that are associated with neuromuscular systems is a characteristic feature that is very common in virtually all forms of neurodegenerative disorders. Progressive nervous system deterioration is a hallmark of neurodegenerative illnesses, which can be spontaneous or genetic. A system of classification for neurodegenerative illnesses based on the accumulation of these proteins, is now possible because to advancements in molecular neuropathology. One protein, Tau linked with microtubules, plays a crucial role in the health of neurons however develops irreversible deposits in conditions now referred to as tauopathies.[Citation36,Citation37] The term “tauopathy” refers to a group of more than 20 clinicopathological conditions, the most prevalent of which being Alzheimer disease, as well as post-encephalitic parkinsonism (PEP), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick’s disease. The array of these illnesses shares significant clinical, pathological, biochemical, and genetic commonalities that have advanced our understanding of the causes of neurodegeneration and Tau.[Citation38,Citation39]
The neurodegenerative disease known as tauopathy is characterized by the abnormal accumulation of tau protein in the form of neurofibrillary or sometimes gliofibrillary tangles inside the brain of human. The tau which is a microtubule-associated protein is excessively phosphorylated to create tangles, which causes the protein to separate from microtubules and aggregate into an insoluble state.[Citation40] Paired helical filaments is another name for these tau protein aggregates that have been hyperphosphorylated. Alois Alzheimer was the first to identify this neurofibrillary form of tangles in an individual living with AD, which was regarded as a secondary Tauopathy.[Citation41] Frontotemporal dementias that run in families have mutations in the gene that encode tau itself, showing that tau abnormalities has the ability of causing neurodegeneration directly.[Citation42] This theory is supported by the fact that the production of both wild-type and mutant forms of human tau in the nervous system of the Drosophila results in a neurodegenerative phenotype that mimics several traits of human tauopathies.[Citation43] Tau regulates microtubule stability in two different ways. The first is based on the extent to which tau has been phosphorylated, with a rise in phosphorylation favoring the microtubules’ depolymerization. Secondly, microtubule stability is regulated by tau through Tau proteins’ isoforms. As a result of alternative splicing of exons two, three, and ten, there are six isoforms.[Citation44] The carboxy-terminal region of the protein has three isoforms with three repeated domains that has the ability of binding to microtubules (3 R) and three isoforms with four domains with which it binds microtubule (4 R). Microtubule stabilization is enhanced by 4 R tau isoforms. Tau proteins indirectly regulate the stability of microtubules, which in turn regulates the movement of cargo-vesicles inside neurons.
The tau protein, which has a molecular weight that ranges from 45 kilodalton to 45 kilodalton and is an essential part of the neuronal cytoskeleton, promotes microtubule assembly in healthy brain tissue. A mixture of protein kinases and phosphatases carefully controls microtubule assembly by balancing tau phosphorylation level.[Citation45] The most prevalent pathology of tau is observed in AD, although it also appears in other conditions like frontotemporal dementias and Parkinson’s disease.[Citation46] Tau is hyperphosphorylated in the AD brain, which results in abnormal secondary structures and subsequent loss of function, which culminates to a diminished capacity to attach effectively to microtubules and to encourage their formation.[Citation47] Other notable cytopathological features seen in AD brain sections include the aberrant translocation of tau from axonal microtubules to neuropil thread inclusions, dendritic processes, where tau clumps and accumulates.[Citation48]
Following initial synthesis as a single chain polypeptide, posttranslational changes are made to the tau protein that change its shape and encourage tau dimerization in an antiparallel fashion.[Citation49] After becoming tau oligomers, stable tau dimers combine to become protomers, which are the building blocks of filaments. When two protomers are twisted around one another with an 80 nm crossover repetition, they create filaments which are paired helix (PHFs), which are a sign of the pathophysiology of AD neuronal cells.[Citation50] Finally, neurofibrillary tangles (NFTs) are formed via PHF assembly and may be seen under a microscope.[Citation51] Normal tau is sequestered by hyperphosphorylated tau along with other proteins associated with the microtubule of neurons e.g. MAP1A, MAP1B and MAP2, which further interrupt microtubules stability, the cytoskeleton of the axon, and transport, ultimately leading to injured neurons.[Citation52] Tau oligomers are liberated into the extracellular compartment after death of neurons, which stimulates the microglia and thus causing further, gradual degeneration of bystander neurons.[Citation53] It has been hypothesized that tau disease is triggered when there is increase in the activity of protein kinase or decrease in the activity of protein phosphatase or both.[Citation54] It has been established that the primary kinases responsible for tau phosphorylation are MAP-kinase, GSK-3, and/or Cdk5. But not all instances of tau phosphorylation in AD can be credited to these kinases.[Citation55]
There is significant debate concerning how tau causes its neuronal toxicity.[Citation56] Numerous degenerative signals, including aggregation of amyloid-beta, cholesterol levels in neural rafts, iron overload, low density lipoprotein species, homocysteine, free radicals have been proposed to trigger the innate immune response.[Citation57] For instance, microglial activation triggers the liberation of pro-inflammatory cytokines that alter behaviors of neurons through abnormal cascades of biochemical reactions, ultimately promoting the hyperphosphorylation of tau protein.[Citation57] Tau, however, appears to be essential for amyloid-beta-induced neurotoxicity in a variety of cellular and transgenic animal models.[Citation57] For instance, tau deficient mice’s cultured hippocampus neurons are resistant to Amyloid-beta disease.[Citation58] Furthermore, tau’s requirement for prefibrillar amyloid-beta-induced microtubule disassembly has been shown by the silencing of tau by siRNA in cultured neurons obtained from wild-type mouse’ hippocampus. Furthermore, it was shown that in AD mouse; triple transgenic, with plaques and tangles, a decrease in soluble amyloid-beta and tau but not amyloid-beta alone induces cognitive deterioration.[Citation59] These findings imply that tau accumulation, even though amyloid-beta is the primary initiator, is crucial to neurodegeneration. Finally, inhibition of endogenic tau protein alleviated amyloid-beta-dependent water maze learning and memory deficits but not rectifying the pathology associated with amyloid in the AD-like genetically modified that expresses amyloid precursor protein (human type) with family mutations.[Citation59] When taken as a whole, these results point to a connection between amyloid-beta and tau as the two factors driving brain abnormalities and clinical symptom presentations. When compared to sporadic AD patients receiving alternative therapy, those treated with methylene blue chloride (MTC) decrease in the deterioration of cognition, suggesting that tau is the primary cause of cognitive abnormalities.[Citation60] More research is required to determine amyloid-beta dependent’s precise function in signal transduction cascades linked to pathogenic tau alterations and its impact to the development of neuronal death.[Citation58]
Phosphorylation of the phosphoprotein tau controls how it binds to microtubules. Tau is hyperphosphorylated in AD, as shown in . Neurons from AD patients have excessively phosphorylated tau proteins that are usually in the form of pair helical/straight filaments (PHF/SF) and soluble species. In situ, oddly hyperphosphorylated tau from AD patients’ brains depolymerizes microtubules and sequesters normal tau, MAP1, and MAP2 from them.[Citation61] Axoplasmic transport is supported by microtubules, and PHFs have taken the place of the microtubule system in the neurons of patients with AD, bearing tangles. Any cell’s health seems to be influenced by the movements of the microtubules, and tau controls these dynamics in a neuron in vivo and in vitro models. According to Alonso et al.,[Citation62] abnormally, excessively phosphorylated tau protein observed in the brains of the sufferer of AD, inhibits the formation of microtubules stimulated in vitro and in removed cells by normal tau and other microtubule-associated proteins.[Citation62] MAPs and regular tau are bound by AD P-tau. Due to this characteristic, hyperphosphorylated tau is a molecular agent that actively obstructs the microtubule system. Upon dephosphorylation, this characteristic is lost[Citation62]; yet, excessively phosphorylated tau does not bind tau or impair microtubules, indicating that the polymerized, excessively tau protein form is inactive. Similar outcomes were reported when Drosophila that expresses human tau on motor neurons was used as a model of neurodegeneration. By causing microtubule disruption, the scientists demonstrated that soluble hyperphosphorylated tau was hazardous.[Citation63] Pseudophosphorylation is a method frequently employed to investigate the significance of tau phosphorylation sites. In C. elegans model of tau hyperphosphorylation; nematode worms that are transgenic for tau pseudophosphorylated at various places had several dorsal and ventral discontinuities, with the dorsal cord appearing to be more seriously impacted. Genetically modified worms were conspicuously impacted more than the control worms and the wild-type tau bioengineered worms at all developmental stages. Neurite degeneration or inadequate neurite expansion during development are both indicated by discontinuities in the nerve cords. The observation that pseudophosphorylated tau eventually not result in the death of neurons showed that changed tau interfered with the mechanisms of development of axons intracellularly.[Citation64]
Figure 1. Kinases and phosphatases control how the tau protein stabilizes microtubules. The production of intractable cytoplasmic tau oligomers and protomers, which assemble to create protomers, are the results of abnormal hyperphosphorylation of tau proteins, which has disastrous effects on microtubule depolymerization. PHFs, which are made up of two protomers coiled around one another, come together to form neurofibrillary tangles (NFTs)[Citation50].
![Figure 1. Kinases and phosphatases control how the tau protein stabilizes microtubules. The production of intractable cytoplasmic tau oligomers and protomers, which assemble to create protomers, are the results of abnormal hyperphosphorylation of tau proteins, which has disastrous effects on microtubule depolymerization. PHFs, which are made up of two protomers coiled around one another, come together to form neurofibrillary tangles (NFTs)[Citation50].](/cms/asset/894471af-42aa-4724-b8cf-7bfe992b9bfb/ljfp_a_2243050_f0001_oc.jpg)
Steinhilb et al.[Citation65] created a genetically modified fly encoding the expression of tau proteins with 14 phosphorylation sites changed to alanine to prevent phosphorylation using Drosophila as a model. The absence of neurotoxicity indicates that tau phosphorylation is necessary for the creation of aberrant conformations as well as for neurotoxicity.[Citation65] However, these scientists demonstrated that multiple sites must be present in concert to exert tau-induced neurotoxicity and that a single site is not to blame for the toxic effect. Blard et al. identified many cytoskeleton-related elements, particularly those from the actin network, as specific modifiers of tau V337M-induced neurotoxicity by screening 1250 mutant Drosophila lines.[Citation66] This result revealed that early events in the pathological process leading to synaptic dysfunction in tau V337M disease could include disruption of the microtubule network in presynaptic nerve terminals.
Amyloid beta (aβ) in AD
It is generally known that the aging process is a key influence in the progression of amyloid plaques, whether or not there is a disease.[Citation67] These extracellular senile plaques, which are instigated by the anomalous cleavage of the transmembrane protein APP, are made up of accumulating A protein that aggregates as
-pleated sheets.[Citation68] Amyloid precursor protein is a protein found on cell surface; it has been hypothesized to play a crucial role in signal transduction, cell migration, and axonal elongation, under normal physiological circumstances.[Citation69] Additionally, it has been shown that the C terminus of amyloid precursor proteins are crucial for both expression of some certain genes and survival of neurons.[Citation70] These physiological mechanisms are operative only when this protein is hydrolyzed by different enzymes, such as intramembranous degradation by beta-site A
PP-cleaving enzyme (BACE1) to form the
C-terminal fragment (
CTF),[Citation71] which is then followed by gammasecretase to produce the small four kilodalton (kDa) amyloid-
(A
) peptides A
1–40 and A
1–42, as shown in .[Citation71]
Figure 2. The processing of APP by beta-site AβPPcleaving enzyme (BACE1) and the mechanisms of actions of phytochemicals in AD.
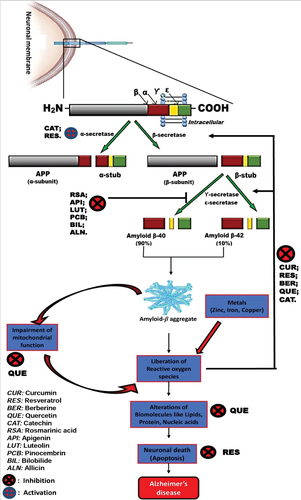
It has been shown that APP hydrolysis is enhanced in the brains of AD sufferers, and that A therapy increases this cleavage even more.[Citation72] Additionally, it has been demonstrated that APP and its breakdown products accumulate in neuritic vesicles in the axons of the brains of AD sufferers and other neurodegenerative illnesses, speculating that APP buildup may be a sign of axonal damage.[Citation73] For instance, it has been shown that in APP genetically modified mice, high A
levels cause synapses to disappear and neuronal transmission to slow down, coupled with behavioral problems, before amyloid plaques form.[Citation72] The unusual early-onset familial AD, which is brought on by mutations in the enzymes that cleave APP, resulting in the accumulation of A
, primarily A
1–42, and is brought on by rapid and abnormal hydrolysis and subsequent overproduction of A
. Conversely, the common form of AD with a late onset is thought to be caused by a combination of factors, including a failure of microglial cells to remove amyloid beta from the brain,[Citation74,Citation75] a decrease in the expression of amyloid beta-degrading proteases like insulysin (the insulin degrading enzyme) and a decrease in the concentration of lipoprotein receptor that are meant to convey A
out of the brain.
The memory-encoding neural network in the entorhinal cortex, the parahippocampal gyrus, and the hippocampus can be disconnected by accumulating soluble A oligomeric forms of the protein, which have been revealed to be synaptotoxic, also capable of pruning dendritic spines.[Citation50] Large insoluble fibrillar masses or plaques that are eventually formed by these oligomers do not directly cause death of neurons, instead attract microglia and astrocytes, which then release proinflammatory cytokines, which are cytotoxic and reactive oxygen species, which may cause death of neuron, indirectly.[Citation76] The susceptibility of cells to subsequent assaults, tau excessive phosphorylation, stimulation of the apoptosome and the activity of protease lysosome, modifications in calcium entry, and direct membrane annihilation (peroxidation) are further suggested processes that contribute to neuronal damage.[Citation77] Despite the fact that the plaques are extracellular, it is believed that the synthesis, oligomerization, and buildup of A
occur within neuronal processes, with the potential that the inclusion of aggregates into plaques happens after the neurites are broken down.[Citation78] A
has undoubtedly been found in multiple neuronal compartments, including the Golgi body, the rough and smooth endoplasmic reticulum, the lysosomes and peroxisomes, and the vacuoles, in investigations using the well-established mice models of AD, suggesting intraneuronal aggregation and disease.[Citation79] Evidence, however, points to A
’s extracellular deposition as the initial pathogenic mechanism in the AD brain, with a connection of anterograde axonal transport inhibition.[Citation80,Citation81] Although there is direct evidence of A
-dependent neurodegeneration, A
pathology develops before clinical symptoms manifest.[Citation82] In order to assess the course of the illness and track patient’s response to antiamyloid medications, it would be helpful to determine the degree of amyloid deposition in the brain of an AD patient (also known as A
load) over time. Interesting studies have shown that the PET Pittsburgh compound B (PiB-PET) binds to A
peptides through amyloid imaging.[Citation83] When compared to controls, this study’s PET amyloid imaging using Pittsburgh compound B (PiB) revealed higher corticaL PiB binding in AD patients, while patients with mild cognitive impairment (MCI) showed intermediate levels of binding.[Citation83] This substance might help with the early diagnosis of AD and monitoring the course of the illness.
Additionally, it was shown through in vivo and in vitro studies that A binding to the cellular prion protein (PrPc), an oligomer-specific high-affinity binding site for A
, can be a key factor in A
-induced memory deficits, loss of synapses, degeneration of axon, and ultimate death of neuron in the AD sufferers’ brain through activation of Fyn kinase.[Citation84] By boosting surface N-methyl-D-aspartate receptor (NMDAR) expression and phosphorylating it, the activation of this kinase alters NMDAR function and finally culminates in dendritic spine in conjunction with surface receptor loss.[Citation84] According to the research, memory deficits in APPswe/PSEN1-M146L double transgenic mice could be reversed, and synaptic density could be restored, by suppressing PrPc.[Citation84] It has been shown that the tau protein and Fyn kinase are associated, and that aberrant Fyn tau connections make synapses more sensitive to glutamate excitotoxicity.[Citation84] Together, these findings imply that PrPc-Fyn signaling may be involved in the diseases of A
and tau, and that inhibiting this signaling could be used as a therapeutic strategy.
Alzheimer’s disease management
As earlier discussed, quite a number of stages are known and utilized to categorize the progression of the pathology based on clinical data. The patients initially exhibit mild cognitive impairment, which is characterized by memory loss with relatively little impairment of other mental and perceptive areas. Mild cognitive impairment may be viewed as a prefatory stage, or a state that exists between healthy aging and extremely mild AD.[Citation85] The subsequent Small short-term memory deficiencies and difficulties recalling specifics (such as obliviousness, time-unconsciousness, and inability to recall familiar places) are the key characteristics of the early stage. This stage is largely disregarded because the symptoms appear gradually and are frequently attributed to stress and aging. As dementia progresses to the middle stage, there is a progressive decline in memory, which worsens language issues and impairs physical coordination, leading to the patient losing their independence. The patient is no longer independent as the Late-stage advances, as evidenced by an increasing influence on locomotor and corporeal capacities, the onset of deranged indications, and total aphasia.[Citation86] Most patients will develop neuropsychiatric symptoms or problematic behaviors throughout the illness, including social and psychical manifestations of lunacy. Changes in perception (delusion, misinterpretation), reasoning, frame of mind (despondency, worry, passivity), and actions (hostility, disquiet, impulsivity that is unsuitable for a social context) are characteristics of social and psychical manifestations of dementia.[Citation87] There is currently no consensus on the potential correlation between social and psychical manifestations of dementia and disease evolution, despite the fact that social and psychical manifestations of dementia are highly widespread in all forms of dementia and may be observed in mild cognitive impairment and all phases of AD. What is certain is that social and psychical manifestations of dementia is linked to more rapid waning, an awful lifestyle, and advanced levels of discomfort; as a result, the social and psychical manifestations of dementia should be closely checked and managed to comprise these measures: examination of the symptoms, recognition and treatment of possible biologic anomalies, non-pharmacological (natural) therapies, pharmaceutical therapies, supplementary and appurtenant medicine, instructive resources, and emotional sustenance for the caregiver.[Citation88]
Non-pharmacological approaches
The most effective substitute for and/or addition to pharmacological treatment, non-pharmacological treatments are often selected as a preferred choice to reduce AD prodrome.[Citation89,Citation90] Non-pharmacological treatments refine the quality of life of AD sufferers and lessen the pressure on caregivers; they comprise quite a number of methods and approaches like workout and motor overhaul, intellective mediation, and psychical therapies; which are usually combined. In addition, the involvement of latest technologies such as telemedicine, assistive devices and domotics, could provide added support to the treatment of AD.[Citation88]
Interventions involving medication (pharmacological approaches)
For AD and other prevalent dementia etiologies, there are sadly no therapeutic medicines. Current pharmaceutical treatments seek to slow down the deterioration of neurocognitive and bodily function, but they do not completely halt the degeneration and death of brain cells. Acetylcholinesterase Inhibitors (AChEIs) and N-methyl-D-aspartate (NMDA) receptor antagonists are the two kinds of drugs that the Food and Drug Administration (FDA) has approved for treating AD. The effectiveness of the former declines as the condition advances, yet it is given to alleviate behavioral symptoms like agitation and lethargy as well as cognitive symptoms like memory and concentration. By blocking acetylcholine (ACh) from being degraded by acetylcholinesterase (AChE), AChEIs increase the concentration of ACh in the synaptic cleft and encourage its communication with the pre- and post-receptors of the synapse. Galantamine, which is taken in mild and moderate AD, and donepezil, which is licensed and marketed to address AD-related dementia that is mild, moderate, and severe, are included in this class. Tacrine, one of the first AD medications approved by the FDA, should be mentioned among the AChEI but it was later discontinued owing to its lethality. All AChEI are “lipid-loving” i.e. lipophilic, and as such, they can easily cross the blood-brain barrier and operate on the central nervous system (CNS) while having limited affinity for peripheral receptors. Galantamine functions as an allosteric regulator of acetylcholine nicotinic receptors, while donepezil and rivastigmine also suppress butyrylcholinesterase (BChE) activity in addition to AChE activity. Despite the variety of additional mechanisms of action, no high-quality data has shown any appreciable variations in their effectiveness.[Citation91]
Memantine, an NMDA-receptor channel blocker with low-to-moderate affinity, belongs to the second class of medications. Its use is supported by research showing that this receptor, which releases glutamate and can lead to neuronal damage, is overstimulated in the brains of AD patients. Memantine was licensed by the FDA in 2002 for use as both a monotherapy and in conjunction with a cholinesterase in patients with moderate to severe AD. Lately, FDA approved the anti-A monoclonal antibody “aducanumab” as a therapy for AD. Early on in the disease’s progression, aducanumab medication seems to be able to halt the disease’s progression. Although the FDA has approved this medication, there is ongoing debate regarding its therapeutic efficacy.[Citation92]
The FDA-approved medications are the only ones that can currently be used for the pharmacological therapy of AD. Rivastigmine, donepezil, and galantamine are three that match the AChEI, while memantine is a NMDA antagonist.[Citation91,Citation93] In AD patients, AChEI may prevent or lessen the cholinesterase enzyme’s catalytic activity, which is responsible for the neurotransmitter ACh breakdown in the synaptic cleft of cholinergic cells.[Citation91] Its pharmacological activity suggests that it enhances central cholinergic neurotransmission, which slows the decline of cognitive function. Clinical trials have demonstrated that all three medications stabilize cognition and forestall neurodegeneration in individuals exhibiting minor to pronounced AD signs. The greatest doses of donepezil 10 and 23 mg tablets and 13.3 mg/24 hr rivastigmine patches have been reported in patients with advanced illness. Despite AChEI’s effectiveness in treating AD, reports show that 5 to 20% of patients experience side effects, which are primarily related to the digestive, cardiac, and pulmonary systems. As a result, it is necessary to develop personalized tolerance surveillance in sufferers based on maximal advocated dose and safety versus dosage.[Citation91,Citation94,Citation95]
These drawbacks are mostly exacerbated by the presence of additional geriatric-specific illnesses such diabetes, hypertension, and hypercholesterolemia in individuals with this form of neurogenerative disease. AChEI also has additional restrictions, such as: its potency and effectiveness is in symptomatic victims regulating a certain activity of the neural network, being merely CNS stabilizers, however, its participation is extremely negligible in ameliorating circumstances that induce AD. Memantine, an NMDA antagonist, works to restore the functional integrity of cognition by lowering the hyper-activation of the receptors of glutamate; expressed in proportion to the development of the disorder.[Citation94,Citation96] This medication is used either alone or in combination with AChEI in therapy. Patients with moderate to severe AD have exhibited positive short- and long-term results from monotherapy, which has improved their lifestyle quality, intelligence, and behavioral and psychical features associated with neurodegenerative illnesses.[Citation91,Citation93] Although it is typically well tolerated and does not cause the most severe side effects like the AChEI does, some people have had significant side effects like sleepiness, dizziness, constipation, headaches, and coughing.[Citation91,Citation94,Citation95] Depending on how the condition is progressing, the dosage might range from 10 mg to 28 mg each day.[Citation93–95] There is a decline in AD when AChEI is used in complementary therapies, although it has been claimed that this condition only becomes apparent for a maximum of three years after which AD continues to worsen.[Citation97] Reducing side effects and lengthening drug action times to improve neurodegeneration decline effectiveness are challenges in present AD therapy methods. Given the multiple pathways that cause AD, one of the principal strategies is the multifaceted and broad-brush therapeutic approach. This involves encouraging the development of novel AChEI or NMDA receptor modulators since these treatments are still neuron-specific and only attempt to stop one of the pathological processes initiated.
Plant chemicals as new potential therapeutic approaches for AD
Experimentally, the pursuit of and classification of beneficial substances under the label of nutraceuticals has been advocated.[Citation90,Citation98,Citation99] When introducing this idea in 1989, Stephen de Felice stated that it included “any material deemed to be food or to be a component of food that offers curative or health benefits.”[Citation100] European and American regulatory governmental agencies have developed a number of definitions for these substances.[Citation101] The descriptions of nutraceuticals are converging, emphasizing that these classes of matters can be intrinsic components of matrices of organic sources from fruits, medicinal herbs, vegetables, microalgae, grains, and leaves, pulps, seeds, stems, and peels from agro-industrial residues. These substances can also be chemically or biologically active.[Citation102–105] They are frequently taken in concentrated amounts and come in non-food forms (pills, powder, liquids, etc.). Due to the presumed positive modulating effect on human health caused by a significant increase in their concentration, they are now regarded as a helpful enhancement for the care and sustentation of healthy people, also, in forestalling diseases that have been officially recognized.[Citation101,Citation106–108] Numerous studies have demonstrated that nutraceuticals can act as antioxidants or anti-neuroinflammatory medications, inhibiting the accretion and accumulation of amyloid peptides; inhibiting the reduction of acetylcholine that facilitates cholinergic neurotransmission; and inhibiting tau protein hyperphosphorylation, all of which are molecular mechanisms that cause AD;[Citation109–111] With the use of this data, we may imagine a situation in which AD might be treatable either as a preventive measure or as a palliative one for those who already experience it.[Citation112–114]
A number of conditions activated in AD, which affect the regulation of genes that encodes amyloidogenic pathway proteins, mitochondrial energy metabolism, and hyperphosphorylation of tau are covered by current trends toward the use of “smart therapeutics,” with supportive and supplementary therapies, using phytopharmaceuticals with antioxidant potentials to confer neuroprotection.[Citation97,Citation99,Citation115] The main drawbacks of standard pharmacology could be mitigated by therapies using phytopharmaceuticals with recognized neuroprotective properties, which could produce neuroenhancers that could significantly increase cognitive function in individuals with AD.[Citation116]
The effectiveness of phytochemicals in the treatment of AD has been shown in numerous experimental preclinical pharmacological research, as summarized in . Lately, hundreds of bioactive substances and extracts of plant origin have been investigated as prospective AD and dementia treatments.[Citation125–127]
Table 1. A Summary of the Information on the Pharmacological Effects of Plant Chemicals as possible agents for the Treatment of AD.
Table 2. A summary of the Information on the Vaccines that might be used to treat AD.
Essential oils extracted from plants like Coriandrum sativum L,[Citation128] Zosima absinthifolia Link,[Citation129] Hedychium gardnerianum Sheppard ex Ker Gawl,[Citation130] Persicaria hydropiper (L.) Delarbre (syn. Polygonum hydropiper L.),[Citation131] and Lavandula spp[Citation132] have been utilized to combat AD. Curcumin,[Citation133] resveratrol,[Citation134] phenolics and flavonoids,[Citation135] coumarin,[Citation136] ginkgolide,[Citation137] quercetin,[Citation138] glycyrrhizin,[Citation139] catechins,[Citation140] monoterpenoid,[Citation141] and S-allylcysteine,[Citation142] which were extracted from some plants like turmeric, Japanese knotweed, spices, Ginkgo biloba leaves, onion, licorice, green tea, and garlic[Citation143–150] have been shown to have the ability to forestall AD progression and prevention. Due to their reducing and stabilizing abilities, phytochemicals are increasingly frequently used in green synthesis, an environmentally beneficial technique.[Citation151,Citation152] The green synthesis of zinc oxide, gold, and silver nanoparticles (NPs) using extracts from Lampranthus coccineus and Malephora lutea Schwantes,[Citation153] Terminalia arjuna,[Citation154] and Sabal palmetto[Citation155] revealed activities that portray phytochemicals as bioactive substances that can inhibit and slow down certain molecular cascades that are implicated in the development of AD.
Curcumin
Curcumin (C21H20O6) is a spice, taste enhancer, and food preservative that is mostly found in turmeric rhizome (Curcuma longa). According to Ringman et al. curcumin possesses anti-amyloidogenic, anti-oxidative, anti-inflammatory and anti-cancer qualities that help it fight off disease and reduce its prevalence.[Citation156] Research has shown that curcumin impedes the development and buildup of Aβ plaques, a characteristic of AD. By preventing BACE1 from expressing, intragastric curcumin reduced the severity of the disease in an AD model. Additionally, it improves spatial learning and memory patterns and slows down synaptic degradation.[Citation157] Aβ’s synthesis has been connected to both β-secretase and glycogen synthase kinase-3β (GSK-3β). Human neuroblastoma SHSY5Y cells exposed to curcumin treatment had much less Aβ production. According to Zhang et al.,[Citation158] curcumin inhibited the activation of GSK-3β dependent PS-1 and thus lowered Aβ production by decreasing PS-1 and GSK-3β protein expression. The elimination of tau tangles and the prevention of neurotoxicity are two additional functions of curcumin. A molecular chaperone called BCL2 associated athanogene 2 (BAG2) carries tau to the proteasome, where it is broken down.[Citation159] After curcumin treatment, there was a considerable increase in BAG2 levels in rat primary cortical neurons whilst hyperphosphorylation of tau was observed to be reduced.[Citation160] Additionally, studies have shown that curcumin exhibits neuroprotection by lowering or blocking the production of reactive oxygen species and neuroinflammation. By increasing the expression of many antioxidant enzymes as well as a few proteins that protect cells, it reduces oxidative assault to the neuron. By producing transcription factors, curcumin also significantly increases the expression of intracellular glutathione.[Citation161]
Resveratrol
Red wine, grapes, and berries contain the polyphenolic phytoalexin resveratrol (C14H12O3). According to Tian and Liu,[Citation162] it has quite a number of cellular, pharmacological, and biological effects. Wine drinking brings about reduction in the risk of AD, according to several epidemiological research. Resveratrol may therefore have therapeutic effects on Alzheimer’s patients, according to speculation.[Citation163] The amyloid precursor protein is encouraged to break down in a non-amyloidogenic way by resveratrol. Additionally, it lessens neuronal damage and enhances amyloid beta-peptide elimination. The majority of experimental studies on resveratrol and AD have recently been carried out in a number of experimental models, in vitro as well as in in vivo. According to Wang et al.,[Citation164] the usage of polyphenols extracted from grape seed decreased the oligomerization of Aβ peptide and ameliorated impairment of cognition in Tg2576 mice. Resveratrol treatment for APP/PS1 mice results in a considerable decrease in the number of activated microglia, it was revealed to impede the production of amyloid.[Citation165] In Sprague-Dawley rat, which were exactly 10 days old, glutamate stimulated the synthesis of monocyte chemical protein-1 (MCP-1). Resveratrol prevented glutamate’s effect on ERK activation, which decreased the production of IL-1β and MCP-1 in the hippocampal region.[Citation166] It prevents memory loss brought on by high-fat diets (HFD). Resveratrol also protects wild-type as well as transgenic 5XFAD strains against tau pathology triggered by high fat diet and lowers the amyloid load that is increased by HFD in the transgenic model. Resveratrol prevented the amyloid precursor protein from being processed in an amyloidogenic manner by HFD in both strains and restored the extremely high proteolytic activity of ubiquitin-proteasome system, suggesting the existence of a compensating mechanism to prevent the buildup of abnormal proteins.[Citation167] Resveratrol enhances SIRT1 activity by stabilizing protein-substrate connections.[Citation168] Resveratrol boosts SIRT1 protein expression and activity, as well as the expression of SIRT1 mRNA.[Citation169] The activation of SIRT1 is regarded to be the cornerstone of resveratrol-mediated protection because it is a key protein thought to be involved in a number of resveratrol effects. Resveratrol decreases apoptosis, blocks the inflammatory response, lowers oxidative stress, and enhances autophagic flux stabilization in human-derived neuroblastoma cell lines via the SIRT1 signaling pathway.[Citation170,Citation171]
Berberine
A yellow plant alkaloid with a bitter taste called berberine (C20H18NO4 +) has been utilized in traditional medicine for up to three thousand years. The nervous system’s sedative effects of berberine were first documented in the 1970s.[Citation172] Numerous neurological conditions, such as cerebral ischemia, depression, Huntington’s disease, Alzheimer’s disease, anxiety, convulsions, Parkinson’s disease, and epilepsy have been the subject of research into the therapeutic potential of berberine. Berberine was shown to impede the progression of AD by preventing the hyperphosphorylation of Tau protein and the production of Aβ. Inhibiting beta-secretase expression, berberine reduces the production of Aβ40/42 by activating extracellular kinase 1/2 signaling pathway.[Citation173] The following enzymes which have been implicated in the etiology of AD, acetylcholinesterase, monoamine oxidase A butyrylcholinesterase, and monoamine oxidase B, were also inhibited by berberine, according to studies.[Citation174] Cholinesterase (ChE), the main enzyme responsible for acetylcholine breakdown, is inhibited, which raises acetylcholine levels in the brain. In order to treat the cognitive symptoms of AD, inhibitors of cholinesterase have been the subject of extensive anti-AD pharmacological study.[Citation175] To determine how berberine impacts ChE activity, several tests have been carried out. Berberine decreased oxidative stress and cholinesterase activity in rats given ethanol treatment.[Citation176] In rats, memory impairment triggered by streptozotocin treatment was ameliorated following a one-month berberine treatment.[Citation177] Berberine treatment during training trials improved cognition whilst reducing oxidative onslaught and hyperglycemia. In an AD transgenic mice model, berberine has been demonstrated to reduce β-amyloid pathology and cognitive decline.[Citation178]
Quercetin
A flavonoid with considerable medicinal and pharmacological characteristics is quercetin (C15H10O7). Quercetin’s ability to protect the brain has been well studied. At low micromolar levels, it lessens the cell toxicity brought on by oxidative stress in neurons. It promotes neuronal regeneration while also preventing neuroinflammation by blocking pro-inflammatory cytokines like NF-kB and iNOS. Quercetin prevents mitochondrial failure and progressive dopaminergic dementia by activating the PKD1-Akt cell survival signaling pathway.[Citation179] It lowers the hyperphosphorylation of tau proteins in HT22 cells by using MAPKs and the PI3K/Akt/GSK3 signaling pathways.[Citation180] In an in vivo study, quercetin was revealed to decrease nitric oxide production, the production of proinflammatory cytokines and the expression of the iNOS gene. In addition, quercetin reduces JNK/Jun phosphorylation and TNF production in rats, shielding neurons from inflammation induced by lipopolysaccharides.[Citation181] In addition, a number of neuroprotective techniques exist, including those for lowering neuroinflammatory mechanisms, neurorepair techniques, antioxidant techniques, hormonal techniques, and oxidative stress.
Green tea catechins
Consuming catechins has been associated with several advantages to health. According to research, green tea catechins were revealed to exhibit neuroprotective properties.[Citation182–184] Green tea catechins are known for their brain permeability.[Citation185] According to studies by Chen et al.[Citation186]; Ishige et al.[Citation187] and Levites et al.,[Citation188] numerous molecular biological activities of green tea catechins have been identified, including the activation of protein kinase C, MAPKs, survival genes, antioxidant enzymes, and APP processing. Extract from green tea shield neurons from Aβ-induced harm, according to a number of in vitro studies.[Citation188,Citation189] In APP695 overexpressing neurons and in Tg2576 AD mice, Epigallocatechin gallate (EGCG), a major catechin derived from the polyphenolic portion of green tea, reduced Amyloid-beta production.[Citation183] It also decreased Aβ levels and plaques. These findings demonstrated that EGCG activates the non-amyloidogenic α-secretase proteolytic pathway by boosting the production of the soluble amyloid precursor protein-C-terminal fragment of APP.[Citation183,Citation188] TNF-α converting enzyme, a candidate for the APP α-secretase, was likewise shown to express itself much more when EGCG was present.[Citation183] Additionally, active α-desintegrin and metalloprotease 10, another APP α-secretase candidate, had their protein levels significantly raised by EGCG, which ultimately resulted in non-amyloidogenic APP processing.[Citation190] It’s also possible that EGCG-mediated reduction of Aβ production is performed via blocking β-secretase activity.[Citation191] Long-term treatment of EGCG to mice has been demonstrated to suppress the production of holo-APP in the hippocampus, indicating that EGCG may lower Aβ levels by doing so.[Citation188]
According to Ono et al.,[Citation192] green tea catechins can also prevent Aβ fibril formation, extension, and stability. By directly interacting with the native unfolded polypeptides and potentially by the creation of stable hydrogen bonds, EGCG has been demonstrated to effectively block the fibrillogenesis of Aβ.[Citation193] This prevents the polypeptides from becoming hazardous, on-pathway aggregation intermediates. Overall, catechins have stronger antioxidant effects than α-tocopherol or ascorbic acid.[Citation194] Catechins limit the production of potentially harmful free radicals through its ability to chelate metals like iron and copper, according to Singh et al..[Citation184] Amyloid precursor protein mRNA translation may be suppressed and APP level may be affected by utilizing EGCG chelation to lower the free iron pool.[Citation184] In order to stop oxidative DNA changes, catechins may potentially transfer an electron to ROS-induced radical sites on DNA.[Citation184] Additionally, EGCG suppresses lipid peroxidation and scavenges ROS.[Citation195] EGCG protects against Aβ-induced apoptosis and improves hippocampus neuronal survival in cells that are simultaneously exposed to Aβ via lowering caspase activity, as well as the level of malondialdehyde.[Citation195] Additionally, rat brain lipid peroxide in the hippocampus region is decreased by EGCG in vivo, lowering Aβ-induced oxidative stress.[Citation196] Catechins’ ability to reduce inflammation is widely known. In human astrocytoma U373MG cells, EGCG suppresses the activation of MAPK and NF-jB and therefore reduces the synthesis of pro-inflammatory markers, as well as endothelial growth factors.[Citation197] Additionally, EGCG inhibits the pro-inflammatory effects of several cytokines[Citation198,Citation199] and reduces COX-2 expression and prostaglandin E2 production that are triggered by IL-1 and Aβ.[Citation197] Additionally, EGCG is said to prevent inflammation-mediated neuronal damage and inhibit LPS-induced microglial activation.[Citation200] Rezai-Zadeh et al. found that EGCG reduces potentially hazardous sarkosyl-soluble phosphotau isoforms via modulating Aβ-mediated tau pathology in Tg2576 mice. In a recent study, it was found that heat-shock protein (HSP) 90 inhibitors reduce the amounts of soluble phospho-tau isoforms.Citation263 According to Palermo et al.,[Citation201] EGCG was discovered to interact directly with HSP90 and stop it from functioning. As a result, EGCG may modify the level of phospho-tau by inhibiting HSP90. The effects of catechins on cognitive function have been studied in several animal models. For instance, according to[Citation200]EGCG lessens cognitive deficits in Tg2576 mice. Rats given water supplemented high amounts of catechins (specifically EGCG) for a period of five months demonstrated decreased memory damage after receiving an intracerebroventricular injection of Aβ1–40.[Citation196] Injections of tea catechins reduced memory impairment and hippocampus neuronal loss in a mouse model of cerebral ischemia.[Citation202,Citation203] Therefore, treatment with green tea catechins, especially EGCG, may be an effective therapeutic strategy for treating cognitive impairment and neurological disorders similar to AD.
Rosmarinic acid
Rosemary (Rosmarinus officinalis), sage (Salvia officinalis), lemon balm (Melissa officinalis), peppermint oregano (Origanum vulgare), and thyme are good sources of rosmarinic acid, a polyphenol.[Citation204] It guards against the toxicity caused by amyloid peptide in PC-12 cells. When rosmarinic acid is taken orally, it goes through sulfate conjugation, methylation, and dehydroxylation. In rats, the half-life of metabolites was estimated to be between 8 and 18 hours, and they were eliminated in the urine.[Citation205,Citation206] By lowering the level of ROS, as well as the level caspase-3, rosmarinic acid inhibits apoptosis as part of its neuroprotective effects. Additionally, it prevents p38 MAPK activation, which contributes to neuronal degeneration.[Citation207] Rosmarinic acid’s structural analysis was examined by NMR tests against the production of amyloid fibrils and toxicity, and the results showed that the compound has a high affinity for β-amyloid, which it can bind to and degrade to prevent plaque development.[Citation208] The data unambiguously point to a successful strategy for inhibiting the development of amyloid fibrils in vitro and for further evaluation in vivo of its efficacy as a therapy for AD. Additionally, it protects memory impairments by focusing on the NF-KB AND TNF-α ameliorated cognitive impairment by focusing on the NF-KB and TNF-α pathway.[Citation209,Citation210]
Apigenin
This flavonoid, apigenin is present in numerous fruits. It is also found in vegetables, including bell pepper, celery, and cabbage.[Citation211] Elsholtzia rugulosa is a plant from which apigenin was extracted. Till present, there is dearth of information to support the idea that apigenin, when ingested as part of a typical diet, encourages unfavorable metabolic processes in vivo.[Citation212,Citation213] It could be efficiently absorbed throughout the whole intestine by a variety of transport systems, although the duodenum is where it is primarily absorbed.[Citation214] It has been discovered that amyloid-β peptide (25–35)-induced rat primary cultures of cerebral microvascular endothelial cells are protected from cytotoxicity and have lower levels of lipid peroxidation and ROS.[Citation215] Apigenin has been shown to have protective effects against amyloid-β peptide (25–35)-induced toxicity in mice by reducing oxidative onslaught and enhancing cognitive functions and memory, as well as improving cerebral blood flow and modulating acetylcholinergic system. According to follow-up research by Liu et al., apigenin treatment of neuronal cells confers protection against toxicity brought on by amyloid through controlling the genes that encodes the homeostasis of copper.[Citation216] It was also able to maintain mitochondrial respiratory activity by preventing the activation of the MAPK pathway.[Citation217]
Luteolin
The plant families Bryophyta, Magnoliophyta, Pinophyta, and Pteridophyta all contain luteolin as a flavonoid. Olive oil, green pepper, celery, carrots, broccoli, and other dietary foods also contain it.[Citation218] Luteolin is sulfated and methylated before being released into the plasma as free Luteolin.[Citation219] According to research, administration of luteolin confers neuroprotection by preventing apoptosis and inhibiting protein-level ERK1/2 activation and Nrf2 pathway in cultures of rat cortical neurons induced by amyloid- β peptide.[Citation220,Citation221] The defense is comparable to the neuroprotective effect brought on by estrogen receptors. By reducing the plaques of amyloid- β peptides, controlling the redox state, mitochondrial activity, and through the induction of the down-regulation of caspases that participates in apoptosis, luteolin also confers neuroprotection in neuroblastoma cells against toxicity induced by β-amyloid (1–42).[Citation222] Luteolin (150–450 mg/kg) can improve learning and memory processes, increase LTP, and protect synapses through controlling CREB protein activity.[Citation223]
Pinocembrin
Pinocembrin is a flavonone that has been isolated from numerous plants, primarily the heartwood of Pinus sylvestris, Eucalyptus globulus, Sparattosperma leucanthum, as well as Populus tremula. Additionally, it comes in finger roots, honey, and propolis.[Citation224] As seen in rats, pinocembrin has a very limited bioavailability. Pinocembrin was quickly absorbed into the large intestine after being given orally, and its half-life is one hour.[Citation225] By blocking the Nrf 2/HO-1 pathway against toxicity caused by β-amyloid in human neuroblastoma cells, it exhibits neuroprotection.[Citation226] Pinocembrin has been demonstrated to block inflammatory pathways such as MAPK and NF-KB in mice when given orally. This, in turn, causes mice with cerebral infusions of amyloid- β peptide (25–35) to undergo apoptosis.[Citation227] Pinocembrin was found to have a similar anti-toxic effect on human brain microvascular endothelial cells when exposed to amyloid-β peptide (1–40).[Citation228]
Bilobalide
A naturally occurring sesqui-terpenoid called bilobalide is most frequently detected in Ginkgo biloba preparations. By expressing soluble APP and lowering the amount of β-APP by up-regulating the PI3-K pathway, bilobalide displays neuroprotective activity against amyloid damage.[Citation229] Additionally, it guards against synaptic loss in neurons and encourages activation of the CREB and BDNF pathways to counteract toxicity caused by amyloid.[Citation230] It demonstrates neuroprotection in primary cultures of hippocampal neurons. A recent study also revealed that it may be able to reduce the toxicity caused by amyloid by having antioxidant characteristics.[Citation231] 148 A different study has demonstrated the method by which it lessens toxicity by preventing the activation of the NF-KB and MAPK pathways. It may also prevent the development of amyloid-β fibrils.[Citation232] The EGb761 decreases the abnormal behavior acquired by toxicity triggered by amyloid in Caenorhabditis elegans in addition to inhibiting the process in vitro.[Citation233] By lowering the amounts of Caspase-3, P53, as well as BAX, in PC12 cells, bilobalide can also decrease the apoptosis that ROS-induced stress causes.[Citation234]
Allicin
Allium sativum, or garlic, is a traditional herb that is grown all over the world. It produces the molecule allicin, which undergoes additional metabolization to create a number of organosulfur compounds.[Citation235] According to research, using garlic extract reduces the oxidative stress caused by amyloid and increases the survival of neurons. A study in neuronal PC-12 cells showed that aged garlic extract administration can, in a concentration-dependent way, decrease the cytotoxicity brought on by amyloid-β peptide (25–35).[Citation236] Similar to this, components of garlic extract containing S-Allyl Cysteine (allicin) can reduce ROS production and death in PC-12 cells that have been infected with amyloid-β peptide (25–35) in vitro.[Citation237] Thio-T-Fluorescence and transmission electron microscope investigations have shown that the organosulfur compounds in garlic have the ability to dismantle the fibril formation in amyloid peptides.[Citation238] In rats with cognitive impairment, aged garlic extract enhanced short-term recognition memory and reduced IL-1β levels by preventing microglia from becoming activated.[Citation239]
Mechanisms of action underlying the role of phytochemicals in AD
Antioxidative potentials
Despite the fact that the effects of AD are complicated and multifaceted, oxidative stress is crucial for the commencement of the pathophysiological activities in cholinergic neurons.[Citation20] Oxidative stress describes the excessive intracellular oxidation of chemicals that damages cells and triggers cellular death processes. Compounds known as reactive oxygen species, like nitric oxide, hydrogen peroxide, peroxynitrite, superoxide radicals, hydroxyl radicals, and their metabolites are responsible for this physiological phenomenon. Within the cell, various ROS exhibit varying degrees of reactivity and toxicity.[Citation240] At the conclusion of aerobic respiration, the mitochondrial crests undergo oxidative phosphorylation, which produces the most detrimental effects.[Citation241,Citation242] DNA can be affected by oxidation processes, which can lead to nucleotide dimerization and replication mistakes. Additionally, critical structural or catalytic proteins may develop a functional change.[Citation243] Finally, they cause cell lysis by affecting the membrane phospholipids’ unsaturated fatty acid chain.[Citation244] Antioxidants, compounds that mitigate the detrimental effects of this kind of disruption at the intracellular level, are used to buffer this oxidative stress in the cell.[Citation245,Citation246] The mode of action of antioxidants on this oxidative stress is classified in accordance with its capacity to:
Prevent the production of free radicals. Antioxidants with this ability are referred to as Indirect antioxidants.
Remove chemically produced free radicals directly. Antioxidants with this ability are referred to as direct antioxidants.
Increase cellular resistance to elevated levels of ROS; detoxify accumulated ROS by the participation of enzymes, or facilitate the repair of oxidative damage. Antioxidants with this ability are referred to as metabolic antioxidants.[Citation247]
It has been suggested that the majority of bioactive compounds function as direct antioxidants and do not rely on endogenous intracellular enzymes to carry out their main function, which is to interact directly with free radicals,[Citation248] as shown in . The nervous system appears to be the most susceptible to ROS at the systemic level in humans, compared to other organs and tissues, primarily because it consumes oxygen at a high rate, has a low endogenously synthesized antioxidant content, has a high concentration of steroid and polyunsaturated lipids, and is exposed to high amount of catalysts which are metals, all of which favor a persistent oxidative physiological event in the cerebral cortex.[Citation248,Citation264] According to theories, ROS are the primary factor contributing to neurological decline and are responsible for diseases like Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease.[Citation110,Citation265]
Excessive oxidative stress, in particular for AD, shows up from the beginning of the illness.[Citation110] It appears that hydrogen peroxide is the key player in encouraging excessive oxidation in neural cells. Thanks to catalase and peroxidase, peroxisomes are in charge of enzymatically regulating superoxide radicals and H2O2 under normal physiological circumstances.[Citation110,Citation217] However, depletion of these organelles has been demonstrated as a result of overexpression of hyperphosphorylated Tau protein (which is a pathophysiological indication of AD), suggesting that is the reason for the rise in H2O2 and ROS.[Citation266] Kou et al.[Citation267] found evidence of diverseness in the distribution of peroxisome in the neurons from AD patients, revealing that Tau, a cytoskeleton protein that is abnormally phosphorylated and may be the reason for the rise in the oxidizing environment, prevents the proper transit of these organelles. However, a connection has been proposed between the rise in ROS, H2O2, metal ions, and the peptide, Amyloid-beta.[Citation268] One of the most commonly used chemicals to induce AD is hydrogen peroxide, it can form neurotoxic aggregates that eventually kill neurons and thus impair cognitive function.[Citation265,Citation269] Peptides (Amyloid-beta) have a strong affinity for copper ions, forming a one-to-one ratio combination that promotes amyloid plaque aggregation. H2O2 and the rise in ROS are related in this process, demonstrating the H2O2 molecule’s critical function in the pathophysiology of AD.[Citation268] H2O2 kills cells by apoptosis, as established by a number of in vitro models using CNS neurons that had been isolated or immortalized cell lines.[Citation270–274]
One of the most popular cell lines is the human neuroblastoma SH-SY5Y,[Citation270,Citation275] which was developed in 1970 from a biopsy metastatic tumor. Their most crucial traits are their capacity to differentiate into specialized neurons through markers and to have a mixed culture (adherent and in suspension), making adrenergic, cholinergic, and dopaminergic models a fundamental model for the study of neurodegenerative disorders like AD.[Citation276,Citation277] Lately, Angeloni et al.[Citation278] assessed the antioxidant and anti-inflammatory effects of coffee grounds (CG) extracts in aqueous, methanolic, and ethanolic solutions on SH-SY5Y cells. After a neuroblastoma culture was exposed to the influence of oxidative stress using H2O2 and then treated with various kinds of CG extracts, a substantial improvement in cell viability was seen. However, the authors came to the conclusion that a deeper examination of extract contents is required for the identification of potential bioactive compounds with potential for neuroprotection. It is hypothesized that the primary chemicals in the proportion of phenolic compounds, primarily caffeic acid, quinic acid, and the family of chlorogenic acids, are what give these qualities.[Citation279–281] By altering lysosomal function in SH-SY5Y cells, Gao et al.[Citation282] showed that upon treatment with chlorogenic acids, there was a significant inhibition in autophagy brought on by Aβ25–35. This attenuated the loss of CA1 neurons and cognitive deficits in APP/PS1 mice, demonstrating the compound’s neuroprotective capabilities against conditions like AD. Utilizing SH-SY5Y, a human neuroblastoma cell line, Kim et al.[Citation283] assessed the effects of two derivatives of caffeoylquinic acids derived from Dipsacus asper Wall. ex C.B. Clarke plant extracts on H2O2-induced cell damage. Their findings revealed reduced Caspase 3 activation, which attenuated neuronal apoptosis. In addition, H2O2-induced intracellular depletion hindered the glutathione concentration’s ability to recover. These phenolic chemicals, according to the authors, might be used therapeutically to treat or ward off neurodegenerative disorders.[Citation283] Izuta et al.[Citation284] investigated how Chinese propolis components affected SH-SY5Y cells’ susceptibility to tunicamycin-induced apoptosis. The authors reported that propolis reduced induction of caspase 3 activation and also reduced the disruption of mitochondrial membrane potential, as well as the suppression of neuronal death. With the help of this empirical data, we may envision a viable solution to the problems caused by the aging population and the emergence of neurological diseases like AD.
Anticholinesterase potentials
The decrease in the neurotransmitter, acetylcholine caused by a rise in BChE and AChE is another crucial sign that AD is being triggered. The catalytic activity of these enzymes can be inhibited by quinic acid and chlorogenic acid, which are derived from plant matrices. This prevents the cholinergic deficiencies that occur in AD and cause memory forfeiture. Using the enzymes AChE and BuChE, Oboh et al.[Citation9] examined the impact of caffeic acid and chlorogenic acid and their co-administration in mouse models. Results indicated that caffeic acid had a higher potential for inhibition than chlorogenic acid, leading researchers to draw the conclusion that these bioactives would confer neuroprotection via their ability to inhibit AChE and BuChE. Arya et al.[Citation285] analysis of the sesquiterpenes lipiodol and amberboin, which are capable of inhibiting cholinesterases with an IC50 <9 M and are found primarily in herbaceous plants, suggests that they may be effective at improving learning and memory in AD patients as well as serving as a potential preventive treatment for this neurodegenerative pathology.
Beta-amyloid peptides anti-aggregation potentials
The development of amyloid plaques in the extracellular matrix of the cerebral cortex, brought on by the excessive liberation of the β-amyloid and facilitated by cells of the cholinergic system, is one of the primary pathogenic features of AD. The APP and intracellular NFT are cleaved by proteolytic enzymes, which results in this event.[Citation286] Miyamae et al.[Citation287] investigated the prevention of the β-amyloid-42 protein’s aggregation in SH-SY5Y cells. The findings demonstrated that caffeoylquinic acids, particularly 3,4,5-tri-O-caffeoylquinic acid and 4,5-di-O-caffeoylquinic acid, significantly prevent the aggregation of β-amyloid-42, indicating that the caffeoyl group in these compounds is necessary for inhibitory actions in the model. Studies carried out recently suggest that the formation of Amyloid-beta peptides is controlled by inhibiting enzymes linked to the amyloidogenic cascade, primarily -secretase.[Citation253,Citation288,Citation289] The BACE protein performs proteolysis to produce “soluble APP,” a fragment that is soluble in the extracellular space, and “CTFβ peptide,” a fragment that is bound to the plasma membrane and goes through yet another proteolytic reaction with γ-secretase to produce toxic neuropeptides Aβ with 40–43 amino acids, which ultimately results in the amyloid plaques that are characteristic of AD.[Citation200,Citation290,Citation291] An important approach for delaying AD in its early stages is the use of bioactive compounds that block the amyloidogenic process.
Anti-neuroinflammatory potentials
Neuroinflammation processes that take place as a brain immune barrier can also cause AD.[Citation292,Citation293] The glia is invaded by circulating immune cells as a result of this immunological response, and pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, prostaglandin E2 (PGE2), interleukin (IL) IL-1β and IL-6, ROS, nitric oxide (NO), and chemokines, are also produced. Apoptotic pathways can be activated by glial cytokines when they bind to certain receptors on the neuronal membrane. According to reports, TNF-α binds to TNFR1, which causes neuronal death.[Citation294,Citation295] According to reports, bioactive molecules including carotenoids, flavonoids and non-flavonoids, phenolic compounds, and others can have neuroprotective effects by preventing the microglia from becoming activated, which in turn regulates inflammatory processes in the CNS.[Citation152,Citation245]
Plant chemicals in AD: delivery and bioavailability
The anti-Amyloid, anticholinesterase, anti-inflammatory, and antioxidant capabilities of phytochemicals all contributed to their anti-AD effects.[Citation296] The rising use of phytochemicals may be attributed to their low toxicity, same synergistic outcomes.[Citation297,Citation298] The amount of a certain substance delivered via various routes that reaches the site of action is reflected by bioavailability, a critical component of pharmacokinetics.[Citation105,Citation298,Citation299] There are a number of reasons why current AD treatments are ineffective, but inadequate bioavailability is one of the most significant ones.[Citation300] Bioavailability determines how quickly phytochemicals will be absorbed following delivery. This area of research is getting harder and harder to study since plants have complex active ingredients.[Citation301] The blood-brain barrier’s permeability is one of the major obstacles to pharmacological penetration of the central nervous system (CNS).[Citation302] In addition to chemical instability in the bloodstream, a high proportion of plant chemicals having high molecular weight and a high level of lipophilicity will not have the ability to penetrate into the brain.[Citation303] Because of this, integrating bioactive molecules into nanoparticles is a tactic that enables the medicine to overcome a number of physical constraints.[Citation299,Citation304] Due to their low bioavailability, phytochemicals do not always conform to the desired drug-like profile; as a result, altering them also attempts to improve their pharmacokinetics.[Citation305–308] Other methods, including the use of drug delivery systems,[Citation309] could be used to reach this scope. It is now feasible to have nanoparticles that enable a sufficient conveyance of plant chemicals in a targeted manner and with a regulator in the release time, after years of study in nanotechnology to deliver medications to the central nervous system.[Citation98] Primarily because the nanoparticle’s sophisticated surface modification permits appropriate transit to the brain.[Citation310] Micro and nanospheres, nanocapsules, polymeric micelles, and nanoparticulate systems, in general, have been demonstrated to improve the exact targeting of bioactives, reduce their systemic lethality, increase therapeutic efficacy, and safeguard active substances from biochemical degradation.[Citation311]
The bioavailability of α-bisabolol, which was extracted from Matricaria chamomilla L. essential oil, is dramatically decreased[Citation312] due to its low solubility. Too little curcumin is absorbed into the brain as a result of poor bioavailability.[Citation313] Natural occurring phytol has a variety of biological effects, but it is difficult to use in therapeutic settings due to its weak solubility and low absorption.[Citation314] Its poor oral bioavailability may be the reason why researchers have been unable to duplicate resveratrol’s protective benefits against AD in humans.[Citation315,Citation316] These examples from scientific investigations demonstrated how limited bioavailability is a barrier to phytochemicals’ beneficial effect. Through delivery technologies such nanoparticles, liposomes, implants, niosomes, and cyclodextrins the bioavailability issues that can hinder the use of phytochemicals against AD can be resolved. Modifying naturally occurring bioactive chemicals, whose polypharmacology has been extensively studied over the years, is a particularly popular technique in the development of new anti-AD drugs. Resveratrol, neoechinulins, genistein, or diosgenin are just a few examples of naturally occurring compounds that have antioxidant, anti-inflammatory, chelating, and/or scavenging activity.[Citation297,Citation306–308] As a result, the combination with other pharmacophores, such as a moiety from an anti-AD medicine that is already in use, results in the creation of a new molecular entity with an extended range of actions.[Citation317]
Potential vaccines for AD
Researchers have recently created vaccines as a novel therapeutic trend to treat AD (). The creation of a novel vaccination against a variety of diseases is challenging, expensive, and labor-intensive.[Citation318–320] Repurposing currently available vaccinations or medications can speed up the procedure because therapies that have been licensed for use in humans have already proven their safety.[Citation321–323] Repurposed treatments make up the AD vaccine development interventions.[Citation124] The buildup of tau proteins, accumulation of beta-amyloid plaques, and neuroinflammation are the most prevalent symptoms of AD; these circumstances are the key targets for potential vaccines against AD.[Citation120,Citation121,Citation124] The creation of AD vaccines aims to slow or stop the disease’s progression. There are numerous methods that researchers are employing to create vaccinations against AD shown in . Beta-amyloid plaques are the focus of some strategies, while tau protein and immunomodulation are the focus of other strategies.[Citation124]
Additional ad interventional strategies
One of the insights from the vast quantity of studies carried out on AD is that this pathology is founded on a number of interrelated biological events, and an all-inclusive approach could help in the creation of highly effective therapeutics. For example, the creation of multitarget ligands (MTDLs), or substances having the ability of engaging multiple disease-related targets, is becoming increasingly important.[Citation324]
One of the most popular MTDL design methodologies is integrating many pharmacophores offered by certain biological activities into a single molecule, either directly or by means of suitable linkers. These compounds may work on numerous fronts to more effectively halt the neurodegenerative process since they are built to maintain their interaction with specific binding sites.
Conclusion
Neurodegenerative illnesses, such as AD, will significantly rise in the ensuing decades. Therefore, it is crucial to conduct research and advance biotechnology in order to develop novel preventative and therapeutic solutions. While current FDA-approved medications offer some benefit, their efficacy diminishes after three years, prompting the search for novel medications. Using phytochemicals, which possess properties addressing key aspects of AD’s pathophysiology, becomes a promising strategy for effective prevention and treatment. The tau protein’s role in promoting microtubule assembly is essential in the healthy brain, while abnormal tau phosphorylation is observed in conditions like AD and other neurological disorders. AD is characterized by excessive tau phosphorylation, leading to structural changes and loss of function. Amyloid plaques, another hallmark of AD, form due to abnormal cleavage of the transmembrane protein APP. The main factor influencing amyloid plaque formation is aging, and they are believed to accumulate both extracellularly and within neural processes.
Acknowledgement
Special thanks to Kampala International University, Uganda and all contributors for their support and valuable input in the creation of this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data available on request from the authors.
Additional information
Funding
References
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer’s Disease, a Multifactorial Disorder Seeking Multitherapies. Alzheimer’s & Dementia. 2010, 6(5), 420–424. DOI: 10.1016/j.jalz.2010.04.006.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019, 24(8), 1583. DOI: 10.3390/molecules24081583.
- Cobley, J. N.; Fiorello, M. L.; Bailey, D. M. 13 Reasons Why the Brain is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. DOI: 10.1016/j.redox.2018.01.008.
- Oboh, G.; Rocha, J. B. T. Antioxidant and Neuroprotective Properties of Sour Tea (Hibiscus Sabdariffa, Calyx) and Green Tea (Camellia Sinensis) on Some Pro-Oxidant-Induced Lipid Peroxidation in Brain In Vitro. Food Biophys. 2008, 3(4), 382–389. DOI: 10.1007/s11483-008-9092-5.
- Isik, A. T. Late Onset Alzheimer’s Disease in Older People. Clin. Interventions Aging. 2010, 307–311. DOI: 10.2147/CIA.S11718.
- Macdonald, I. R.; Rockwood, K.; Martin, E.; Darvesh, S. Cholinesterase Inhibition in Alzheimer’s Disease: Is Specificity the Answer? J. Alzheimer’s Dis. 2014, 42(2), 379–384. DOI: 10.3233/JAD-140219.
- Webber, K. M.; Raina, A. K.; Marlatt, M. W.; Zhu, X.; Prat, M. I.; Morelli, L.; Smith, M. A. The Cell Cycle in Alzheimer Disease: A Unique Target for Neuropharmacology. Mech. Ageing Dev. 2005, 126(10), 1019–1025. DOI: 10.1016/j.mad.2005.03.024.
- Michalke, B.; Willkommen, D.; Drobyshev, E.; Solovyev, N. The Importance of Speciation Analysis in Neurodegeneration Research. TrAc Trends Anal. Chem. 2018, 104, 160–170. DOI: 10.1016/j.trac.2017.08.008.
- Oboh, G.; Agunloye, O. M.; Akinyemi, A. J.; Ademiluyi, A. O.; Adefegha, S. A. Comparative Study on the Inhibitory Effect of Caffeic and Chlorogenic Acids on Key Enzymes Linked to Alzheimer’s Disease and Some Pro-Oxidant Induced Oxidative Stress in rats’ Brain-In Vitro. Neurochem. Res. 2013a, 38(2), 413–419. DOI: 10.1007/s11064-012-0935-6.
- Zago, M. P.; Verstraeten, S. V.; Oteiza, P. I. Zinc in the Prevention of Fe2+ initiated Lipid and Protein Oxidation. Biol. Res. 2000, 33, 143–150. DOI: 10.4067/S0716-97602000000200014.
- Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, A. M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The Worldwide Costs of Dementia 2015 and Comparisons with 2010. Alzheimer’s & Dementia. 2017, 13, 1–7. DOI: 10.1016/j.jalz.2016.07.150.
- Herrmann, L. K.; Welter, E.; Leverenz, J.; Lerner, A. J.; Udelson, N.; Kanetsky, C.; Sajatovic, M. A Systematic Review of Dementia-Related Stigma Research: Can We Move the Stigma Dial? Am. J. Geriatric Psychiatry. 2018, 26(3), 316–331. DOI: 10.1016/j.jagp.2017.09.006.
- Podcasy, J. L.; Epperson, C. N. Considering Sex and Gender in Alzheimer Disease and Other Dementias. Dialogues Clin. Neurosci. 2022, 18, 2016(4), 437–446.
- Mahdi, O.; Baharuldin, M. T. H.; Nor, N. H. M.; Chiroma, S. M.; Jagadeesan, S.; Moklas, M. A. M. Chemicals Used for the Induction of Alzheimer’s Disease-Like Cognitive Dysfunctions in Rodents. Biomed. Res. Ther. 2019, 6, 3460–3484. DOI: 10.15419/bmrat.v6i11.575.
- Nakanishi, M.; Igarashi, A.; Ueda, K.; Brnabic, A. J. M.; Treuer, T.; Sato, M.; Kahle-Wrobleski, K.; Meguro, K.; Yamada, M.; Mimura, M. Costs and Resource Use Associated with Community-Dwelling Patients with Alzheimer’s Disease in Japan: Baseline Results from the Prospective Observational GERAS-J Study. J. Alzheimer’s Dis. 2020, 74, 127–138. DOI: 10.3233/JAD-190811.
- Ferreira, M. E. S.; de Vasconcelos, A. S.; da Costa Vilhena, T.; da Silva, T. L.; da Silva Barbosa, A.; Gomes, A. R. Q.; Dolabela, M. F.; Percário, S. Oxidative Stress in Alzheimer’s Disease: Should We Keep Trying Antioxidant Therapies? Cell Mol. Neurobiol. 2015, 35, 595–614. DOI: 10.1007/s10571-015-0157-y.
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 29. DOI: 10.1038/s41392-019-0063-8.
- Peera, K.; Yellamma, K. Sericin as a Chlinergic Modulator in Alzaeimer’s Disease Induced Rat. Int. J. Pharm. Pharm. Sci. 2015, 7, 108–112.
- Wu, T.; Dejanovic, B.; Gandham, V. D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M. A.; Wang, Y.; Wang, T.-M. Complement C3 is Activated in Human AD Brain and is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell. Rep. 2019, 28, 2111–2123. DOI: 10.1016/j.celrep.2019.07.060.
- Butterfield, D. A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20(3), 148–160. DOI: 10.1038/s41583-019-0132-6.
- Markesbery, W. R.; Lovell, M. A. Damage to Lipids, Proteins, DNA, and RNA in Mild Cognitive Impairment. Arch. Neurol. 2007, 64(7), 954–956. DOI: 10.1001/archneur.64.7.954.
- Waldemar, G.; Dubois, B.; Emre, M.; Georges, J.; McKeith, I. G.; Rossor, M.; Scheltens, P.; Tariska, P.; Winblad, B. Recommendations for the Diagnosis and Management of Alzheimer’s Disease and Other Disorders Associated with Dementia: EFNS Guideline. Eur. J. Neurol. 2007, 14(1), e1–e26. DOI: 10.1111/j.1468-1331.2006.01605.x.
- Ritchie, C.; Smailagic, N.; Noel‐Storr, A. H.; Takwoingi, Y.; Flicker, L.; Mason, S. E.; McShane, R. Plasma and Cerebrospinal Fluid Amyloid Beta for the Diagnosis of Alzheimer’s Disease Dementia and Other Dementias in People with Mild Cognitive Impairment (MCI). Cochrane Database Syst. Rev. 2014, 6. DOI: 10.1002/14651858.CD008782.pub4.
- Bäckman, L.; Jones, S.; Berger, A.; Laukka, E. J.; Small, B. J. Multiple Cognitive Deficits During the Transition to Alzheimer’s Disease. J. Intern. Med. 2004, 256(3), 195–204. DOI: 10.1111/j.1365-2796.2004.01386.x.
- Stella, F.; Radanovic, M.; Balthazar, M. L. F.; Canineu, P. R.; de Souza, L. C.; Forlenza, O. V. Neuropsychiatric Symptoms in the Prodromal Stages of Dementia. Curr. Opin. Psychiatry. 2014, 27, 230–235. DOI: 10.1097/YCO.0000000000000050.
- Anderson, N. D. State of the Science on Mild Cognitive Impairment (MCI). CNS Spectr. 2019, 24(1), 78–87. DOI: 10.1017/S1092852918001347.
- Petersen, S. L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFα Signaling Renders Human Cancer Cells Susceptible to Smac-Mimetic-Induced Apoptosis. Cancer Cell. 2007, 12(5), 445–456. DOI: 10.1016/j.ccr.2007.08.029.
- Perrotta, G. Alzheimer’s Disease: Definition, Contexts, Neural Correlates, Strategies and Clinical Approaches. J. Aging Stud. Ther. 2019, 1, 15.
- Doolen, A. C.; Radvansky, G. A. A Novel Study: Long-Lasting Event Memory. Memory. 2021, 29(8), 963–982. DOI: 10.1080/09658211.2021.1953079.
- Fraser, K. C.; Meltzer, J. A.; Rudzicz, F. Linguistic Features Identify Alzheimer’s Disease in Narrative Speech. J. Alzheimer’s Dis. 2016, 49, 407–422. DOI: 10.3233/JAD-150520.
- Svanström, R.; Sundler, A. J. Gradually Losing One’s Foothold–A Fragmented Existence When Living Alone with Dementia. Dementia. 2015, 14(2), 145–163. DOI: 10.1177/1471301213494510.
- Nishio, Y.; Yokoi, K.; Hirayama, K.; Ishioka, T.; Hosokai, Y.; Gang, M.; Uchiyama, M.; Baba, T.; Suzuki, K.; Takeda, A. Defining Visual Illusions in Parkinson’s Disease: Kinetopsia and Object Misidentification Illusions. Parkinsonism Relat. Disord. 2018, 55, 111–116. DOI: 10.1016/j.parkreldis.2018.05.023.
- Wimo, A.; Reed, C. C.; Dodel, R.; Belger, M.; Jones, R. W.; Happich, M.; Argimon, J. M.; Bruno, G.; Novick, D.; Vellas, B. The GERAS Study: A Prospective Observational Study of Costs and Resource Use in Community Dwellers with Alzheimer’s Disease in Three European Countries–Study Design and Baseline Findings. J. Alzheimer’s Dis. 2013, 36, 385–399. DOI: 10.3233/JAD-122392.
- Tarawneh, R.; Holtzman, D. M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2(5), a006148. DOI: 10.1101/cshperspect.a006148.
- Sayeed Ahmad, S.; Akhtar, S.; Mohammad Sajid Jamal, Q.; A Kamal, M.; Khan, K. A.; Siddiqui, H. Multiple Targets for the Management of Alzheimer’s Disease. CNS Neurol. Disord-Drug Targets (Formerly Current Drug Targets-CNS & Neurol. Disord). 2016, 15, 1279–1289. DOI: 10.2174/1871527315666161003165855.
- Goedert, M.; Spillantini, M. G. Propagation of Tau Aggregates. Mol. Brain. 2017, 10(1), 1–9. DOI: 10.1186/s13041-017-0298-7.
- Onatsu, J.; Vanninen, R.; JÄkÄlÄ, P.; Mustonen, P.; Pulkki, K.; Korhonen, M.; Hedman, M.; Höglund, K.; Blennow, K.; Zetterberg, H. Tau, S100B and NSE as Blood Biomarkers in Acute Cerebrovascular Events. Vivo (Brooklyn). 2020, 34, 2577–2586. DOI: 10.21873/invivo.12075.
- Chornenkyy, Y.; Fardo, D. W.; Nelson, P. T. Tau and TDP-43 Proteinopathies: Kindred Pathologic Cascades and Genetic Pleiotropy. Lab. Invest. 2019, 99(7), 993–1007. DOI: 10.1038/s41374-019-0196-y.
- Williams, D. R. Tauopathies: Classification and Clinical Update on Neurodegenerative Diseases Associated with Microtubule‐Associated Protein Tau. Intern. Med. J. 2006, 36(10), 652–660. DOI: 10.1111/j.1445-5994.2006.01153.x.
- Hensley, K.; Kursula, P. Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau. J. Alzheimer’s Dis. 2016, 53, 1–14. DOI: 10.3233/JAD-160076.
- Dickson, D. W.; Braak, H.; Duda, J. E.; Duyckaerts, C.; Gasser, T.; Halliday, G. M.; Hardy, J.; Leverenz, J. B.; Del Tredici, K.; Wszolek, Z. K. Neuropathological Assessment of Parkinson’s Disease: Refining the Diagnostic Criteria. Lancet. Neurol. 2009, 8, 1150–1157. DOI: 10.1016/S1474-4422(09)70238-8.
- Goedert, M. Tau Gene Mutations and Their Effects. Mov. Disord. 2005, 20(S12), S45–S52. DOI: 10.1002/mds.20539.
- Kadas, D.; Papanikolopoulou, K.; Xirou, S.; Consoulas, C.; Skoulakis, E. M. C. Human Tau Isoform-Specific Presynaptic Deficits in a Drosophila Central Nervous System Circuit. Neurobiol. Dis. 2019, 124, 311–321. DOI: 10.1016/j.nbd.2018.12.004.
- Sergeant, J. A. Modeling Attention-Deficit/hyperactivity Disorder: A Critical Appraisal of the Cognitive-Energetic Model. Biol. Psychiatry. 2005, 57(11), 1248–1255. DOI: 10.1016/j.biopsych.2004.09.010.
- Iqbal, K.; Gong, C.-X.; Liu, F. Microtubule-Associated Protein Tau as a Therapeutic Target in Alzheimer’s Disease. Expert Opin. Ther. Targets. 2014, 18, 307–318. DOI: 10.1517/14728222.2014.870156.
- Henderson, M. X.; Sengupta, M.; Trojanowski, J. Q.; Lee, V. M. Y. Alzheimer’s Disease Tau is a Prominent Pathology in LRRK2 Parkinson’s Disease. Acta Neuropathol. Commun. 2019, 7, 1–16. DOI: 10.1186/s40478-019-0836-x.
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 2016, 6(1), 6. DOI: 10.3390/biom6010006.
- Castellani, R. J.; Perry, G. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J. Alzheimer’s Dis. 2019, 67, 447–467. DOI: 10.3233/JAD-180721.
- Nizynski, B.; Dzwolak, W.; Nieznanski, K. Amyloidogenesis of Tau Protein. Protein Sci. 2017, 26(11), 2126–2150. DOI: 10.1002/pro.3275.
- Mokhtar, S. H.; Bakhuraysah, M. M.; Cram, D. S.; Petratos, S. The Beta-Amyloid Protein of Alzheimer’s Disease: Communication Breakdown by Modifying the Neuronal Cytoskeleton. Int. J. Alzheimers Dis. 2013, 2013, 1–15. DOI: 10.1155/2013/910502.
- Luna-Viramontes, N. I.; Campa-Córdoba, B. B.; Ontiveros-Torres, M. Á.; Harrington, C. R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; de la Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 247. DOI: 10.3389/fncel.2020.00247.
- De Vos, K. J.; Hafezparast, M. Neurobiology of Axonal Transport Defects in Motor Neuron Diseases: Opportunities for Translational Research? Neurobiol. Dis. 2017, 105, 283–299. DOI: 10.1016/j.nbd.2017.02.004.
- Ising, C.; Heneka, M. T. Functional and Structural Damage of Neurons by Innate Immune Mechanisms During Neurodegeneration. Cell Death Dis. 2018, 9(2), 120. DOI: 10.1038/s41419-017-0153-x.
- Thornton, C.; Bright, N. J.; Sastre, M.; Muckett, P. J.; Carling, D. AMP-Activated Protein Kinase (AMPK) is a Tau Kinase, Activated in Response to Amyloid β-Peptide Exposure. Biochem. J. 2011, 434(3), 503–512. DOI: 10.1042/BJ20101485.
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and Pathological Phosphorylation of Tau by Cdk5. Front Mol. Neurosci. 2014, 7, 65. DOI: 10.3389/fnmol.2014.00065.
- Forner, S.; Baglietto-Vargas, D.; Martini, A. C.; Trujillo-Estrada, L.; LaFerla, F. M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017, 40, 347–357. DOI: 10.1016/j.tins.2017.04.002.
- Maccioni, R. B.; Farías, G.; Morales, I.; Navarrete, L. The Revitalized Tau Hypothesis on Alzheimer’s Disease. Arch. Med. Res. 2010, 41(3), 226–231. DOI: 10.1016/j.arcmed.2010.03.007.
- Amadoro, G.; Corsetti, V.; Ciotti, M. T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogenous Aβ Causes Cell Death via Early Tau Hyperphosphorylation. Neurobiol. Aging. 2011, 32(6), 969–990. DOI: 10.1016/j.neurobiolaging.2009.06.005.
- Roberson, E. D.; Scearce-Levie, K.; Palop, J. J.; Yan, F.; Cheng, I. H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid ß-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science. 2007, 316, 750–754. DOI: 10.1126/science.1141736.
- Bulic, B.; Pickhardt, M.; Mandelkow, E.-M.; Mandelkow, E. Tau Protein and Tau Aggregation Inhibitors. Neuropharmacology. 2010, 59(4–5), 276–289. DOI: 10.1016/j.neuropharm.2010.01.016.
- Pîrşcoveanu, D. F. V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.-A.; Albu, V.-C.; Bondari, S.; Bumbea, A. M.; Pîrşcoveanu, M. Tau Protein in Neurodegenerative Diseases-A Review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150.
- Alonso, A. D. C.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. Polymerization of Hyperphosphorylated Tau into Filaments Eliminates Its Inhibitory Activity. Proc. Natl. Acad. Sci. USA. 2006, 103(23), 8864–8869. DOI: 10.1073/pnas.0603214103.
- Cowan, C. M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble Hyper-Phosphorylated Tau Causes Microtubule Breakdown and Functionally Compromises Normal Tau In Vivo. Acta. Neuropathol. 2010, 120(5), 593–604. DOI: 10.1007/s00401-010-0716-8.
- Corbo, C. P.; Alonso, A. D. C. Therapeutic Targets in Alzheimer’s Disease and Related Tauopathies. Prog. mol. biol. transl. sci. 2011, 98, 47–83.
- Steinhilb, M. L.; Dias‐Santagata, D.; Mulkearns, E. E.; Shulman, J. M.; Biernat, J.; Mandelkow, E.; Feany, M. B. S/P and T/P Phosphorylation is Critical for Tau Neurotoxicity in Drosophila. J. Neurosci. Res. 2007, 85(6), 1271–1278. DOI: 10.1002/jnr.21232.
- Blard, O.; Feuillette, S.; Bou, J.; Chaumette, B.; Frébourg, T.; Campion, D.; Lecourtois, M. Cytoskeleton Proteins are Modulators of Mutant Tau-Induced Neurodegeneration in Drosophila. Hum. Mol. Genet. 2007, 16(5), 555–566. DOI: 10.1093/hmg/ddm011.
- Bilkei-Gorzo, A. Genetic Mouse Models of Brain Ageing and Alzheimer’s Disease. Pharmacol. Ther. 2014, 142(2), 244–257. DOI: 10.1016/j.pharmthera.2013.12.009.
- Takahashi, R. H.; Nagao, T.; Gouras, G. K. Plaque Formation and the Intraneuronal Accumulation of β‐Amyloid in Alzheimer’s Disease. Pathol. Int. 2017, 67, 185–193. DOI: 10.1111/pin.12520.
- Soldano, A.; Hassan, B. A. Beyond Pathology: APP, Brain Development and Alzheimer’s Disease. Curr. Opin. Neurobiol. 2014, 27, 61–67. DOI: 10.1016/j.conb.2014.02.003.
- Ryu, J.; Yu, H.-N.; Cho, H.; Kim, H.-S.; Baik, T.-K.; Lee, S.-J.; Woo, R.-S. Neuregulin-1 Exerts Protective Effects Against Neurotoxicities Induced by C-Terminal Fragments of APP via ErbB4 Receptor. J. Pharmacol. Sci. 2012, 119(1), 73–81. DOI: 10.1254/jphs.12057FP.
- Barthelson, K.; Newman, M.; Lardelli, M. Sorting Out the Role of the Sortilin-Related Receptor 1 in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2020, 4, 123–140. DOI: 10.3233/ADR-200177.
- Nguyen, T. V.; Galvan, V.; Huang, W.; Banwait, S.; Tang, H.; Zhang, J.; Bredesen, D. E. Signal Transduction in Alzheimer Disease: P21‐Activated Kinase Signaling Requires C‐Terminal Cleavage of APP at Asp664. J. Neurochem. 2008, 104, 1065–1080. DOI: 10.1111/j.1471-4159.2007.05031.x.
- Muresan, V.; Varvel, N. H.; Lamb, B. T.; Muresan, Z. The Cleavage Products of Amyloid-β Precursor Protein are Sorted to Distinct Carrier Vesicles That are Independently Transported within Neurites. J. Neurosci. 2009, 29, 3565–3578. DOI: 10.1523/JNEUROSCI.2558-08.2009.
- Chakravarthy, B.; Gaudet, C.; Ménard, M.; Atkinson, T.; Brown, L.; LaFerla, F. M.; Armato, U.; Whitfield, J. Amyloid-β Peptides Stimulate the Expression of the p75 NTR Neurotrophin Receptor in SHSY5Y Human Neuroblastoma Cells and AD Transgenic Mice. J. Alzheimer’s Dis. 2010, 19, 915–925. DOI: 10.3233/JAD-2010-1288.
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin Response Mediator Protein-2: An Emerging Pathologic Feature and Therapeutic Target for Neurodisease Indications. Mol. Neurobiol. 2011, 43(3), 180–191. DOI: 10.1007/s12035-011-8166-4.
- Heneka, M. T.; Carson, M. J.; El Khoury, J.; Landreth, G. E.; Brosseron, F.; Feinstein, D. L.; Jacobs, A. H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R. M. Neuroinflammation in Alzheimer’s Disease. Lancet. Neurol. 2015, 14, 388–405. DOI: 10.1016/S1474-4422(15)70016-5.
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 1–13. DOI: 10.1155/2019/5080843.
- Busche, M. A.; Hyman, B. T. Synergy Between Amyloid-β and Tau in Alzheimer’s Disease. Nat. Neurosci. 2020, 23(10), 1183–1193. DOI: 10.1038/s41593-020-0687-6.
- Muresan, Z.; Muresan, V. Neuritic Deposits of Amyloid-β Peptide in a Subpopulation of Central Nervous System-Derived Neuronal Cells. Mol. Cell. Biol. 2006, 26, 4982–4997. DOI: 10.1128/MCB.00371-06.
- Kanaan, N. M.; Morfini, G.; Pigino, G.; LaPointe, N. E.; Andreadis, A.; Song, Y.; Leitman, E.; Binder, L. I.; Brady, S. T. Phosphorylation in the Amino Terminus of Tau Prevents Inhibition of Anterograde Axonal Transport. Neurobiol. Aging. 2012, 33, 826–e15. DOI: 10.1016/j.neurobiolaging.2011.06.006.
- Kumar, S.; Walter, J. Phosphorylation of Amyloid Beta (Aβ) Peptides–A Trigger for Formation of Toxic Aggregates in Alzheimer’s Disease. Aging (Albany N.Y). 2011, 3, 803. DOI: 10.18632/aging.100362.
- Morris, G. P.; Clark, I. A.; Vissel, B. Questions Concerning the Role of Amyloid-β in the Definition, Aetiology and Diagnosis of Alzheimer’s Disease. Acta. Neuropathol. 2018, 136(5), 663–689. DOI: 10.1007/s00401-018-1918-8.
- Devanand, D. P.; Schupf, N.; Stern, Y.; Parsey, R.; Pelton, G. H.; Mehta, P.; Mayeux, R. Plasma Aβ and PET PiB Binding are Inversely Related in Mild Cognitive Impairment. Neurology. 2011, 77, 125–131. DOI: 10.1212/WNL.0b013e318224afb7.
- Um, J. W.; Nygaard, H. B.; Heiss, J. K.; Kostylev, M. A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E. C.; Strittmatter, S. M. Alzheimer Amyloid-β Oligomer Bound to Postsynaptic Prion Protein Activates Fyn to Impair Neurons. Nat. Neurosci. 2012, 15(9), 1227–1235. DOI: 10.1038/nn.3178.
- Grundman, M.; Petersen, R. C.; Ferris, S. H.; Thomas, R. G.; Aisen, P. S.; Bennett, D. A.; Foster, N. L.; Jack, C. R., Jr; Galasko, D. R.; Doody, R. Mild Cognitive Impairment Can Be Distinguished from Alzheimer Disease and Normal Aging for Clinical Trials. Arch. Neurol. 2004, 61, 59–66. DOI: 10.1001/archneur.61.1.59.
- Arnsten, A. F. T.; Datta, D.; Preuss, T. M. Studies of Aging Nonhuman Primates Illuminate the Etiology of Early‐Stage Alzheimer’s‐Like Neuropathology: An Evolutionary Perspective. Am. J. Primatol. 2021, 83(11), e23254. DOI: 10.1002/ajp.23254.
- Nussbaum, L.; Hogea, L. M.; Calina, D.; Andreescu, N.; Gradinaru, R.; Stefanescu, R.; Puiu, M. Modern Treatment Approaches in Psychoses. Pharmacogenetic, Neuroimagistic and Clinical Implications. Farmacia. 2017, 65, 75–81.
- Zucchella, C.; Sinforiani, E.; Tamburin, S.; Federico, A.; Mantovani, E.; Bernini, S.; Casale, R.; Bartolo, M. The Multidisciplinary Approach to Alzheimer’s Disease and Dementia. A Narrative Review of Non-Pharmacological Treatment. Front. Neurol. 2018, 9, 1058. DOI: 10.3389/fneur.2018.01058.
- Calina, D.; Buga, A. M.; Mitroi, M.; Buha, A.; Caruntu, C.; Scheau, C.; Bouyahya, A.; El Omari, N.; El Menyiy, N.; Docea, A. O. The Treatment of Cognitive, Behavioural and Motor Impairments from Brain Injury and Neurodegenerative Diseases Through Cannabinoid System Modulation—Evidence from In Vivo Studies. J. Clin. Med. 2020a, 9, 2395. DOI: 10.3390/jcm9082395.
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D. A.; Docea, A. O.; Mahomoodally, M. F.; Lobine, D.; Chazot, P. L.; Kurt, B.; Boyunegmez Tumer, T.; Catarina Moreira, A. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9(4), 1061. DOI: 10.3390/jcm9041061.
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. North America. 2019, 103(2), 263–293. DOI: 10.1016/j.mcna.2018.10.009.
- Howard, R.; Liu, K. Y. Questions EMERGE as Biogen Claims Aducanumab Turnaround. Nat. Rev. Neurol. 2020, 16(2), 63–64. DOI: 10.1038/s41582-019-0295-9.
- Yiannopoulou, K. G.; Papageorgiou, S. G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. DOI: 10.1177/1179573520907397.
- Finn, L. A. Current Medications for the Treatment of Alzheimer’s Disease: Acetylcholinesterase Inhibitors and NMDA Receptor Antagonist, in Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders; Elsevier, 2017; pp. 49–58.
- Tricco, A. C.; Ashoor, H. M.; Soobiah, C.; Rios, P.; Veroniki, A. A.; Hamid, J. S.; Ivory, J. D.; Khan, P. A.; Yazdi, F.; Ghassemi, M. Comparative Effectiveness and Safety of Cognitive Enhancers for Treating Alzheimer’s Disease: Systematic Review and Network Metaanalysis. J. Am. Geriatr. Soc. 2018, 66, 170–178. DOI: 10.1111/jgs.15069.
- Wang, R.; Reddy, P. H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. DOI: 10.3233/JAD-160763.
- Korábečný, J.; Nepovimova, E.; Cikankova, T.; Špilovská, K.; Vašková, L.; Mezeiova, E.; Kuča, K.; Hroudova, J. Newly Developed Drugs for Alzheimer’s Disease in Relation to Energy Metabolism, Cholinergic and Monoaminergic Neurotransmission. Neuroscience. 2018, 370, 191–206. DOI: 10.1016/j.neuroscience.2017.06.034.
- Islam, M. S.; Quispe, C.; Hossain, R.; Islam, M. T.; Al-Harrasi, A.; Al-Rawahi, A.; Martorell, M.; Mamurova, A.; Seilkhan, A.; Altybaeva, N. Neuropharmacological Effects of Quercetin: A Literature-Based Review. Front. Pharmacol. 2021, 12, 665031. DOI: 10.3389/fphar.2021.665031.
- Tsoukalas, D.; Zlatian, O.; Mitroi, M.; Renieri, E.; Tsatsakis, A.; Izotov, B. N.; Burada, F.; Sosoi, S.; Burada, E.; Buga, A. M. A Novel Nutraceutical Formulation Can Improve Motor Activity and Decrease the Stress Level in a Murine Model of Middle-Age Animals. J. Clin. Med. 2021b, 10(4), 624. DOI: 10.3390/jcm10040624.
- Aronson, J. K. Defining ‘Nutraceuticals’: Neither Nutritious nor Pharmaceutical. Br.J. Clin. Pharmacol. 2017, 83(1), 8–19. DOI: 10.1111/bcp.12935.
- Santini, A.; Cammarata, S. M.; Capone, G.; Ianaro, A.; Tenore, G. C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the Debate for a Regulatory Framework. Br.J. Clin. Pharmacol. 2018, 84(4), 659–672. DOI: 10.1111/bcp.13496.
- Ayaz, M.; Ullah, F.; Sadiq, A.; Kim, M. O.; Ali, T. Natural Products-Based Drugs: Potential Therapeutics Against Alzheimer’s Disease and Other Neurological Disorders; Frontiers Media SA, 2019a. DOI:10.3389/978-2-88963-348-7.
- Cassidy, L.; Fernandez, F.; Johnson, J. B.; Naiker, M.; Owoola, A. G.; Broszczak, D. A. Oxidative Stress in Alzheimer’s Disease: A Review on Emergent Natural Polyphenolic Therapeutics. Complement Ther. Med. 2020, 49, 102294. DOI: 10.1016/j.ctim.2019.102294.
- Gustavsson, J.; Cederberg, C.; Sonesson, U.; Van Otterdijk, R.; Meybeck, A. Global food losses and food waste: Extent causes and prevention. In Extent of food losses and waste. Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2011; pp 4–10.
- Sharifi-Rad, J.; Herrera-Bravo, J.; Kamiloglu, S.; Petroni, K.; Mishra, A. P.; Monserrat-Mesquida, M.; Sureda, A.; Martorell, M.; Aidarbekovna, D. S.; Yessimsiitova, Z. Recent Advances in the Therapeutic Potential of Emodin for Human Health. Biomed. Pharmacother. 2022a, 154, 113555. DOI: 10.1016/j.biopha.2022.113555.
- Gul, K.; Singh, A. K.; Jabeen, R. Nutraceuticals and Functional Foods: The Foods for the Future World. Crit. Rev. Food Sci. Nutr. 2016, 56(16), 2617–2627. DOI: 10.1080/10408398.2014.903384.
- Kitic, D.; Miladinovic, B.; Randjelovic, M.; Szopa, A.; Sharifi-Rad, J.; Calina, D.; Seidel, V. Anticancer Potential and Other Pharmacological Properties of Prunus armeniaca L.: An Updated Overview. Plants. 2022, 11(14), 1885. DOI: 10.3390/plants11141885.
- Taroncher, M.; Vila-Donat, P.; Tolosa, J.; Ruiz, M. J.; Rodríguez-Carrasco, Y. Biological Activity and Toxicity of Plant Nutraceuticals: An Overview. Curr. Opin. Food Sci. 2021, 42, 113–118. DOI: 10.1016/j.cofs.2021.05.008.
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S. Endoplasmic Reticulum Stress Signalling–From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286(2), 241–278. DOI: 10.1111/febs.14608.
- Kim, G. H.; Kim, J. E.; Rhie, S. J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24(4), 325. DOI: 10.5607/en.2015.24.4.325.
- Reyes-Fermín, L. M.; Aparicio-Trejo, O. E.; Avila-Rojas, S. H.; Gómez-Sierra, T.; Martínez-Klimova, E.; Pedraza-Chaverri, J. Natural antioxidants’ Effects on Endoplasmic Reticulum Stress-Related Diseases. Food Chem. Toxicol. 2020, 138, 111229. DOI: 10.1016/j.fct.2020.111229.
- Chen, X.; Drew, J.; Berney, W.; Lei, W. Neuroprotective Natural Products for Alzheimer’s Disease. Cells. 2021, 10(6), 1309. DOI: 10.3390/cells10061309.
- Tsoukalas, D.; Buga, A. M.; Docea, A. O.; Sarandi, E.; Mitrut, R.; Renieri, E.; Spandidos, D. A.; Rogoveanu, I.; Cercelaru, L.; Niculescu, M. Reversal of Brain Aging by Targeting Telomerase: A Nutraceutical Approach. Int. J. Mol. Med. 2021a, 48(5), 1–11. DOI: 10.3892/ijmm.2021.5032.
- Venkatesan, R.; Ji, E.; Kim, S. Y. Phytochemicals That Regulate Neurodegenerative Disease by Targeting Neurotrophins: A Comprehensive Review. Biomed Res. Int. 2015, 2015, 1–22. DOI: 10.1155/2015/814068.
- Tsoukalas, D.; Fragkiadaki, P.; Docea, A. O.; Alegakis, A. K.; Sarandi, E.; Thanasoula, M.; Spandidos, D. A.; Tsatsakis, A.; Razgonova, M. P.; Calina, D. Discovery of Potent Telomerase Activators: Unfolding New Therapeutic and Anti-Aging Perspectives. Mol. Med. Rep. 2019, 20, 3701–3708. DOI: 10.3892/mmr.2019.10614.
- Calina, D.; Buga, A. M.; Mitroi, M.; Buha, A.; Caruntu, C.; Scheau, C.; Bouyahya, A.; El Omari, N.; El Menyiy, N.; Docea, A. O. The Treatment of Cognitive, Behavioural and Motor Impairments from Brain Injury and Neurodegenerative Diseases Through Cannabinoid System Modulation—Evidence from In Vivo Studies. J. Clin. Med. 2020b, 9, 2395. DOI: 10.3390/jcm9082395.
- Kwan, P.; Konno, H.; Chan, K. Y.; Baum, L. Rationale for the Development of an Alzheimer’s Disease Vaccine. Hum. Vaccin. Immunother. 2020, 16(3), 645–653. DOI: 10.1080/21645515.2019.1665453.
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P. FUNDAMANT: An Interventional 72-Week Phase 1 Follow-Up Study of AADvac1, an Active Immunotherapy Against Tau Protein Pathology in Alzheimer’s Disease. Alzheimers Res. Ther. 2018, 10, 1–16. DOI: 10.1186/s13195-018-0436-1.
- Wang, C. Y.; Wang, P.-N.; Chiu, M.-J.; Finstad, C. L.; Lin, F.; Lynn, S.; Tai, Y.-H.; De Fang, X.; Zhao, K.; Hung, C.-H. UB-311, a Novel UBITh® Amyloid β Peptide Vaccine for Mild Alzheimer’s Disease. Alzheimer’s & Dementia: Transl. Res. Clin. Interventions. 2017a, 3, 262–272. DOI: 10.1016/j.trci.2017.03.005.
- Molina, E. P.; Pesini, P.; Sarasa‐SanJose, M.; Marcos, I.; Lacosta, A. M.; Allué, J. A.; Fandos, N.; Sarasa, M.; Boada, M.; Group, A. P. S. Safety, Tolerability and Immunogenicity of an Active Anti‐Aβ40 Vaccine (ABvac40) in Patients with Amnestic Mild Cognitive Impairment (A‐MCI) or Very Mild Alzheimer’s Disease (VM‐AD): A Randomized, Double‐Blind, Placebo‐Controlled, Phase II Trial: Human/Human Trials: Anti‐Amyloid. Alzheimer’s & Dementia. 2020, 16, e045720.
- Rafii, M. S.; Sol, O.; Mobley, W. C.; Delpretti, S.; Skotko, B. G.; Burke, A. D.; Sabbagh, M. N.; Yuan, S. H.; Rissman, R. A.; Pulsifer, M. Safety, Tolerability, and Immunogenicity of the ACI-24 Vaccine in Adults with Down Syndrome: A Phase 1b Randomized Clinical Trial. JAMA Neurol. 2022, 79(6), 565–574. DOI: 10.1001/jamaneurol.2022.0983.
- Koh, S.-H.; Kwon, H. S.; Choi, S. H.; Jeong, J. H.; Na, H. R.; Lee, C. N.; Yang, Y.; Lee, A. Y.; Lee, J.-H.; Park, K. W. Efficacy and Safety of GV1001 in Patients with Moderate-To-Severe Alzheimer’s Disease Already Receiving Donepezil: A Phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. Alzheimers Res. Ther. 2021, 13, 1–11. DOI: 10.1186/s13195-021-00803-w.
- Dow, C. T.; Greenblatt, C. L.; Chan, E. D.; Dow, J. F. Evaluation of BCG Vaccination and Plasma Amyloid: A Prospective, Pilot Study with Implications for Alzheimer’s Disease. Microorganisms. 2022, 10(2), 424. DOI: 10.3390/microorganisms10020424.
- Cacabelos, R. How Plausible is an Alzheimer’s Disease Vaccine? Expert Opin. Drug Discov. 2020, 15(1), 1–6. DOI: 10.1080/17460441.2019.1667329.
- Farooqui, A. A. Therapeutic Potentials of Curcumin for Alzheimer Disease; Springer International Publishing, 2016. DOI:10.1007/978-3-319-15889-1.
- Libro, R.; Giacoppo, S.; Soundara Rajan, T.; Bramanti, P.; Mazzon, E. Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview. Molecules. 2016, 21(4), 518. DOI: 10.3390/molecules21040518.
- Piccialli, I.; Tedeschi, V.; Caputo, L.; D’Errico, S.; Ciccone, R.; De, F. V.; Secondo, A.; Pannaccione, A. Exploring the Therapeutic Potential of Phytochemicals in Alzheimer’s Disease: Focus on Polyphenols and Monoterpenes. Front. Pharmacol. 2022, 13. DOI: 10.3389/fphar.2022.876614.
- Cioanca, O.; Hritcu, L.; Mihasan, M.; Hancianu, M. Cognitive-Enhancing and Antioxidant Activities of Inhaled Coriander Volatile Oil in Amyloid β (1–42) Rat Model of Alzheimer’s Disease. Physiol. Behav. 2013, 120, 193–202. DOI: 10.1016/j.physbeh.2013.08.006.
- Karakaya, S.; Koca, M.; Yılmaz, S. V.; Yıldırım, K.; Pınar, N. M.; Demirci, B.; Brestic, M.; Sytar, O. Molecular Docking Studies of Coumarins Isolated from Extracts and Essential Oils of Zosima Absinthifolia Link as Potential Inhibitors for Alzheimer’s Disease. Molecules. 2019, 24, 722. DOI: 10.3390/molecules24040722.
- Arruda, M.; Viana, H.; Rainha, N.; Neng, N. R.; Rosa, J. S.; Nogueira, J. M. F.; Barreto, M. D. C. Anti-Acetylcholinesterase and Antioxidant Activity of Essential Oils from Hedychium Gardnerianum Sheppard Ex Ker-Gawl. Molecules. 2012, 17(3), 3082–3092. DOI: 10.3390/molecules17033082.
- Ayaz, M.; Junaid, M.; Ullah, F.; Sadiq, A.; Khan, M. A.; Ahmad, W.; Shah, M. R.; Imran, M.; Ahmad, S. Comparative Chemical Profiling, Cholinesterase Inhibitions and Anti-Radicals Properties of Essential Oils from Polygonum hydropiper L: A Preliminary Anti-Alzheimer’s Study. Lipids Health Dis. 2015, 14(1), 1–12. DOI: 10.1186/s12944-015-0145-8.
- Hancianu, M.; Cioanca, O.; Mihasan, M.; Hritcu, L. Neuroprotective Effects of Inhaled Lavender Oil on Scopolamine-Induced Dementia via Anti-Oxidative Activities in Rats. Phytomedicine. 2013, 20(5), 446–452. DOI: 10.1016/j.phymed.2012.12.005.
- Baum, L.; Lam, C. W. K.; Cheung, S.-K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients with Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. DOI: 10.1097/jcp.0b013e318160862c.
- Turner, R. S.; Thomas, R. G.; Craft, S.; Van Dyck, C. H.; Mintzer, J.; Reynolds, B. A.; Brewer, J. B.; Rissman, R. A.; Raman, R.; Aisen, P. S. A Randomized, Double-Blind, Placebo-Controlled Trial of Resveratrol for Alzheimer Disease. Neurology. 2015, 85, 1383–1391. DOI: 10.1212/WNL.0000000000002035.
- Kamal, Z.; Ullah, F.; Ayaz, M.; Sadiq, A.; Ahmad, S.; Zeb, A.; Hussain, A.; Imran, M. Anticholinesterse and Antioxidant Investigations of Crude Extracts, Subsequent Fractions, Saponins and Flavonoids of Atriplex Laciniata L.: Potential Effectiveness in Alzheimer’s and Other Neurological Disorders. Biol. Res. 2015, 48(1), 1–11. DOI: 10.1186/s40659-015-0011-1.
- Chen, Z.; Mao, X.; Liu, A.; Gao, X.; Chen, X.; Ye, M.; Ye, J.; Liu, P.; Xu, S.; Liu, J. Osthole, a Natural Coumarin Improves Cognitive Impairments and BBB Dysfunction After Transient Global Brain Ischemia in C57 BL/6J Mice: Involvement of Nrf2 Pathway. Neurochem. Res. 2015, 40(1), 186–194. DOI: 10.1007/s11064-014-1483-z.
- Kaur, N.; Dhiman, M.; Perez‐Polo, J. R.; Mantha, A. K. Ginkgolide B Revamps Neuroprotective Role of Apurinic/Apyrimidinic Endonuclease 1 and Mitochondrial Oxidative Phosphorylation Against Aβ25–35‐induced Neurotoxicity in Human Neuroblastoma Cells. J. Neurosci. Res. 2015, 93, 938–947. DOI: 10.1002/jnr.23565.
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W. S.; Akkol, E. K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules. 2019, 10(1), 59. DOI: 10.3390/biom10010059.
- Kong, Z.-H.; Chen, X.; Hua, H.-P.; Liang, L.; Liu, L.-J. The Oral Pretreatment of Glycyrrhizin Prevents Surgery-Induced Cognitive Impairment in Aged Mice by Reducing Neuroinflammation and Alzheimer’s-Related Pathology via HMGB1 Inhibition. J. Mol. Neurosci. 2017, 63, 385–395. DOI: 10.1007/s12031-017-0989-7.
- Geiser, R. J.; Chastain, S. E.; Moss, M. A. Regulation of Bace1 mRNA Expression in Alzheimer’s Disease by Green Tea Catechins and Black Tea Theaflavins. Biophys. J. 2017, 112(3), 362a. DOI: 10.1016/j.bpj.2016.11.1965.
- Videira, R.; Castanheira, P.; Graos, M.; Resende, R.; Salgueiro, L.; Faro, C.; Cavaleiro, C. Dose-Dependent Inhibition of BACE-1 by the Monoterpenoid 2, 3, 4, 4-Tetramethyl-5-Methylenecyclopent-2-Enone in Cellular and Mouse Models of Alzheimer’s Disease. J. Nat. Prod. 2014, 77(6), 1275–1279. DOI: 10.1021/np400903w.
- Pérez-Severiano, F.; Salvatierra-Sánchez, R.; Rodrıguez-Pérez, M.; Cuevas-Martınez, E. Y.; Guevara, J.; Limón, D.; Maldonado, P. D.; Medina-Campos, O. N.; Pedraza-Chaverrı, J.; Santamarıa, A. S-Allylcysteine Prevents Amyloid-β Peptide-Induced Oxidative Stress in Rat Hippocampus and Ameliorates Learning Deficits. Eur. J. Pharmacol. 2004, 489, 197–202. DOI: 10.1016/j.ejphar.2004.03.001.
- Amagase, H.; Petesch, B. L.; Matsuura, H.; Kasuga, S.; Itakura, Y. Intake of Garlic and Its Bioactive Components. J. Nutr. 2001, 131(3), 955S–962S. DOI: 10.1093/jn/131.3.955S.
- Beňová, B.; Adam, M.; Pavlíková, P.; Fischer, J. Supercritical Fluid Extraction of Piceid, Resveratrol and Emodin from Japanese Knotweed. J. Supercrit. Fluids. 2010, 51(3), 325–330. DOI: 10.1016/j.supflu.2009.10.009.
- Ko, M. J.; Cheigh, C. I.; Cho, S. W.; Chung, M. S. Subcritical Water Extraction of Flavonol Quercetin from Onion Skin. J. Food Eng. 2011, 102(4), 327–333. DOI: 10.1016/j.jfoodeng.2010.09.008.
- Lichtblau, D.; Berger, J. M.; Nakanishi, K. Efficient Extraction of Ginkgolides and Bilobalide from Ginkgo Biloba Leaves. J. Nat. Prod. 2002, 65(10), 1501–1504. DOI: 10.1021/np0201974.
- Lončar, M.; Jakovljević, M.; Šubarić, D.; Pavlić, M.; Buzjak Služek, V.; Cindrić, I.; Molnar, M. Coumarins in Food and Methods of Their Determination. Foods. 2020, 9(5), 645. DOI: 10.3390/foods9050645.
- Perva-Uzunalić, A.; Škerget, M.; Knez, Ž.; Weinreich, B.; Otto, F.; Grüner, S. Extraction of Active Ingredients from Green Tea (Camellia Sinensis): Extraction Efficiency of Major Catechins and Caffeine. Food Chem. 2006, 96(4), 597–605. DOI: 10.1016/j.foodchem.2005.03.015.
- Priyadarsini, K. I. The Chemistry of Curcumin: From Extraction to Therapeutic Agent. Molecules. 2014, 19(12), 20091–20112. DOI: 10.3390/molecules191220091.
- Taarji, N.; Bouhoute, M.; Fainassi, F.; Hafidi, A.; Kobayashi, I.; Neves, M. A.; Tominaga, K.; Isoda, H.; Nakajima, M. Interfacial and Emulsifying Properties of Purified Glycyrrhizin and Non-Purified Glycyrrhizin-Rich Extracts from Liquorice Root (Glycyrrhiza Glabra). Food Chem. 2021, 337, 127949. DOI: 10.1016/j.foodchem.2020.127949.
- Ali, E. S.; Akter, S.; Ramproshad, S.; Mondal, B.; Riaz, T. A.; Islam, M. T.; Khan, I. N.; Docea, A. O.; Calina, D.; Sharifi-Rad, J. Targeting Ras-ERK Cascade by Bioactive Natural Products for Potential Treatment of Cancer: An Updated Overview. Cancer Cell Int. 2022, 22(1), 246. DOI: 10.1186/s12935-022-02666-z.
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A. O. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. DOI: 10.3389/fphar.2020.592654.
- Youssif, K. A.; Haggag, E. G.; Elshamy, A. M.; Rabeh, M. A.; Gabr, N. M.; Seleem, A.; Salem, M. A.; Hussein, A. S.; Krischke, M.; Mueller, M. J., et al. Anti-Alzheimer Potential, Metabolomic Profiling and Molecular Docking of Green Synthesized Silver Nanoparticles of Lampranthus Coccineus and Malephora Lutea Aqueous Extracts. Plos One. 2019, 14(11), e0223781. DOI: 10.1371/journal.pone.0223781.
- Suganthy, N.; Sri Ramkumar, V.; Pugazhendhi, A.; Benelli, G.; Archunan, G. Biogenic Synthesis of Gold Nanoparticles from Terminalia Arjuna Bark Extract: Assessment of Safety Aspects and Neuroprotective Potential via Antioxidant, Anticholinesterase, and Antiamyloidogenic Effects. Environ. Sci. Pollut. Res. 2018, 25(11), 10418–10433. DOI: 10.1007/s11356-017-9789-4.
- El-Hawwary, S. S.; Abd Almaksoud, H. M.; Saber, F. R.; Elimam, H.; Sayed, A. M.; Raey, M. A. E.; Abdelmohsen, U. R. Green-Synthesized Zinc Oxide Nanoparticles, Anti-Alzheimer Potential and the Metabolic Profiling of Sabal Blackburniana Grown in Egypt Supported by Molecular Modelling. R.S.C. Adv. 2021, 11(29), 18009–18025. DOI: 10.1039/D1RA01725J.
- Ringman, J. M.; Frautschy, S. A.; Cole, G. M.; Masterman, D. L.; Cummings, J. L. A Potential Role of the Curry Spice Curcumin in Alzheimer’s Disease. Curr. Alzheimer Res. 2005, 2, 131–136. DOI: 10.2174/1567205053585882.
- Zheng, K.; Dai, X.; Xiao, N.; Wu, X.; Wei, Z.; Fang, W.; Zhu, Y.; Zhang, J.; Chen, X. Curcumin Ameliorates Memory Decline via Inhibiting BACE1 Expression and β-Amyloid Pathology in 5× FAD Transgenic Mice. Mol. Neurobiol. 2017, 54(3), 1967–1977. DOI: 10.1007/s12035-016-9802-9.
- Zhang, X.; Zhang, H.; Si, L.; Li, Y. Curcumin Mediates Presenilin-1 Activity to Reduce β-Amyloid Production in a Model of Alzheimer’s Disease. Pharmacol. Rep. 2011, 63, 1101–1108. DOI: 10.1016/S1734-1140(11)70629-6.
- Carrettiero, D. C.; Hernandez, I.; Neveu, P.; Papagiannakopoulos, T.; Kosik, K. S. The Cochaperone BAG2 Sweeps Paired Helical Filament-Insoluble Tau from the Microtubule. J. Neurosci. 2009, 29, 2151–2161. DOI: 10.1523/JNEUROSCI.4660-08.2009.
- Patil, S. P.; Tran, N.; Geekiyanage, H.; Liu, L.; Chan, C. Curcumin-Induced Upregulation of the Anti-Tau Cochaperone BAG2 in Primary Rat Cortical Neurons. Neurosci. Lett. 2013, 554, 121–125. DOI: 10.1016/j.neulet.2013.09.008.
- Mani, R.; Sha Sulthana, A.; Muthusamy, G.; Elangovan, N. Progress in the Development of Naturally Derived Active Metabolites‐Based Drugs: Potential Therapeutics for Alzheimer’s Disease. Biotechnol. Appl. Biochem. 2022, 69, 2713–2732. DOI: 10.1002/bab.2317.
- Tian, B.; Liu, J. Resveratrol: A Review of Plant Sources, Synthesis, Stability, Modification and Food Application. J. Sci. Food Agric. 2020, 100(4), 1392–1404. DOI: 10.1002/jsfa.10152.
- Rahman, M.; Akter, R.; Bhattacharya, T.; Abdel-Daim, M. M.; Alkahtani, S.; Arafah, M. W.; Al-Johani, N. S.; Alhoshani, N. M.; Alkeraishan, N.; Alhenaky, A. Resveratrol and Neuroprotection: Impact and Its Therapeutic Potential in Alzheimer’s Disease. Front. Pharmacol. 2020, 2272. DOI: 10.3389/fphar.2020.619024.
- Wang, J.; Ho, L.; Zhao, W.; Ono, K.; Rosensweig, C.; Chen, L.; Humala, N.; Teplow, D. B.; Pasinetti, G. M. Grape-Derived Polyphenolics Prevent Aβ Oligomerization and Attenuate Cognitive Deterioration in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2008, 28, 6388–6392. DOI: 10.1523/JNEUROSCI.0364-08.2008.
- Jeon, B. T.; Jeong, E. A.; Shin, H. J.; Lee, Y.; Lee, D. H.; Kim, H. J.; Kang, S. S.; Cho, G. J.; Choi, W. S.; Roh, G. S. Resveratrol Attenuates Obesity-Associated Peripheral and Central Inflammation and Improves Memory Deficit in Mice Fed a High-Fat Diet. Diabetes. 2012, 61(6), 1444–1454. DOI: 10.2337/db11-1498.
- Lee, E. O.; Park, H. J.; Kang, J. L.; Kim, H.; Chong, Y. H. Resveratrol Reduces Glutamate‐Mediated Monocyte Chemotactic Protein‐1 Expression via Inhibition of Extracellular Signal‐Regulated Kinase 1/2 Pathway in Rat Hippocampal Slice Cultures. J. Neurochem. 2010, 112(6), 1477–1487. DOI: 10.1111/j.1471-4159.2009.06564.x.
- Sarroca, S.; Gatius, A.; Rodríguez-Farré, E.; Vilchez, D.; Pallàs, M.; Griñán-Ferré, C.; Sanfeliu, C.; Corpas, R. Resveratrol Confers Neuroprotection Against High-Fat Diet in a Mouse Model of Alzheimer’s Disease via Modulation of Proteolytic Mechanisms. J. Nutr. Biochem. 2021, 89, 108569. DOI: 10.1016/j.jnutbio.2020.108569.
- Lin, C.-H.; Lin, C.-C.; Ting, W.-J.; Pai, P.-Y.; Kuo, C.-H.; Ho, T.-J.; Kuo, W.-W.; Chang, C.-H.; Huang, C.-Y.; Lin, W.-T. Resveratrol Enhanced FOXO3 Phosphorylation via Synergetic Activation of SIRT1 and PI3K/Akt Signaling to Improve the Effects of Exercise in Elderly Rat Hearts. Age (Omaha). 2014, 36(5), 1–10. DOI: 10.1007/s11357-014-9705-5.
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T. G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M. P. Involvement of PGC-1α in the Formation and Maintenance of Neuronal Dendritic Spines. Nat. Commun. 2012, 3(1), 1250. DOI: 10.1038/ncomms2238.
- Li, X.; Yang, S.; Wang, L.; Liu, P.; Zhao, S.; Li, H.; Jiang, Y.; Guo, Y.; Wang, X. resveratrol Inhibits Paclitaxel-Induced Neuropathic Pain by the Activation of PI3K/Akt and SIRT1/PGC1α Pathway. J. Pain Res. 2019, Volume 12, 879–890. DOI: 10.2147/JPR.S185873.
- Wang, B.; Yang, Q.; Sun, Y.; Xing, Y.; Wang, Y.; Lu, X.; Bai, W.; Liu, X.; Zhao, Y. Resveratrol‐Enhanced Autophagic Flux Ameliorates Myocardial Oxidative Stress Injury in Diabetic Mice. J. Cell Mol. Med. 2014, 18(8), 1599–1611. DOI: 10.1111/jcmm.12312.
- Shanbhag, S. M.; Kulkarni, H. J.; Gaitonde, B. B. Pharmacological Actions of Berberine on the Central Nervous System. Japanese J. Pharmacol. 1970, 20, 482–487. DOI: 10.1254/jjp.20.482.
- Durairajan, S. S. K.; Liu, L.-F.; Lu, J.-H.; Chen, L.-L.; Yuan, Q.; Chung, S. K.; Huang, L.; Li, X.-S.; Huang, J.-D.; Li, M. Berberine Ameliorates β-Amyloid Pathology, Gliosis, and Cognitive Impairment in an Alzheimer’s Disease Transgenic Mouse Model. Neurobiol. Aging. 2012, 33, 2903–2919. DOI: 10.1016/j.neurobiolaging.2012.02.016.
- Ji, H.-F.; Shen, L. Molecular Basis of Inhibitory Activities of Berberine Against Pathogenic Enzymes in Alzheimer’s Disease. Sci. World J. 2012, 2012, 1–4. DOI: 10.1100/2012/823201.
- Cummings, J. L. Cholinesterase Inhibitors: A New Class of Psychotropic Compounds. Am. j. psychiatry. 2000, 157(1), 4–15. DOI: 10.1176/ajp.157.1.4.
- Yu, G.; Li, Y.; Tian, Q.; Liu, R.; Wang, Q.; Wang, J.-Z.; Wang, X. Berberine Attenuates Calyculin A-Induced Cytotoxicity and Tau Hyperphosphorylation in HEK293 Cells. J. Alzheimer’s Dis. 2011, 24, 525–535. DOI: 10.3233/JAD-2011-101779.
- de Oliveira, J. S.; Abdalla, F. H.; Dornelles, G. L.; Adefegha, S. A.; Palma, T. V.; Signor, C.; da Silva Bernardi, J.; Baldissarelli, J.; Lenz, L. S.; Magni, L. P. Berberine Protects Against Memory Impairment and Anxiogenic-Like Behavior in Rats Submitted to Sporadic Alzheimer’s-Like Dementia: Involvement of Acetylcholinesterase and Cell Death. Neurotoxicology. 2016, 57, 241–250. DOI: 10.1016/j.neuro.2016.10.008.
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. I. Berberine Improves Cognitive Impairment by Promoting Autophagic Clearance and Inhibiting Production of β-Amyloid in APP/Tau/PS1 Mouse Model of Alzheimer’s Disease. Exp. Gerontol. 2017, 91, 25–33. DOI: 10.1016/j.exger.2017.02.004.
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A. G. Molecular Mechanisms Underlying Protective Effects of Quercetin Against Mitochondrial Dysfunction and Progressive Dopaminergic Neurodegeneration in Cell Culture and MitoPark Transgenic Mouse Models of Parkinson’s Disease. J. Neurochem. 2017, 141, 766–782. DOI: 10.1111/jnc.14033.
- Jiang, W.; Luo, T.; Li, S.; Zhou, Y.; Shen, X.-Y.; He, F.; Xu, J.; Wang, H.-Q.; Arai, K. Quercetin Protects Against Okadaic Acid-Induced Injury via MAPK and PI3K/Akt/GSK3β Signaling Pathways in HT22 Hippocampal Neurons. Plos One. 2016, 11(4), e0152371. DOI: 10.1371/journal.pone.0152371.
- Qureshi, A. A.; Tan, X.; Reis, J. C.; Badr, M. Z.; Papasian, C. J.; Morrison, D. C.; Qureshi, N. Inhibition of Nitric Oxide in LPS-Stimulated Macrophages of Young and Senescent Mice by δ-Tocotrienol and Quercetin. Lipids Health Dis. 2011, 10(1), 1–22. DOI: 10.1186/1476-511X-10-239.
- Rezai-Zadeh, K.; Arendash, G. W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R. D.; Tan, J. Green Tea Epigallocatechin-3-Gallate (EGCG) Reduces β-Amyloid Mediated Cognitive Impairment and Modulates Tau Pathology in Alzheimer Transgenic Mice. Brain. Res. 2008a, 1214, 177–187. DOI: 10.1016/j.brainres.2008.02.107.
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D. Green Tea Epigallocatechin-3-Gallate (EGCG) Modulates Amyloid Precursor Protein Cleavage and Reduces Cerebral Amyloidosis in Alzheimer Transgenic Mice. J. Neurosci. 2005, 25, 8807–8814. DOI: 10.1523/JNEUROSCI.1521-05.2005.
- Singh, M.; Arseneault, M.; Sanderson, T.; Murthy, V.; Ramassamy, C. Challenges for Research on Polyphenols from Foods in Alzheimer’s Disease: Bioavailability, Metabolism, and Cellular and Molecular Mechanisms. J. Agric. Food. Chem. 2008, 56(13), 4855–4873. DOI: 10.1021/jf0735073.
- Mandel, S.; Amit, T.; Reznichenko, L.; Weinreb, O.; Youdim, M. B. H. Green Tea Catechins as Brain‐Permeable, Natural Iron Chelators‐Antioxidants for the Treatment of Neurodegenerative Disorders. Mol. Nutr Food Res. 2006, 50, 229–234. DOI: 10.1002/mnfr.200500156.
- Chen, C.; Yu, R.; Owuor, E. D.; Tony Kong, A.-N. Activation of Antioxidant-Response Element (ARE), Mitogen-Activated Protein Kinases (MAPKs) and Caspases by Major Green Tea Polyphenol Components During Cell Survival and Death. Arch. Pharm. Res. 2000, 23, 605–612. DOI: 10.1007/BF02975249.
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids Protect Neuronal Cells from Oxidative Stress by Three Distinct Mechanisms. Free Radic. Biol. Med. 2001, 30(4), 433–446. DOI: 10.1016/S0891-5849(00)00498-6.
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M. B. H. Neuroprotection and Neurorescue Against Aβ Toxicity and PKC‐Dependent Release of Non‐Amyloidogenic Soluble Precursor Protein by Green Tea Polyphenol (‐)‐Epigallocatechin‐3‐Gallate. FASEB. J. 2003, 17, 1–23. DOI: 10.1096/fj.02-0881fje.
- Bastianetto, S.; Yao, Z.; Papadopoulos, V.; Quirion, R. Neuroprotective Effects of Green and Black Teas and Their Catechin Gallate Esters Against β‐Amyloid‐Induced Toxicity. European J. Neurosci. 2006, 23(1), 55–64. DOI: 10.1111/j.1460-9568.2005.04532.x.
- Obregon, D. F.; Rezai-Zadeh, K.; Bai, Y.; Sun, N.; Hou, H.; Ehrhart, J.; Zeng, J.; Mori, T.; Arendash, G. W.; Shytle, D. ADAM10 Activation is Required for Green Tea (–)-epigallocatechin-3-gallate-induced α-Secretase Cleavage of Amyloid Precursor Protein. J. Biol. Chem. 2006, 281(24), 16419–16427. DOI: 10.1074/jbc.M600617200.
- Jeon, S.-Y.; Bae, K.; Seong, Y.-H.; Song, K.-S. Green Tea Catechins as a BACE1 (β-Secretase) Inhibitor. Bioorg. Med. Chem. Lett. 2003, 13(22), 3905–3908. DOI: 10.1016/j.bmcl.2003.09.018.
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti‐Amyloidogenic Effects for Alzheimer’s β‐Amyloid Fibrils In Vitro. J. Neurosci. Res. 2004, 75, 742–750. DOI: 10.1002/jnr.20025.
- Ehrnhoefer, D. E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E. E. EGCG Redirects Amyloidogenic Polypeptides into Unstructured, Off-Pathway Oligomers. Nat. Struct. Mol. Biol. 2008, 15(6), 558–566. DOI: 10.1038/nsmb.1437.
- Zhao, B.; Li, X.; He, R.; Cheng, S.; Wenjuan, X. Scavenging Effect of Extracts of Green Tea and Natural Antioxidants on Active Oxygen Radicals. Cell Biophys. 1989, 14, 175–185. DOI: 10.1007/BF02797132.
- Choi, Y.-T.; Jung, C.-H.; Lee, S.-R.; Bae, J.-H.; Baek, W.-K.; Suh, M.-H.; Park, J.; Park, C.-W.; Suh, S.-I. The Green Tea Polyphenol (−)-Epigallocatechin Gallate Attenuates β-Amyloid-Induced Neurotoxicity in Cultured Hippocampal Neurons. Life. sci. 2001, 70(5), 603–614. DOI: 10.1016/S0024-3205(01)01438-2.
- Haque, A. M.; Hashimoto, M.; Katakura, M.; Hara, Y.; Shido, O. Green Tea Catechins Prevent Cognitive Deficits Caused by Aβ1–40 in Rats. J. Nutr Biochem. 2008, 19(9), 619–626. DOI: 10.1016/j.jnutbio.2007.08.008.
- Kim, S.-J.; Jeong, H.-J.; Lee, K.-M.; Myung, N.-Y.; An, N.-H.; Yang, W. M.; Park, S. K.; Lee, H.-J.; Hong, S.-H.; Kim, H.-M. Epigallocatechin-3-Gallate Suppresses NF-Κb Activation and Phosphorylation of p38 MAPK and JNK in Human Astrocytoma U373MG Cells. J. Nutr Biochem. 2007, 18(9), 587–596. DOI: 10.1016/j.jnutbio.2006.11.001.
- Ahmed, S.; Rahman, A.; Hasnain, A.; Lalonde, M.; Goldberg, V. M.; Haqqi, T. M. Green Tea Polyphenol Epigallocatechin-3-Gallate Inhibits the IL-1β-Induced Activity and Expression of Cyclooxygenase-2 and Nitric Oxide Synthase-2 in Human Chondrocytes. Free Radic. Biol. Med. 2002, 33(8), 1097–1105. DOI: 10.1016/S0891-5849(02)01004-3.
- Han, M.-K. Epigallocatechin Gallate, a Constituent of Green Tea, Suppresses Cytokine-Induced Pancreatic β-Cell Damage. Exp. Mol. Med. 2003, 35(2), 136–139. DOI: 10.1038/emm.2003.19.
- Rezai-Zadeh, K.; Arendash, G. W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R. D.; Tan, J. Green Tea Epigallocatechin-3-Gallate (EGCG) Reduces β-Amyloid Mediated Cognitive Impairment and Modulates Tau Pathology in Alzheimer Transgenic Mice. Brain. Res. 2008b, 1214, 177–187. DOI: 10.1016/j.brainres.2008.02.107.
- Palermo, C. M.; Westlake, C. A.; Gasiewicz, T. A. Epigallocatechin Gallate Inhibits Aryl Hydrocarbon Receptor Gene Transcription Through an Indirect Mechanism Involving Binding to a 90 kDa Heat Shock Protein. Biochemistry. 2005, 44(13), 5041–5052. DOI: 10.1021/bi047433p.
- Lee, S.-Y.; Kim, C.-Y.; Lee, J.-J.; Jung, J.-G.; Lee, S.-R. Effects of Delayed Administration of (−)-Epigallocatechin Gallate, a Green Tea Polyphenol on the Changes in Polyamine Levels and Neuronal Damage After Transient Forebrain Ischemia in Gerbils. Brain Res. Bull. 2003, 61(4), 399–406. DOI: 10.1016/S0361-9230(03)00139-4.
- Matsuoka, Y.; Hasegawa, H.; Okuda, S.; Muraki, T.; Uruno, T.; Kubota, K. Ameliorative Effects of Tea Catechins on Active Oxygen-Related Nerve Cell Injuries. J. Pharmacol. Exp. Ther. 1995, 274, 602–608.
- Shekarchi, M.; Hajimehdipoor, H.; Saeidnia, S.; Gohari, A. R.; Hamedani, M. P. Comparative Study of Rosmarinic Acid Content in Some Plants of Labiatae Family. Pharmacogn. Mag. 2012, 8, 37. DOI: 10.4103/0973-1296.93316.
- Baba, S.; Osakabe, N.; Natsume, M.; Terao, J. Orally Administered Rosmarinic Acid is Present as the Conjugated And/Or Methylated Forms in Plasma, and is Degraded and Metabolized to Conjugated Forms of Caffeic Acid, Ferulic Acid and M-Coumaric Acid. Life. sci. 2004, 75(2), 165–178. DOI: 10.1016/j.lfs.2003.11.028.
- Nakazawa, T.; Ohsawa, K. Metabolism of Rosmarinic Acid in Rats. J. Nat. Prod. 1998, 61(8), 993–996. DOI: 10.1021/np980072s.
- Iuvone, T.; De Filippis, D.; Esposito, G.; D’Amico, A.; Izzo, A. A. The Spice Sage and Its Active Ingredient Rosmarinic Acid Protect PC12 Cells from Amyloid-β Peptide-Induced Neurotoxicity. J. Pharmacol. Exp. Ther. 2006, 317, 1143–1149. DOI: 10.1124/jpet.105.099317.
- Airoldi, C.; Sironi, E.; Dias, C.; Marcelo, F.; Martins, A.; Rauter, A. P.; Nicotra, F.; Jimenez‐Barbero, J. Natural Compounds Against Alzheimer’s Disease: Molecular Recognition of Aβ1-42 Peptide by Salvia Sclareoides Extract and Its Major Component, Rosmarinic Acid, as Investigated by NMR. Chem.–An Asian J. 2013, 8(3), 596–602. DOI: 10.1002/asia.201201063.
- Alkam, T.; Nitta, A.; Mizoguchi, H.; Itoh, A.; Nabeshima, T. A Natural Scavenger of Peroxynitrites, Rosmarinic Acid, Protects Against Impairment of Memory Induced by Aβ25–35. Behav. Brain Res. 2007, 180(2), 139–145. DOI: 10.1016/j.bbr.2007.03.001.
- Bulgakov, V. P.; Inyushkina, Y. V.; Fedoreyev, S. A. Rosmarinic Acid and Its Derivatives: Biotechnology and Applications. Crit. Rev. Biotechnol. 2012, 32(3), 203–217. DOI: 10.3109/07388551.2011.596804.
- Shukla, S.; Gupta, S. Apigenin: A Promising Molecule for Cancer Prevention. Pharm. Res. 2010, 27(6), 962–978. DOI: 10.1007/s11095-010-0089-7.
- Hollman, P. H.; Katan, M. B. Dietary Flavonoids: Intake, Health Effects and Bioavailability. Food Chem. Toxicol. 1999, 37, 937–942. DOI: 10.1016/S0278-6915(99)00079-4.
- Zhang, J.; Liu, D.; Huang, Y.; Gao, Y.; Qian, S. Biopharmaceutics Classification and Intestinal Absorption Study of Apigenin. Int. J. Pharm. 2012, 436(1–2), 311–317. DOI: 10.1016/j.ijpharm.2012.07.002.
- Gradolatto, A.; Canivenc-Lavier, M.-C.; Basly, J.-P.; Siess, M.-H.; Teyssier, C. Metabolism of Apigenin by Rat Liver Phase I and Phase II Enzymes and by Isolated Perfused Rat Liver. Drug Metab. Dispos. 2004, 32(1), 58–65. DOI: 10.1124/dmd.32.1.58.
- Zhao, L.; Hou, L.; Sun, H.; Yan, X.; Sun, X.; Li, J.; Bian, Y.; Chu, Y.; Liu, Q. Apigenin Isolated from the Medicinal Plant Elsholtzia Rugulosa Prevents β-Amyloid 25–35-induces Toxicity in Rat Cerebral Microvascular Endothelial Cells. Molecules. 2011, 16(5), 4005–4019. DOI: 10.3390/molecules16054005.
- Liu, R.; Zhang, T.; Yang, H.; Lan, X.; Ying, J.; Du, G. The Flavonoid Apigenin Protects Brain Neurovascular Coupling Against Amyloid-β 25-35-Induced Toxicity in Mice. J. Alzheimer’s Dis. 2011b, 24(1), 85–100. DOI: 10.3233/JAD-2010-101593.
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2013, 2013, 1–10. DOI: 10.1155/2013/316523.
- López-Lázaro, M. Distribution and Biological Activities of the Flavonoid Luteolin. Mini. Rev. Med. Chem. 2009, 9(1), 31–59. DOI: 10.2174/138955709787001712.
- Shimoi, K.; Okada, H.; Furugori, M.; Goda, T.; Takase, S.; Suzuki, M.; Hara, Y.; Yamamoto, H.; Kinae, N. Intestinal Absorption of Luteolin and Luteolin 7-O-β-Glucoside in Rats and Humans. FEBS Lett. 1998, 438(3), 220–224. DOI: 10.1016/S0014-5793(98)01304-0.
- Cheng, H.; Hsieh, M.; Tsai, F.; Wu, C.; Chiu, C.; Lee, M.; Xu, H.; Zhao, Z.; Peng, W. Neuroprotective Effect of Luteolin on Amyloid β Protein (25–35)‐induced Toxicity in Cultured Rat Cortical Neurons. Phytotherapy Res. 2010, 24, S102–S108. DOI: 10.1002/ptr.2940.
- Wruck, C. J.; Claussen, M.; Fuhrmann, G.; Römer, L.; Schulz, A.; Pufe, T.; Waetzig, V.; Peipp, M.; Herdegen, T.; Götz, M. E. Luteolin Protects Rat PC 12 and C6 Cells Against MPP+ Induced Toxicity via an ERK Dependent Keapl-Nrf2-ARE Pathway; Springer, 2007. DOI:10.1007/978-3-211-73574-9_9.
- Liu, R.; Meng, F.; Zhang, L.; Liu, A.; Qin, H.; Lan, X.; Li, L.; Du, G. Luteolin Isolated from the Medicinal Plant Elsholtzia Rugulosa (Labiatae) Prevents Copper-Mediated Toxicity in β-Amyloid Precursor Protein Swedish Mutation Overexpressing SH-SY5Y Cells. Molecules. 2011a, 16(3), 2084–2096. DOI: 10.3390/molecules16032084.
- Xu, B.; Li, X.-X.; He, G.-R.; Hu, J.-J.; Mu, X.; Tian, S.; Du, G.-H. Luteolin Promotes Long-Term Potentiation and Improves Cognitive Functions in Chronic Cerebral Hypoperfused Rats. Eur. J. Pharmacol. 2010, 627(1–3), 99–105. DOI: 10.1016/j.ejphar.2009.10.038.
- Rasul, A.; Millimouno, F. M.; Ali Eltayb, W.; Ali, M.; Li, J.; Li, X. Pinocembrin: A Novel Natural Compound with Versatile Pharmacological and Biological Activities. Biomed Res. Int. 2013, 2013, 1–9. DOI: 10.1155/2013/379850.
- Guo, W.-W.; Qiu, F.; Chen, X.-Q.; Ba, Y.-Y.; Wang, X.; Wu, X. In-Vivo Absorption of Pinocembrin-7-O-β-D-Glucoside in Rats and Its in-Vitro Biotransformation. Sci. Rep. 2016, 6(1), 1–8. DOI: 10.1038/srep29340.
- Wang, Y.; Miao, Y.; Mir, A. Z.; Cheng, L.; Wang, L.; Zhao, L.; Cui, Q.; Zhao, W.; Wang, H. Inhibition of Beta-Amyloid-Induced Neurotoxicity by Pinocembrin Through Nrf2/HO-1 Pathway in SH-SY5Y Cells. J. Neurol. Sci. 2016, 368, 223–230. DOI: 10.1016/j.jns.2016.07.010.
- Liu, R.; Wu, C.; Zhou, D.; Yang, F.; Tian, S.; Zhang, L.; Zhang, T.; Du, G. Pinocembrin Protects Against β-Amyloid-Induced Toxicity in Neurons Through Inhibiting Receptor for Advanced Glycation End Products (RAGE)-Independent Signaling Pathways and Regulating Mitochondrion-Mediated Apoptosis. BMC Med. 2012, 10(1), 1–21. DOI: 10.1186/1741-7015-10-105.
- Liu, R.; Li, J.; Song, J.; Sun, J.; Li, Y.; Zhou, S.; Zhang, T.; Du, G. Pinocembrin Protects Human Brain Microvascular Endothelial Cells Against Fibrillar Amyloid-β1− 40 Injury by Suppressing the MAPK/NF-κ B Inflammatory Pathways. Biomed Res. Int. 2014, 2014, 1–14. DOI: 10.1155/2014/470393.
- Shi, C.; Wu, F.; Xu, J.; Zou, J. Bilobalide Regulates Soluble Amyloid Precursor Protein Release via Phosphatidyl Inositol 3 Kinase-Dependent Pathway. Neurochem. Int. 2011, 59, 59–64. DOI: 10.1016/j.neuint.2011.03.028.
- Tchantchou, F.; Lacor, P. N.; Cao, Z.; Lao, L.; Hou, Y.; Cui, C.; Klein, W. L.; Luo, Y. Stimulation of Neurogenesis and Synaptogenesis by Bilobalide and Quercetin via Common Final Pathway in Hippocampal Neurons. J. Alzheimer’s Dis. 2009, 18, 787–798. DOI: 10.3233/JAD-2009-1189.
- Bastianetto, S.; Ramassamy, C.; Doré, S.; Christen, Y.; Poirier, J.; Quirion, R. The Ginkgo Biloba Extract (EGb 761) Protects Hippocampal Neurons Against Cell Death Induced by β‐Amyloid. European J. Neurosci. 2000, 12(6), 1882–1890. DOI: 10.1046/j.1460-9568.2000.00069.x.
- Longpré, F.; Garneau, P.; Ramassamy, C. Protection by EGb 761 Against β-Amyloid-Induced Neurotoxicity: Involvement of NF-κB, SIRT1, and MAPKs Pathways and Inhibition of Amyloid Fibril Formation. Free Radic. Biol. Med. 2006, 41, 1781–1794. DOI: 10.1016/j.freeradbiomed.2006.08.015.
- Wu, Y.; Wu, Z.; Butko, P.; Christen, Y.; Lambert, M. P.; Klein, W. L.; Link, C. D.; Luo, Y. Amyloid-β-Induced Pathological Behaviors are Suppressed by Ginkgo Biloba Extract EGb 761 and Ginkgolides in Transgenic Caenorhabditis Elegans. J. Neurosci. 2006, 26, 13102–13113. DOI: 10.1523/JNEUROSCI.3448-06.2006.
- Zhou, L.-J.; Zhu, X.-Z. Reactive Oxygen Species-Induced Apoptosis in PC12 Cells and Protective Effect of Bilobalide. J. Pharmacol. Exp. Ther. 2000, 293, 982–988.
- Block, E. The Chemistry of Garlic and Onions. Sci. Am. 1985, 252, 114–121. DOI: 10.1038/scientificamerican0385-114.
- Selassie, M.; Griffin, B.; Gwebu, N.; Gwebu, E. T. Aged Garlic Extract Attenuates the Cytotoxicity of (Beta-Amyloid) on Undifferentiated PC12 Cells. Vitro Cell. Dev. Biol. 1999, 35, 369. DOI: 10.1007/s11626-999-0109-2.
- Peng, Q.; Buz’zard, A. R.; Lau, B. H. S. Neuroprotective Effect of Garlic Compounds in Amyloid-Beta Peptide-Induced Apoptosis In Vitro. Med. Sci. Monit. 2002, 8, BR328–37.
- Gupta, V. B.; Indi, S. S.; Rao, K. S. J. Garlic Extract Exhibits Antiamyloidogenic Activity on Amyloid‐Beta Fibrillogenesis: Relevance to Alzheimer’s Disease. Phytother. Res. 2009, 23, 111–115. DOI: 10.1002/ptr.2574.
- Nillert, N.; Pannangrong, W.; Welbat, J. U.; Chaijaroonkhanarak, W.; Sripanidkulchai, K.; Sripanidkulchai, B. Neuroprotective Effects of Aged Garlic Extract on Cognitive Dysfunction and Neuroinflammation Induced by β-Amyloid in Rats. Nutrients. 2017, 9(1), 24. DOI: 10.3390/nu9010024.
- Alshehri, M. M.; Quispe, C.; Herrera-Bravo, J.; Sharifi-Rad, J.; Tutuncu, S.; Aydar, E. F.; Topkaya, C.; Mertdinc, Z.; Ozcelik, B.; Aital, M. A Review of Recent Studies on the Antioxidant and Anti-Infectious Properties of Senna Plants. Oxid. Med. Cell Longev. 2022, 2022, 1–38. DOI: 10.1155/2022/6025900.
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C. J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38(7), 592–607. DOI: 10.1016/j.tips.2017.04.005.
- Zhang, J.; Duan, D.; Song, Z.; Liu, T.; Hou, Y.; Fang, J. Small Molecules Regulating Reactive Oxygen Species Homeostasis for Cancer Therapy. Med. Res. Rev. 2021, 41(1), 342–394. DOI: 10.1002/med.21734.
- Sharifi-Rad, J.; Rapposelli, S.; Sestito, S.; Herrera-Bravo, J.; Arancibia-Diaz, A.; Salazar, L. A.; Yeskaliyeva, B.; Beyatli, A.; Leyva-Gómez, G.; González-Contreras, C. Multi-Target Mechanisms of Phytochemicals in Alzheimer’s Disease: Effects on Oxidative Stress, Neuroinflammation and Protein Aggregation. J. Pers. Med. 2022c, 12, 1515. DOI: 10.3390/jpm12091515.
- Escobar, S. J. D. M.; Fong, G. M.; Winnischofer, S. M. B.; Simone, M.; Munoz, L.; Dennis, J. M.; Rocha, M. E. M.; Witting, P. K. Anti-Proliferative and Cytotoxic Activities of the Flavonoid Isoliquiritigenin in the Human Neuroblastoma Cell Line SH-SY5Y. Chem. Biol. Interact. 2019, 299, 77–87. DOI: 10.1016/j.cbi.2018.11.022.
- Amin, R.; Quispe, C.; Docea, A. O.; Alibek, Y.; Kulbayeva, M.; Daştan, S. D.; Calina, D.; Sharifi-Rad, J. The Role of Tumour Necrosis Factor in Neuroinflammation Associated with Parkinson’s Disease and Targeted Therapies. Neurochem. Int. 2022, 158, 105376. DOI: 10.1016/j.neuint.2022.105376.
- Taheri, Y.; Quispe, C.; Herrera-Bravo, J.; Sharifi-Rad, J.; Ezzat, S. M.; Merghany, R. M.; Shaheen, S.; Azmi, L.; Prakash Mishra, A.; Sener, B., et al. Urtica Dioica-Derived Phytochemicals for Pharmacological and Therapeutic Applications. Evid. Based Complement. Altern. Med. 2022, 2022, 1–30. DOI: 10.1155/2022/4024331.
- Behl, C.; Moosmann, B. Oxidative Nerve Cell Death in Alzheimer's Disease and Stroke: Antioxidants as Neuroprotective Compounds. Biol. Chem. 2002, 383(3–4). DOI: 10.1515/BC.2002.053.
- Dhyani, P.; Quispe, C.; Sharma, E.; Bahukhandi, A.; Sati, P.; Attri, D. C.; Szopa, A.; Sharifi-Rad, J.; Docea, A. O.; Mardare, I. Anticancer Potential of Alkaloids: A Key Emphasis to Colchicine, Vinblastine, Vincristine, Vindesine, Vinorelbine and Vincamine. Cancer Cell Int. 2022, 22(1), 1–20. DOI: 10.1186/s12935-022-02624-9.
- Suganthy, N.; Devi, K. P. Protective Effect of Catechin Rich Extract of Rhizophora Mucronata Against β-Amyloid-Induced Toxicity in PC12 Cells. J. Appl. Biomed. 2016, 14(2), 137–146. DOI: 10.1016/j.jab.2015.10.003.
- Ide, K.; Matsuoka, N.; Yamada, H.; Furushima, D.; Kawakami, K. Effects of Tea Catechins on Alzheimer’s Disease: Recent Updates and Perspectives. Molecules. 2018, 23(9), 2357. DOI: 10.3390/molecules23092357.
- Okello, E. J.; Mather, J. Comparative Kinetics of Acetyl-And Butyryl-Cholinesterase Inhibition by Green Tea Catechins| Relevance to the Symptomatic Treatment of Alzheimer’s Disease. Nutrients. 2020, 12, 1090. DOI: 10.3390/nu12041090.
- Ayaz, M.; Ullah, F.; Sadiq, A.; Kim, M. O.; Ali, T. Natural Products-Based Drugs: Potential Therapeutics Against Alzheimer’s Disease and Other Neurological Disorders; Frontiers Media SA, 2019b. DOI:10.3389/978-2-88963-348-7.
- Chougle, S.; Kumar, D.; Andleeb, K.; Zehra, S.; Ahmad, A. L. İ. Treatment of Alzheimer’s Disease by Natural Products. J. Exp. Clin. Med. 2021, 38, 634–644. DOI: 10.52142/omujecm.38.4.42.
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of the Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029112. DOI: 10.1177/11795735211029113.
- Cai, Z.; Wang, C.; Yang, W. Role of Berberine in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2016, 2509–2520. DOI: 10.2147/NDT.S114846.
- Yuan, N.-N.; Cai, C.-Z.; Wu, M.-Y.; Su, H.-X.; Li, M.; Lu, J.-H. Neuroprotective Effects of Berberine in Animal Models of Alzheimer’s Disease: A Systematic Review of Pre-Clinical Studies. BMC Complement. Altern. Med. 2019, 19(1), 1–10. DOI: 10.1186/s12906-019-2510-z.
- Giacomeli, R.; Izoton, J. C.; Dos Santos, R. B.; Boeira, S. P.; Jesse, C. R.; Haas, S. E. Neuroprotective Effects of Curcumin Lipid-Core Nanocapsules in a Model Alzheimer’s Disease Induced by β-Amyloid 1-42 Peptide in Aged Female Mice. Brain. Res. 2019, 1721, 146325. DOI: 10.1016/j.brainres.2019.146325.
- Wu, P.; Li, B.; Yu, Y.; Su, P.; Liu, X.; Zhang, Z.; Zhi, D.; Qi, F.; Fei, D.; Zhang, Z. Isolation, Characterization, and Possible Anti‐Alzheimer’s Disease Activities of Bisabolane‐Type Sesquiterpenoid Derivatives and Phenolics from the Rhizomes of Curcuma Longa. Chem. Biodivers. 2020, 17, e2000067. DOI: 10.1002/cbdv.202000067.
- Seo, E.-J.; Fischer, N.; Efferth, T. Phytochemicals as Inhibitors of NF-Κb for Treatment of Alzheimer’s Disease. Pharmacol. Res. 2018, 129, 262–273. DOI: 10.1016/j.phrs.2017.11.030.
- Gomes, B. A. Q.; Silva, J. P. B.; Romeiro, C. F. R.; Dos Santos, S. M.; Rodrigues, C. A.; Gonçalves, P. R.; Sakai, J. T.; Mendes, P. F. S.; Varela, E. L. P.; Monteiro, M. C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 1–15. DOI: 10.1155/2018/8152373.
- Yamakawa, M. Y.; Uchino, K.; Watanabe, Y.; Adachi, T.; Nakanishi, M.; Ichino, H.; Hongo, K.; Mizobata, T.; Kobayashi, S.; Nakashima, K. Anthocyanin Suppresses the Toxicity of Aβ Deposits Through Diversion of Molecular Forms in In Vitro and In Vivo Models of Alzheimer’s Disease. Nutr. Neurosci. 2016, 19(1), 32–42. DOI: 10.1179/1476830515Y.0000000042.
- Ali, T.; Kim, T.; Rehman, S. U.; Khan, M. S.; Amin, F. U.; Khan, M.; Ikram, M.; Kim, M. O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55(7), 6076–6093. DOI: 10.1007/s12035-017-0798-6.
- Dickey, C. A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R. M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Petrucelli, L. et al, The High-Affinity HSP90-CHIP Complex Recognizes and Selectively Degrades Phosphorylated Tau Client Proteins. J. Clin. Invest. 2007, 117(3), 648–658.
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms Behind ROS/RNS Generation. J. Biomed. Sci. 2017, 24(1), 1–10. DOI: 10.1186/s12929-017-0379-z.
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53(6), 4094–4125. DOI: 10.1007/s12035-015-9337-5.
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in Brain Development and Function. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863(5), 934–955. DOI: 10.1016/j.bbamcr.2015.12.005.
- Kou, J.; Kovacs, G. G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R. Peroxisomal Alterations in Alzheimer’s Disease. Acta. Neuropathol 2011, 122(3), 271–283. DOI: 10.1007/s00401-011-0836-9.
- Prosdocimi, T.; De Gioia, L.; Zampella, G.; Bertini, L. On the Generation of OH· Radical Species from H 2 O 2 by Cu (I) Amyloid Beta Peptide Model Complexes: A DFT Investigation. JBIC J. Biol. Inorg. Chem. 2016, 21, 197–212. DOI: 10.1007/s00775-015-1322-y.
- Selkoe, D. J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. DOI: 10.15252/emmm.201606210.
- Huang, B.; Liu, J.; Fu, S.; Zhang, Y.; Li, Y.; He, D.; Ran, X.; Yan, X.; Du, J.; Meng, T. α-Cyperone Attenuates H2O2-Induced Oxidative Stress and Apoptosis in SH-SY5Y Cells via Activation of Nrf2. Front. Pharmacol. 2020, 11, 281. DOI: 10.3389/fphar.2020.00281.
- Othman, S. B.; Yabe, T. Use of Hydrogen Peroxide and Peroxyl Radicals to Induce Oxidative Stress in Neuronal Cells. RAS. 2015, 3, 40–45. DOI: 10.7831/ras.3.40.
- Park, H. R.; Lee, H.; Park, H.; Jeon, J. W.; Cho, W.-K.; Ma, J. Y. Neuroprotective Effects of Liriope Platyphylla Extract Against Hydrogen Peroxide-Induced Cytotoxicity in Human Neuroblastoma SH-SY5Y Cells. BMC Complement. Altern. Med. 2015, 15(1), 1–11. DOI: 10.1186/s12906-015-0679-3.
- Wang, J.; Liu, H.; Zhang, X.; Li, X.; Geng, L.; Zhang, H.; Zhang, Q. Sulfated Hetero-Polysaccharides Protect SH-SY5Y Cells from H2O2-Induced Apoptosis by Affecting the PI3K/Akt Signaling Pathway. Mar. Drugs. 2017b, 15(4), 110. DOI: 10.3390/md15040110.
- Yao, D.; Wang, J.; Wang, G.; Jiang, Y.; Shang, L.; Zhao, Y.; Huang, J.; Yang, S.; Wang, J.; Yu, Y. Design, Synthesis and Biological Evaluation of Coumarin Derivatives as Novel Acetylcholinesterase Inhibitors That Attenuate H2O2-Induced Apoptosis in SH-SY5Y Cells. Bioorg. Chem. 2016, 68, 112–123. DOI: 10.1016/j.bioorg.2016.07.013.
- Juan-García, A.; Caprioli, G.; Sagratini, G.; Mañes, J.; Juan, C. Coffee Silverskin and Spent Coffee Suitable as Neuroprotectors Against Cell Death by Beauvericin and α-Zearalenol: Evaluating Strategies of Treatment. Toxins (Basel). 2021, 13(2), 132. DOI: 10.3390/toxins13020132.
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Neuronal Cell Cult. Methods Protoc. 2013, 1078, 9–21.
- Shipley, M. M.; Mangold, C. A.; Szpara, M. L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. JoVE (J. Vis. Exp.). 2016, 108(108), e53193. DOI: 10.3791/53193.
- Angeloni, S.; Freschi, M.; Marrazzo, P.; Hrelia, S.; Beghelli, D.; Juan-García, A.; Juan, C.; Caprioli, G.; Sagratini, G.; Angeloni, C.; et al. Antioxidant and Anti-Inflammatory Profiles of Spent Coffee Ground Extracts for the Treatment of Neurodegeneration. Oxid. Med. Cell Longev. 2021, 2021, 1–19. DOI: 10.1155/2021/6620913.
- Amato, A.; Terzo, S.; Mulè, F. Natural Compounds as Beneficial Antioxidant Agents in Neurodegenerative Disorders: A Focus on Alzheimer’s Disease. Antioxidants. 2019, 8(12), 608. DOI: 10.3390/antiox8120608.
- Naveed, M.; Hejazi, V.; Abbas, M.; Kamboh, A. A.; Khan, G. J.; Shumzaid, M.; Ahmad, F.; Babazadeh, D.; FangFang, X.; Modarresi-Ghazani, F. Chlorogenic Acid (CGA): A Pharmacological Review and Call for Further Research. Biomed. Pharmacother. 2018, 97, 67–74. DOI: 10.1016/j.biopha.2017.10.064.
- Oboh, G.; Agunloye, O. M.; Akinyemi, A. J.; Ademiluyi, A. O.; Adefegha, S. A. Comparative Study on the Inhibitory Effect of Caffeic and Chlorogenic Acids on Key Enzymes Linked to Alzheimer’s Disease and Some Pro-Oxidant Induced Oxidative Stress in rats’ Brain-In Vitro. Neurochem. Res. 2013b, 38(2), 413–419. DOI: 10.1007/s11064-012-0935-6.
- Gao, L.; Li, X.; Meng, S.; Ma, T.; Wan, L.; Xu, S. chlorogenic Acid Alleviates Aβ25-35-Induced Autophagy and Cognitive Impairment via the mTOR/TFEB Signaling Pathway. Drug Des. Devel. Ther. 2020, Volume 14, 1705–1716. DOI: 10.2147/DDDT.S235969.
- Kim, S.; Park, R.; Jeon, H.; Kwon, Y.; Chun, W. Neuroprotective Effects of 3, 5‐Dicaffeoylquinic Acid on Hydrogen Peroxide‐Induced Cell Death in SH‐SY5Y Cells. Phytother. Res. 2005, 19, 243–245. DOI: 10.1002/ptr.1652.
- Izuta, H.; Shimazawa, M.; Tazawa, S.; Araki, Y.; Mishima, S.; Hara, H. Protective Effects of Chinese Propolis and Its Component, Chrysin, Against Neuronal Cell Death via Inhibition of Mitochondrial Apoptosis Pathway in SH-SY5Y Cells. J. Agric. Food. Chem. 2008, 56(19), 8944–8953. DOI: 10.1021/jf8014206.
- Arya, A.; Chahal, R.; Rao, R.; Rahman, M. H.; Kaushik, D.; Akhtar, M. F.; Saleem, A.; Khalifa, S. M. A.; El-Seedi, H. R.; Kamel, M. Acetylcholinesterase Inhibitory Potential of Various Sesquiterpene Analogues for Alzheimer’s Disease Therapy. Biomolecules. 2021, 11, 350. DOI: 10.3390/biom11030350.
- Cheng, K.-C.; Chiang, H.-C. XBP1 and PERK Have Distinct Roles in Aβ-Induced Pathology. Mol. Neurobiol. 2018, 55(9), 7523–7532. DOI: 10.1007/s12035-018-0942-y.
- Miyamae, Y.; Han, J.; Sasaki, K.; Terakawa, M.; Isoda, H.; Shigemori, H. 3, 4, 5-Tri-O-Caffeoylquinic Acid Inhibits Amyloid β-Mediated Cellular Toxicity on SH-SY5Y Cells Through the Upregulation of PGAM1 and G3PDH. Cytotechnology. 2011, 63(2), 191–200. DOI: 10.1007/s10616-011-9341-1.
- Ma, E.-H.; Rathnayake, A. U.; Lee, J. K.; Lee, S.-M.; Byun, H.-G. Characterization of β-Secretase Inhibitory Extracts from Sea Cucumber (Stichopus Japonicus) Hydrolysis with Their Cellular Level Mechanism in SH-SY5Y Cells. Eur. Food Res. Technol. 2021, 247(8), 2039–2052. DOI: 10.1007/s00217-021-03770-6.
- Rathnayake, A. U.; Abuine, R.; Palanisamy, S.; Lee, J. K.; Byun, H.-G. Characterization and Purification of β− Secretase Inhibitory Peptides Fraction from Sea Cucumber (Holothuria Spinifera) Enzymatic Hydrolysates. Process Biochem. 2021, 111, 86–96. DOI: 10.1016/j.procbio.2021.10.007.
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9(1), 1–13. DOI: 10.1186/s40035-020-00189-z.
- Williams, R. J.; Spencer, J. P. E. Flavonoids, Cognition, and Dementia: Actions, Mechanisms, and Potential Therapeutic Utility for Alzheimer Disease. Free Radic. Biol. Med. 2012, 52, 35–45. DOI: 10.1016/j.freeradbiomed.2011.09.010.
- Agostinho, P.; Cunha, R. A.; Oliveira, C. Neuroinflammation, Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Curr. Pharm. Des. 2010, 16(25), 2766–2778. DOI: 10.2174/138161210793176572.
- Ceulemans, A.-G.; Zgavc, T.; Kooijman, R.; Hachimi-Idrissi, S.; Sarre, S.; Michotte, Y. The Dual Role of the Neuroinflammatory Response After Ischemic Stroke: Modulatory Effects of Hypothermia. J. Neuroinflammation. 2010, 7(1), 1–18. DOI: 10.1186/1742-2094-7-74.
- MacEwan, D. J. TNF Receptor Subtype Signalling: Differences and Cellular Consequences. Cell. Signal. 2002, 14(6), 477–492. DOI: 10.1016/S0898-6568(01)00262-5.
- Taylor, D. L.; Jones, F.; Kubota, E. S. F. C. S.; Pocock, J. M. Stimulation of Microglial Metabotropic Glutamate Receptor mGlu2 Triggers Tumor Necrosis Factor α-Induced Neurotoxicity in Concert with Microglial-Derived Fas Ligand. J. Neurosci. 2005, 25, 2952–2964. DOI: 10.1523/JNEUROSCI.4456-04.2005.
- Walker, D.; Lue, L.-F. Anti-Inflammatory and Immune Therapy for Alzheimer’s Disease: Current Status and Future Directions. Curr. Neuropharmacol. 2007, 5, 232–243. DOI: 10.2174/157015907782793667.
- Semwal, P.; Painuli, S.; Abu-Izneid, T.; Rauf, A.; Sharma, A.; Daştan, S. D.; Kumar, M.; Alshehri, M. M.; Taheri, Y.; Das, R. Diosgenin: An Updated Pharmacological Review and Therapeutic Perspectives. Oxid. Med. Cell Longev. 2022, 2022, 1–17. DOI: 10.1155/2022/1035441.
- Sharifi-Rad, J.; Herrera-Bravo, J.; Semwal, P.; Painuli, S.; Badoni, H.; Ezzat, S. M.; Farid, M. M.; Merghany, R. M.; Aborehab, N. M.; Salem, M. A. Artemisia Spp.: An Update on Its Chemical Composition, Pharmacological and Toxicological Profiles. Oxid. Med. Cell Longev. 2022b, 2022, 1–23. DOI: 10.1155/2022/5628601.
- Javed, Z.; Khan, K.; Herrera-Bravo, J.; Naeem, S.; Iqbal, M. J.; Raza, Q.; Sadia, H.; Raza, S.; Bhinder, M.; Calina, D. Myricetin: Targeting Signaling Networks in Cancer and Its Implication in Chemotherapy. Cancer Cell Int. 2022, 22(1), 239. DOI: 10.1186/s12935-022-02663-2.
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s Disease: Clinical Trials and Drug Development. Lancet. Neurol. 2010, 9, 702–716. DOI: 10.1016/S1474-4422(10)70119-8.
- Ahmad, I.; Aqil, F.; Owais, M. Modern Phytomedicine: Turning Medicinal Plants into Drugs; John Wiley & Sons, 2006. DOI:10.1002/9783527609987.
- Salehi, B.; Sharifi-Rad, J.; Cappellini, F.; Reiner, Ž.; Zorzan, D.; Imran, M.; Sener, B.; Kilic, M.; El-Shazly, M.; Fahmy, N. M. The Therapeutic Potential of Anthocyanins: Current Approaches Based on Their Molecular Mechanism of Action. Front. Pharmacol. 2020, 11, 1300. DOI: 10.3389/fphar.2020.01300.
- Leyva-Gomez, G.; Cortes, H.; Magana, J. J.; Leyva-García, N.; Quintanar-Guerrero, D.; Florán, B. Nanoparticle Technology for Treatment of Parkinson’s Disease: The Role of Surface Phenomena in Reaching the Brain. Drug Discov. Today. 2015, 20(7), 824–837. DOI: 10.1016/j.drudis.2015.02.009.
- Quispe, C.; Herrera-Bravo, J.; Khan, K.; Javed, Z.; Semwal, P.; Painuli, S.; Kamiloglu, S.; Martorell, M.; Calina, D.; Sharifi-Rad, J. Therapeutic Applications of Curcumin Nanomedicine Formulations in Cystic Fibrosis. Prog. Biomater. 2022, 11(4), 321–329. DOI: 10.1007/s40204-022-00198-3.
- Docea, A. O.; Calina, D.; Buga, A. M.; Zlatian, O.; Paoliello, M. M. B.; Mogosanu, G. D.; Streba, C. T.; Popescu, E. L.; Stoica, A. E.; Bîrcă, A. C. The Effect of Silver Nanoparticles on Antioxidant/pro-Oxidant Balance in a Murine Model. Int. J. Mol. Sci. 2020, 21, 1233. DOI: 10.3390/ijms21041233.
- Sharifi-Rad, J.; Bahukhandi, A.; Dhyani, P.; Sati, P.; Capanoglu, E.; Docea, A. O.; Al-Harrasi, A.; Dey, A.; Calina, D. Therapeutic Potential of Neoechinulins and Their Derivatives: An Overview of the Molecular Mechanisms Behind Pharmacological Activities. Front. Nutr. 2021a, 8, 664197. DOI: 10.3389/fnut.2021.664197.
- Sharifi-Rad, J.; Dey, A.; Koirala, N.; Shaheen, S.; El Omari, N.; Salehi, B.; Goloshvili, T.; Cirone Silva, N. C.; Bouyahya, A.; Vitalini, S. Cinnamomum Species: Bridging Phytochemistry Knowledge, Pharmacological Properties and Toxicological Safety for Health Benefits. Front. Pharmacol. 2021b, 12, 600139. DOI: 10.3389/fphar.2021.600139.
- Sharifi-Rad, J.; Quispe, C.; Imran, M.; Rauf, A.; Nadeem, M.; Gondal, T. A.; Ahmad, B.; Atif, M.; Mubarak, M. S.; Sytar, O. Genistein: An Integrative Overview of Its Mode of Action, Pharmacological Properties, and Health Benefits. Oxid. Med. Cell Longev. 2021c, 2021, 1–36. DOI: 10.1155/2021/3268136.
- Ramalho, M. J.; Andrade, S.; Loureiro, J. A.; Carmo Pereira, M. D. Nanotechnology to Improve the Alzheimer’s Disease Therapy with Natural Compounds. Drug Deliv. Transl. Res. 2020, 10, 380–402. DOI: 10.1007/s13346-019-00694-3.
- Del Prado-Audelo, M. L.; Caballero-Florán, I. H.; Meza-Toledo, J. A.; Mendoza-Muñoz, N.; González-Torres, M.; Florán, B.; Cortés, H.; Leyva-Gómez, G. Formulations of Curcumin Nanoparticles for Brain Diseases. Biomolecules. 2019, 9, 56. DOI: 10.3390/biom9020056.
- Naqvi, S.; Panghal, A.; Flora, S. J. S. Nanotechnology: A Promising Approach for Delivery of Neuroprotective Drugs. Front. Neurosci. 2020, 14, 494. DOI: 10.3389/fnins.2020.00494.
- Kamatou, G. P. P.; Viljoen, A. M. A Review of the Application and Pharmacological Properties of α-Bisabolol and α-Bisabolol-Rich Oils. J. Am. Oil Chem. Soc. 2010, 87(1), 1–7. DOI: 10.1007/s11746-009-1483-3.
- Anand, P.; Kunnumakkara, A. B.; Newman, R. A.; Aggarwal, B. B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4(6), 807–818. DOI: 10.1021/mp700113r.
- Islam, M. T.; Ali, E. S.; Uddin, S. J.; Shaw, S.; Islam, M. A.; Ahmed, M. I.; Shill, M. C.; Karmakar, U. K.; Yarla, N. S.; Khan, I. N. Phytol: A Review of Biomedical Activities. Food Chem. Toxicol. 2018, 121, 82–94. DOI: 10.1016/j.fct.2018.08.032.
- Mohar, D. S.; Malik, S. The Sirtuin System: The Holy Grail of Resveratrol? J. Clin. Exp. Cardiolog. 2012, 3. DOI: 10.4172/2155-9880.1000216.
- Walle, T. Bioavailability of Resveratrol. Ann. N Y Acad. Sci. 2011, 1215(1), 9–15. DOI: 10.1111/j.1749-6632.2010.05842.x.
- Bacci, A.; Runfola, M.; Sestito, S.; Rapposelli, S. Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants. 2021, 10(3), 367. DOI: 10.3390/antiox10030367.
- Calina, D.; Hartung, T.; Docea, A. O.; Spandidos, D. A.; Egorov, A. M.; Shtilman, M. I.; Carvalho, F.; Tsatsakis, A. COVID-19 Vaccines: Ethical Framework Concerning Human Challenge Studies. DARU J. Pharm. Sci. 2020c, 28(2), 807–812. DOI: 10.1007/s40199-020-00371-8.
- Calina, D.; Sarkar, C.; Arsene, A. L.; Salehi, B.; Docea, A. O.; Mondal, M.; Islam, M. T.; Zali, A.; Sharifi-Rad, J. Recent Advances, Approaches and Challenges in Targeting Pathways for Potential COVID-19 Vaccines Development. Immunol. Res. 2020d, 68(6), 315–324. DOI: 10.1007/s12026-020-09154-4.
- Torequl Islam, M.; Nasiruddin, M.; Khan, I. N.; Mishra, S. K.; Kudrat-E-Zahan, M.; Alam Riaz, T.; Ali, E. S.; Rahman, M. S.; Mubarak, M. S.; Martorell, M. A Perspective on Emerging Therapeutic Interventions for COVID-19. Front. Public Health. 2020, 8, 281. DOI: 10.3389/fpubh.2020.00281.
- Calina, D.; Hernández, A. F.; Hartung, T.; Egorov, A. M.; Izotov, B. N.; Nikolouzakis, T. K.; Tsatsakis, A.; Vlachoyiannopoulos, P. G.; Docea, A. O. Challenges and Scientific Prospects of the Newest Generation of mRNA-Based Vaccines Against SARS-CoV-2. Life. 2021, 11, 907. DOI: 10.3390/life11090907.
- Kostoff, R. N.; Kanduc, D.; Porter, A. L.; Shoenfeld, Y.; Calina, D.; Briggs, M. B.; Spandidos, D. A.; Tsatsakis, A. Vaccine-And Natural Infection-Induced Mechanisms That Could Modulate Vaccine Safety. Toxicol. Rep. 2020, 7, 1448–1458. DOI: 10.1016/j.toxrep.2020.10.016.
- Neagu, M.; Calina, D.; Docea, A. O.; Constantin, C.; Filippini, T.; Vinceti, M.; Drakoulis, N.; Poulas, K.; Nikolouzakis, T. K.; Spandidos, D. A. Back to Basics in COVID‐19: Antigens and Antibodies—Completing the Puzzle. J. Cell Mol. Med. 2021, 25(10), 4523–4533. DOI: 10.1111/jcmm.16462.
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M. L. From Combinations to Multitarget‐Directed Ligands: A Continuum in Alzheimer’s Disease Polypharmacology. Med. Res. Rev. 2021, 41(5), 2606–2633. DOI: 10.1002/med.21699.