
Abstract
Recent studies have identified that constitutively active phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling is an important feature of osteosarcoma, where it promotes cell proliferation, survival, and chemo-resistance. Here, we studied the therapeutic potential of NVP-BEZ235, a novel dual PI3K/mTOR dual inhibitor, on osteosarcoma cells in vivo and in vitro. NVP-BEZ235 was cytotoxic and cytostatic to a panel of osteosarcoma lines (MG-63, U2OS and SaOs-2), where it induce apoptosis and cell-cycle arrest. At the molecular level, NVP-BEZ235 inhibited PI3K-AKT-mTORC1 activation and downregulated cyclin D1/cyclin B1 expressions, while increasing MEK/Erk phosphorylation in osteosarcoma cells. MEK/Erk inhibitors PD98059 and MEK-162 increased NVP-BEZ235 activity on osteosarcoma cells. In vivo, oral NVP-BEZ235 inhibited U2OS xenograft in SCID mice, and its anti-tumor efficiency was further enhanced by MEK-162 co-administration. Taken together, our findings indicate that dual inhibition of PI3K and mTOR with NVP-BEZ235, either alone or in combination with MEK/Erk inhibitors, may be an efficient treatment for osteosarcoma.
Abbreviations
| (mTOR) | = | mammalian target of rapamycin |
| (mTORC1) | = | mTOR complex 1 |
| (mTORC2) | = | mTOR complex 2 |
| (PDK1) | = | phosphoinositide-dependent protein kinase-1 |
| (PI3K) | = | phosphatidylinositol 3-kinase |
| (RTKs) | = | receptor tyrosine kinases |
Introduction
Malignant osteosarcoma is typically diagnosed in teenagers and young adults, causing many mortalities.Citation1 Chemotherapy has dramatically improved the overall survival of osteosarcoma patients from 11% at the 1960s, to 70% by the mid-1980s.Citation2 Since then, it has not been significantly ameliorated.Citation3,4 The malignant osteosarcoma is among the most intrinsically resistant malignancies to almost all chemotherapeutic drugs.Citation5,6 As such, recent research effects have been focusing on searching for the novel and more efficient agents against this disease.Citation5
The mechanisms of osteosarcoma carcinogenesis, progression and chemo-resistance have not been fully understood, and several factors are known to play critical roles.Citation7-10 For example, studies have shown that over-activation of phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is an important contributor of progression and chemo-resistance of malignant osteosarcoma.Citation7-11 PI3K, a heterodimer consisting of the p85 regulatory and p110 catalytic subunits, is a major signaling component downstream of receptor tyrosine kinases (RTKs).Citation12 PI3K activates its downstream Akt, which phosphorylates several substrates involved in various key cellular processes including cell growth, proliferation, survival, apoptosis-resistance and metabolism.Citation13 All of these cellular processes are being considered as crucial features for the establishment and the maintenance of the tumorigenic phenotype.Citation13,14 A number of agents targeting this pathway have been developed to potentially treat cancer.Citation13,14
In an initial medicinal chemistry efforts to develop phosphoinositide-dependent protein kinase-1 (PDK1) inhibitors, several lead compounds were developed which inhibited class I PI3K.Citation15 Further optimization for these class I PI3K inhibitors has led to the identification of NVP-BEZ235 as a novel and efficient dual PI3K and mTOR kinase inhibitor.Citation16,17 In the current study, we tested the potential activity of NVP-BEZ235 against osteosarcoma both in vivo and in vitro, and studied the underlying signaling mechanisms.
Results
The cytotoxic effect of NVP-BEZ235 against osteosarcoma cells
We first tested in vitro activity of NVP-BEZ235 in cultured osteosarcoma cells. MTT assay results in demonstrated that NVP-BEZ235 dose-dependently inhibited MG-63 osteosarcoma cell growth. Results also showed that it took at least 48 hrs to see a significant inhibitory effect by NVP-BEZ235 on MG-63 cells (). Results in showed that NVP-BEZ235 inhibited growth of 2 other osteosarcoma cell lines: U2OS and SaOs-2. Its effect was again dose- and time-dependent (). Among the cell lines tested, NVP-BEZ235 showed highest efficiency against MG-63 cells, with lowest 50% growth inhibition (GI-50) (). Further, NVP-BEZ235 treatment increased the number of trypan blue positive (“dead”) MG-63 cells (). The cytotoxic effect of NVP-BEZ235 on osteosarcoma cells was further demonstrated by the “clonogenicity” assay, as the number of survival MG-63 colonies was significantly decreased after NVP-BEZ235 stimulation (). Notably, NVP-BEZ235 only exerted a minor inhibition on osteoblastic MC3T3-E1 cell growth (), suggesting its unique cytotoxic effect in cancerous cells. Together, these results display the cytotoxic effect of NVP-BEZ235 against osteosarcoma cells in vitro.
Figure 1. NVP-BEZ235 is cytotoxic only against osteosarcoma cells. Growth of MG-63 (A and B), U2OS (C and D) and SaOs-2 (E and F) osteosarcoma cells or MC3T3-E1 osteoblastic cells (I) with or without indicated NVP-BEZ235 (NVP) treatment was analyzed by MTT assay. MG-63 cells were treated with indicated concentration of NVP-BEZ235, trypan blue staining (G, 72 hrs) and “clonogenicity” (I, 10 days) were performed to test cell death. The data in this and all following figures were representatives of 3 different experiments. n=5 for each assay. The values were expressed as the means ±SD (Same for all figures). “C” stands for untreated control group (Same for all figures). *P < 0.05 vs. group “C.”
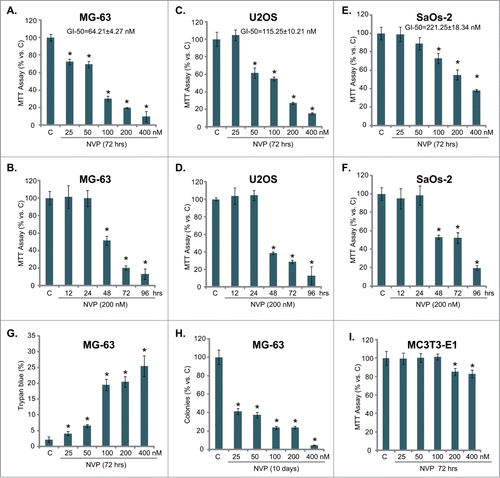
Caspase-dependent apoptosis contributes to NVP-BEZ235-induced cytotoxicity against osteosarcoma cells
Next, we tested whether cell apoptosis contributed to NVP-BEZ235-induced cytotoxicity against osteosarcoma cells. Cell apoptosis after NVP-BEZ235 treatment was tested by 3 independent assays including caspase-3 activity assay, Histone-DNA ELISA assay and TUNEL staining assay. Results from all these 3 assays demonstrated that NVP-BEZ235 dose-dependently induced MG-63 cell apoptosis (). Western blots further confirmed caspase-3 cleavage by NVP-BEZ235 in MG-63 cells (, upper panel). Significantly, the pan-caspase inhibitor (z-VAD-fmk, vad), the caspase-3 inhibitor (z-DVED-fmk, dved) or the caspase-8 inhibitor (z-ITED-fmk, ited) dramatically inhibited NVP-BEZ235 (200 nM)-induced MG-63 cell growth inhibition (MTT assay, ) and cell death (clonogenicity assay, ). Together, these results demonstrate that NVP-BEZ235 induces caspase-3 activation and osteosarcoma cell apoptosis, which contributes to its cytotoxicity. TUNEL assay results in showed apoptosis activation by NVP-BEZ235 in U2OS and SaOs-2 osteosarcoma cell lines.
Figure 2. NVP-BEZ235 activates apoptosis in osteosarcoma cells. MG-63 cells were treated with indicated concentration of NVP-BEZ235 (NVP) for 42 hrs, cell apoptosis was tested by caspase-3 activity assay (A), Histone-DNA ELISA assay (B) and TUNEL staining assay (C). The expression of caspase-3 (regular and cleaved) and tubulin (equal loading) was tested by Western blotting (A, upper panel). MG-63 cells were pretreated with z-VAD-fmk (zvad, 25 μM), z-DVED-fmk (dved, 25 μM) or z-ITED-fmk (ited, 25 μM) for 1 hr, followed by NVP-BEZ235 (200 nM) stimulation, cells were further cultured, before cell growth (D) and colony formation (E) were tested. Apoptosis in NVP-BEZ235 (200 nM, 42 hrs)-treated U2OS and SaOs-2 cells was analyzed by TUNEL staining assay (F). n = 5 for each assay. *P < 0.05 vs. group “C” (A-C, F). #P < 0.05 vs. NVP-BEZ235 only group (D-E).
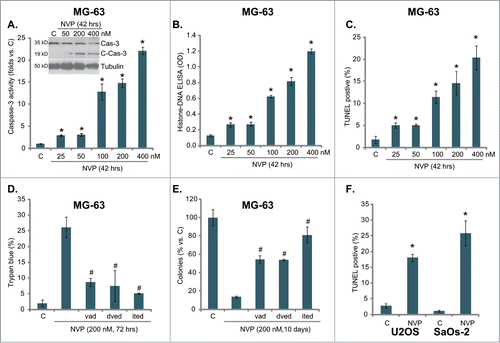
NVP-BEZ235 inhibits osteosarcoma cell cycle progression
Next we examined NVP-BEZ235s effect on osteosarcoma cell cycle progression. As shown in , NVP-BEZ235 (200 nM) significantly disrupted osteosarcoma cell cycle progression. The cell cycle distribution in NVP-BEZ235-treated cells was significantly different from that of untreated control cells, and a clear G1-S arrest was observed (). The effect of NVP-BEZ235 on osteosarcoma cell cycle progression was again dose-dependent (Data not shown). Thus, NVP-BEZ235 exerts a cytostatic effect on osteosarcoma cells, causing G1-S cycle arrest.
Figure 3. NVP-BEZ235 exerts a cytostatic effect in osteosarcoma cells. Quantitative cell cycle distribution of MG-63 cells (A) or U2OS cells (B) treated with or without NVP-BEZ235 (NVP, 200 nM, 42 hrs) was examined by FACS assay. n = 5 for each assay. *P < 0.05 vs. group “C.”
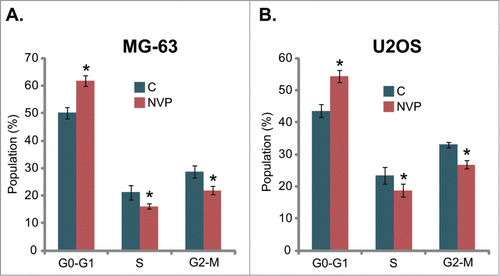
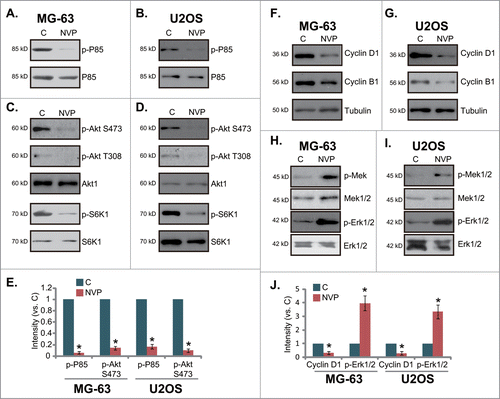
NVP-BEZ235 inhibits PI3K-AKT-mTORC1 activation and cyclins expression, while activating MEK/Erk in osteosarcoma cells
NVP-BEZ235 is a PI3K-mTOR dual inhibitor,Citation16,17 the effect of NVP-BEZ235 PI3K-Akt-mTOR activation in cultured osteosarcoma cells was tested. As shown in , the phosphorylation of p85, an indicator of PI3K activation, was almost blocked by NVP-BEZ235 treatment in both MG-63 cells and U2OS cells. Meanwhile, Akt activation, evidenced by phosphorylation at both Ser-473 and Thr-308, was similarly inhibited by NVP-BEZ235 (). Further, S6K1 phosphorylation (Thr-308), or mTORC1 activation, was also suppressed by NVP-BEZ235 treatment (). P85 and Akt phosphorylation was quantified (). Significantly, the expression of cell cycle associated proteins including cyclin D1 and cyclin B1 was downregulated by NVP-BEZ235 in both osteosarcoma cell lines (, cyclin D1 expression was quantified in ). Interestingly, phosphorylation of MEK and Erk was increased in NVP-BEZ235-treated MG-63 cells and U2OS cells ( and 4J). Similar signaling changes were also obtained in SaOs-2 cells (Data not shown). Together, these results show that NVP-BEZ235 inhibits PI3K-AKT-mTORC1 activation and cyclins expression, while activating MEK/Erk signaling in cultured osteosarcoma cells.
Figure 4. Signaling changes by NVP-BEZ235 in osteosarcoma cells. MG-63 cells or U2OS cells were left untreated (“C”), or stimulated with NVP-BEZ235 (NVP, 200 nM) for 12 hrs, expression of listed proteins was tested by Western blots using corresponding antibodies (A-D, F-I). Phosphorylation of P85 and Akt was quantified and normalized to “C” (E), Phosphorylation of Erk and cyclin D1 expression was similar quantified (J). *P < 0.05 vs. group “C.”
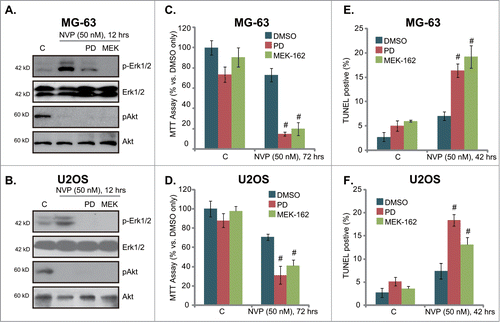
MEK/Erk inhibitors increase the sensitivity of NVP-BEZ235 in osteosarcoma cells
Above results showed that NVP-BEZ235 treatment in osteosarcoma cells activates MEK/Erk signaling, which is a well-known pro-survival signaling. We thus tested the role of Erk activation in NVP-BEZ235-induced activity. Two MEK/Erk inhibitors, including PD98059 and MEK-162Citation18 were utilized. Results demonstrated that both PD98059 and MEK-162 prevented NVP-BEZ235-induced Erk activation, without affecting Akt signaling in MG-63 cells () and U2OS cells (). Significantly, in both cell lines, PD98059 or MEK-162 dramatically enhanced NVP-BEZ235-induced growth inhibition (MTT assay, and 5D), as well as cell apoptosis (). These results indicate that activation of MEK/Erk by NVP-BEZ235 exerts a pro-growth and anti-apoptosis role, and NVP-BEZ235s activity against osteosarcoma cells could be further enhanced by MEK/Erk inhibitors.
Figure 5. MEK/Erk inhibitors increase the sensitivity of NVP-BEZ235 in osteosarcoma cells. MG-63 cells or U2OS cells, pretreated with MEK/Erk inhibitor PD98059 (PD, 1 μM) or MEK-162 (MEK, 10 nM) for 30 min, were stimulated with a low concentration of NVP-BEZ235 (NVP, 50 nM) for indicated time, expression of listed proteins was tested by Western blots (A)and B), cell growth was tested by MTT assay (C)and D), while cell apoptosis was also tested by TUNEL staining (E)and F). # P < 0.05 vs. NVP-BEZ235 only group.
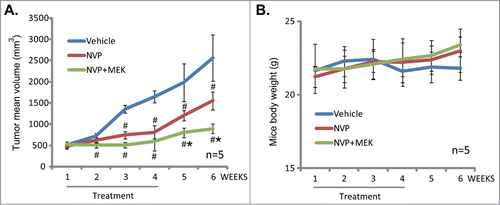
NVP-BEZ235 inhibits U2OS cell growth in vivo, its efficiency is further enhanced by MEK-162 co-administration
At last, by using a mice U2OS OS xenograft model,Citation19 we tested the anti-osteosarcoma activity of NVP-BEZ235 in vivo. As shown in , the mice that received NVP-BEZ235 (20 mg/kg, oral gavage daily, for 21 days) showed a significantly reduced tumor growth as compared to mice that received vehicle control (10% NMP–90% PEG). The mice body weight was not significantly affected by NVP-BEZ235 administration, indicating the relative safety of this regimen (). Note that co-administration of MEK/Erk inhibitor MEK-162 (10 mg/kg, oral gavage daily, for 21 days) further increased the efficiency of NVP-BEZ235 in vivo (). Once again, the body weight was not affected by the co-administration (). No apparent toxicity was observed. MEK-162 at the concentration alone had no effect on U2OS cell growth in vivo (Data not shown). Note that the concentration and treatment duration of NVP-BEZ235 or MEK-162 were chosen based on published literaturesCitation16,20 and pre-experimental results. These results show that NVP-BEZ235 inhibits U2OS osteosarcoma cell growth in vivo, its efficiency could be further enhanced by MEK-162 co-administration.
Figure 6. The activity of NVP-BEZ235 in vivo. The U2OS cell xenografted mice (5 mice per group) were treated with once daily with vehicle (10% NMP–90% PEG), NVP-BEZ235 (20 mg/kg, oral gavage, 21 days), or plus MEK-162 (MEK, 10 mg/kg, oral gavage, 21 days), tumor size was measured every week for a total of 6 weeks (A). Mice body weight was also recorded (B). #P < 0.05 vs. vehicle control group (A). *P < 0.05 vs. NVP-BEZ235 only group (A).
Discussions
Malignant osteosarcoma is one of the leading causes of cancer-related mortality among teenagers and young adults. Five-year survival is relative low for this disease, as it has a propensity to invade surrounding tissue and organs, which prevents surgical resection.Citation2,10,21,22 Further, malignant osteosarcoma tend to be resistant to the conventional chemotherapies due to constitutively active factors that promote cancer progression.Citation2,10,21,22 For example, hyper-activation or over-expression of PI3K-Akt-mTOR pathway contributes to osteosarcoma cell growth, proliferation, survival, and apoptosis-resistance.Citation9,10 In the current study, we found that due inhibition of PI3K and mTOR by NVP-BEZ235 efficiently induced growth arrest and apoptosis in cultured osteosarcoma cells. Further, it downregulated cyclin D1/B1 expression, and disrupted cell cycle progression, causing G1-S arrest. In vivo, oral NVP-BEZ235 inhibited U2OS xenograft growth in SCID mice. Thus, we provided evidence to support that NVP-BEZ235 could be investigated as a novel and efficient anti-osteosarcoma agent.
Signaling through the mTOR pathway contributes to growth, progression, and chemo-resistance of osteosarcoma and many other cancers. Accordingly, inhibitors have been developed to treat this and other malignancies clinically and pre-clinically.Citation23-25 MTOR exists in at least 2 distinct complexes, mTOR complex 1 (mTORC1) and mTORC2, with each complex containing at least several distinctive proteins.Citation26,27 MTORC1, or the rapamycin-sensitive mTOR complex, is assembled by mTOR, raptor, and G protein β subunit-like (GβL) among others. Whereas mTORC2 contains mTOR, GβL, Sin1, Rictor and others.Citation26,27 The functions of these 2 complexes are strikingly distinct. MTORC1 activation regulates protein synthesis, cell growth, and proliferation via phosphorylating at least 2 characterized downstream targets, 4E-BP1 and S6K1. While the Rictor–mTOR complex (mTORC2) phosphorylates Akt at the Ser-473 position, leading to full activation of Akt.Citation26,27
The activity of traditional mTORC1 inhibitors (rapamycin and its analogs, rapalogs) was limited.Citation28 Further, mTORC1 inhibition by rapalogs will result in a feedback activation of the PI3K-Akt pathway.Citation29,30 Thus, the advantage of NVP-BEZ235, besides being a more efficiency mTORC1 inhibitor, is simultaneous PI3K-Akt and mTORC2 inhibition. Here, we saw that NVP-BEZ235 inhibited PI3K-Akt-mTORC1/2 signalings in cultured osteosarcoma cells. This might explain why NVP-BEZ235 exerted significant cytostatic and cytotoxic activities on osteosarcoma cells, while rapalogs (i.e. RAD001) only showed cytostatic activity, without inducing osteosarcoma cell death.Citation31 Indeed, protein synthesis, which is required for cell growth and cell cycle progression, is regulated by mTORC1, while PI3K-Akt as well as mTORC2 activation is also important for cell survival.Citation32,33
Interestingly, in consistent with recent studies,Citation34,35 we found that NVP-BEZ235 caused increased phosphorylation of MEK/Erk, although it appeared to be less significant compared with mTOR or PI3K inhibitors.Citation34,35 It has been suggested that activation of MEK/Erk signaling is a resistance mechanism in mTOR inhibitor-based therapy. Consequently MAPK/Erk inhibitors can improve anti-tumor effect of PI3K-mTOR inhibitors.Citation34-36 Here we observed that MEK/Erk inhibitors enhanced the activity of NVP-BEZ235 both in vivo and in vitro. Thus these findings suggest that concomitant targeting of MAPK/Erk signaling is a promising strategy to enhance antitumor effect of NVP-BEZ235 against osteosarcoma and possible other cancers.
Taken together, our findings indicate dual inhibition of PI3K and mTOR with NVP-BEZ235, either alone or in combination with MEK/Erk inhibitors, may be an efficient treatment for osteosarcoma.
Material and Methods
Reagents and chemicals
NVP-BEZ235, PD98059 and MEK-162 were obtained from Selleck China (Shanghai, China). The broad caspase inhibitor z-VAD-fmk, the caspase-3 specific inhibitor z-DVED-fmk, and the caspase-8 specific inhibitor z-ITED-fmk were all from Calbiochem (Darmstadt, Germany). Antibodies for p-Akt (Ser-473), p-Akt (Thr-308), Akt1, p-PI3K p85 (Tyr-458), p85, p-Erk1/2 (Thr-202/Tyr-204), Erk1/2, p-MEK1/2 (Ser-217/Ser-221), MEK1/2, p-S6K1 (Thr-389), S6K1, cleaved-caspase 3, cyclin D1, cyclin B1 and tubulin were obtained from Cell Signaling Technologies (Beverly, MA).
Cell Culture
U2OS, SaOs-2 and MG-63 osteosarcoma cell lines were maintained and cultured as reported.Citation19 The murine calvaria-derived osteoblastic MC3T3-E1 cells were cultured in α-MEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were then subjected to l-ascorbic acid (50 μg/ml, Sigma Chemical Company, St. Louis, Mo.) and β-glycerol phosphate (5 mM, Sigma) for differentiation.Citation19
Cell growth assay (MTT assay)
As reported,Citation19 cells (0.5×10 4/well) were seeded in 96-well plates. After treatment, 30 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, sigma) solution (5 mg/ml) were added into each well, and incubated for an additional 4 hrs. The formazan crystal formed was dissolved with 100 μl of DMSO. Absorbance was measured at 490 nm by a microplate reader (Bio-Rad, Nanjing, China).
Trypan blue staining of “dead” cells
Dead cells were stained by trypan blue dye (Sigma), and the percentage (%) was calculated by the number of the trypan blue stained cells dividing by the total cell number.
Clonogenicity assay
Osteosarcoma cells (5 × 104) were suspended in 1 ml of DMEM containing 1% agar (Sigma), 10 % FBS and with indicated treatment. The cell suspension was then added on top of a pre-solidified 1% agar in a 100 mm culture dish. The drug containing medium was refreshed every 2 d After 10-day of incubation, colonies were photographed. The number of surviving colonies were counted manually.
Western blotting and data quantification
After treatments, cells were lysed using lysis buffer described.Citation19 Samples (20–30 μg) were separated by 10% SDS-polyacrylamide gel, and electro-transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). Afterwards, the membranes were blocked with blocking buffer (10% (w/v) milk in PBS Tween-20 (PBST), incubated overnight at 4°C with the indicated primary antibodies, and then incubated with HRP-conjugated second antibodies (Santa Cruz Biotech, Santa Cruz, CA). The detection was performed by ECL Supersingnal West Pico Chemiluminescent Substrate. Blot quantification was performed as described.Citation19
TUNEL assay of apoptosis
Cell apoptosis was detected by the TUNEL (Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling) In Situ Cell Apoptosis Detection Kit (Roche Molecular Biochemicals, Indianapolis, IN), according to the manufacturer's instructions. Cells were also stained with 4′,6′-diamino-2-phenylin-dole (DAPI, blue fluorescence; Molecular Probes) to visualize the cell nuclei. The apoptosis rate was determined by TUNEL percentage, which was calculated by the number of TUNEL-positive cells divided by the number of DAPI-stained cells. At least 1000 total cells in 10 views from 10 repeat wells (1 × 100) of each condition were included for counting the TUNEL percentage.
Cell cycle analysis
Cells were seeded in 60-mm dishes (5 × 105/dish) and treated with NVP-BEZ235. Floating and adherent cells were collected by trypsinization and washed once with PBS. Cells were incubated in 70% ethanol at −20°C overnight, treated with 20 μg/mL RNase A, then stained with 0.5 μg/mL of propidium iodide (PI), and evaluated by flow cytometry (Beckman Coulter, Miami, FL).
Caspase-3 activity assay
After treatment, cytosolic proteins from approximately 2–3 × 106 cells were extracted in hypotonic cell lysis buffer described in.Citation19 Twenty μg of cytosolic extracts were added to caspase assay buffer (312.5 mM HEPES, pH 7.5, 31.25% sucrose, 0.3125% CHAPS) with Ac-DEVD-AFC (15 μg/ml) (Calbiochem, Darmstadt, Germany) as the substrate. After incubation at 37°C for 1 hr, the amount of AFC liberated was measured using a spectrofluorometer (Thermo-Labsystems, Helsinki, Finland) with excitation of 380 nm and emission wavelength of 460 nm.
Histone/DNA ELISA for detection of apoptosis
The Cell Death Detection ELISA Kit (Roche) was used for assessing osteosarcoma cell apoptosis according to the protocol provided by the manufacturer. Detailed procedures can be found in other studies.Citation37,38 Briefly, the cells were lysed and the cell lysates were overlaid and incubated in microtiter plate modules coated with anti-Histone antibody. Samples were then incubated with anti-DNA peroxidase followed by color development with ABTS substrate. The absorbance of the samples were determined with a microplate reader at 405 nm.
Mice U2OS xenograft
CB.17 severe combined immuno-deficient (SCID) male mice (4–6 weeks old) were maintained at the animal facility of Southeast University of China. Mice were then injected subcutaneously (s.c.) into the right flank with 2 × 106 U2OS cells in 0.2 ml DMEM/10 % FBS. When the right flank xenografts were established at about 500 mm3 (considered as established growing xenografts), the animals (5 mice per group) were administrated with NVP-BEZ235 (20 mg/kg in 10% NMP–90% PEG), or plus MEK-162 (10 mg/kg in 10% NMP–90% PEG), which were both freshly prepared and given oral gavage once every 24 hrs in 100 μL of volume for 21 consecutive days. Control mice received vehicle (10% NMP–90% PEG) only. The xenografted tumor diameter was measured every 7 d using calipers. Tumor volumes (mm3) and mice body weight (g) were recorded as described.Citation19 All animals were maintained in accordance with the guidelines of NIH.
Statistics
The data presented were mean ± standard deviation (SD). Statistical differences were analyzed by one-way ANOVA followed by multiple comparisons performed with post hoc Bonferroni test (SPSS version 16). Fifty percent growth inhibition (GI-50) was also calculated by SPSS. The duration of treatment and concentration of agents were chosen based on pre-experiment results. Values of P < 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Funding
This work is supported by the National Natural Science Foundation of China.
References
- Ham SJ, Schraffordt Koops H, van der Graaf WT, van Horn JR, Postma L, Hoekstra HJ. Historical, current and future aspects of osteosarcoma treatment. Eur J Surg Oncol 1998; 24: 584–600; PMID:9870738; http://dx.doi.org/10.1016/S0748-7983(98)93896-3.
- Chou AJ, Gorlick R. Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther 2006; 6: 1075–85; PMID:16831079; http://dx.doi.org/10.1586/14737140.6.7.1075.
- Berberoglu S, Oguz A, Aribal E, Ataoglu O. Osteoblastoma response to radiotherapy and chemotherapy. Med Pediatr Oncol 1997; 28: 305–9; PMID:9078333.
- Camitta B, Wells R, Segura A, Unni KK, Murray K, Dunn D. Osteoblastoma response to chemotherapy. Cancer 1991; 68: 999–1003; PMID:1913494; http://dx.doi.org/10.1002/1097-0142(19910901)68:5%3c999::AID-CNCR2820680515%3e3.0.CO;2-Z.
- Zardawi SJ, O'Toole SA, Sutherland RL, Musgrove EA. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol Histopathol 2009; 24: 385–98; PMID:19130408.
- Gorelik L, Flavell RA. Transforming growth factor-β in T-cell biology. Nat Rev Immunol 2002; 2: 46–53; PMID:11905837; http://dx.doi.org/10.1038/nri704.
- Fukaya Y, Ishiguro N, Senga T, Ichigotani Y, Sohara Y, Tsutsui M, Shioura T, Iwamoto T, Hamaguchi M. A role for PI3K-Akt signaling in pulmonary metastatic nodule formation of the osteosarcoma cell line, LM8. Oncol Rep 2005; 14: 847–52; PMID:16142341.
- He JP, Hao Y, Wang XL, Yang XJ, Shao JF, Guo FJ, Feng JX. Review of the molecular pathogenesis of osteosarcoma. Asian Pac J Cancer Prev 2014; 15: 5967–76; PMID:25124559; http://dx.doi.org/10.7314/APJCP.2014.15.15.5967.
- Broadhead ML, Clark JC, Myers DE, Dass CR, Choong PF. The molecular pathogenesis of osteosarcoma: a review. Sarcoma 2011; 2011: 959248; PMID:21559216; http://dx.doi.org/10.1155/2011/959248.
- Yang J, Zhang W. New molecular insights into osteosarcoma targeted therapy. Curr Opin Oncol 2013; 25: 398–406; PMID:23666471; http://dx.doi.org/10.1097/CCO.0b013e3283622c1b.
- Dai X, Ma W, He X, Jha RK. Review of therapeutic strategies for osteosarcoma, chondrosarcoma, and Ewing's sarcoma. Med Sci Monit 2011; 17: RA177–90; PMID:21804475; http://dx.doi.org/10.12659/MSM.881893.
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009; 8: 627–44; PMID:19644473; http://dx.doi.org/10.1038/nrd2926.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501; PMID:12094235; http://dx.doi.org/10.1038/nrc839.
- Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005; 24: 7455–64; PMID:16288292; http://dx.doi.org/10.1038/sj.onc.1209085.
- Stauffer F, Maira SM, Furet P, Garcia-Echeverria C. Imidazo[4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg Med Chem Lett 2008; 18: 1027–30; PMID:18248814; http://dx.doi.org/10.1016/j.bmcl.2007.12.018.
- Cho DC, Cohen MB, Panka DJ, Collins M, Ghebremichael M, Atkins MB, Signoretti S, Mier JW. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin Cancer Res 2010; 16: 3628–38; PMID:20606035; http://dx.doi.org/10.1158/1078-0432.CCR-09-3022.
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008; 7: 1851–63; PMID:18606717; http://dx.doi.org/10.1158/1535-7163.MCT-08-0017.
- Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, Linnartz R, Zubel A, Slamon DJ, Finn RS. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer 2014; PMID:25167228.
- Zhu YR, Xu Y, Fang JF, Zhou F, Deng XW, Zhang YQ. Bufotalin-induced apoptosis in osteoblastoma cells is associated with endoplasmic reticulum stress activation. Biochem Biophys Res Commun 2014; 451: 112–8; PMID:25068992; http://dx.doi.org/10.1016/j.bbrc.2014.07.077.
- Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, Bastian BC. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014; 33: 4724–34; PMID:24141786; http://dx.doi.org/10.1038/onc.2013.418.
- Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer 2011; 47: 2431–45; PMID:21703851; http://dx.doi.org/10.1016/j.ejca.2011.05.030.
- Ritter J, Bielack SS. Osteosarcoma. Ann Oncol 2010; 21 Suppl 7: vii320-5; PMID:20943636.
- Garcia-Echeverria C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg Med Chem Lett 2010; 20: 4308–12; PMID:20561789; http://dx.doi.org/10.1016/j.bmcl.2010.05.099.
- Konings IR, Verweij J, Wiemer EA, Sleijfer S. The applicability of mTOR inhibition in solid tumors. Curr Cancer Drug Targets 2009; 9: 439–50; PMID:19442061; http://dx.doi.org/10.2174/156800909788166556.
- Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer 2012; 31: 8–18; PMID:22059905; http://dx.doi.org/10.5732/cjc.011.10282.
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12: 9–22; PMID:17613433; http://dx.doi.org/10.1016/j.ccr.2007.05.008.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149: 274–93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017.
- Vilar E, Perez-Garcia J, Tabernero J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther 2011; 10: 395–403; PMID:21216931; http://dx.doi.org/10.1158/1535-7163.MCT-10-0905.
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66: 1500–8; PMID:16452206; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925.
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26: 1932–40; PMID:17001314; http://dx.doi.org/10.1038/sj.onc.1209990.
- Moriceau G, Ory B, Mitrofan L, Riganti C, Blanchard F, Brion R, Charrier C, Battaglia S, Pilet P, Denis MG, et al. Zoledronic acid potentiates mTOR inhibition and abolishes the resistance of osteosarcoma cells to RAD001 (Everolimus): pivotal role of the prenylation process. Cancer Res 2010; 70: 10329–39; PMID:20971812; http://dx.doi.org/10.1158/0008-5472.CAN-10-0578.
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005; 9: 59–71; PMID:15784165; http://dx.doi.org/10.1111/j.1582-4934.2005.tb00337.x.
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4: 988–1004; PMID:16341064; http://dx.doi.org/10.1038/nrd1902.
- Roccaro AM, Sacco A, Husu EN, Pitsillides C, Vesole S, Azab AK, Azab F, Melhem M, Ngo HT, Quang P, et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood 2010; 115: 559–69; PMID:19965685; http://dx.doi.org/10.1182/blood-2009-07-235747.
- Moon du G, Lee SE, Oh MM, Lee SC, Jeong SJ, Hong SK, Yoon CY, Byun SS, Park HS, Cheon J. NVP-BEZ235, a dual PI3K/mTOR inhibitor synergistically potentiates the antitumor effects of cisplatin in bladder cancer cells. Int J Oncol 2014; 45: 1027–35; PMID:24969552.
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118: 3065–74; PMID:18725988.
- Chen MB, Shen WX, Yang Y, Wu XY, Gu JH, Lu PH. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. J Cell Physiol 2010; 226: 1915–25; http://dx.doi.org/10.1002/jcp.22522.
- Chen MB, Wu XY, Gu JH, Guo QT, Shen WX, Lu PH. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem Biophys 2011; 60: 311–22; PMID:21274754; http://dx.doi.org/10.1007/s12013-011-9153-0.