
Abstract
A growing amount of evidence reported in the literature in recent years strongly supports the relevance of the interplay between autophagy and other pathways. In this context, the study of the link between autophagy and cell proliferation regulation has been among the most challenging. In our recent publications, we finely characterize a role for the pro-autophagic protein AMBRA1 in the regulation of cell proliferation. AMBRA1 modulates autophagy and interacts with PPP2/PP2A (protein phosphatase 2), thus also modulating MYC protein levels and the cell proliferation rate. Interestingly, this pathway of regulation is controlled by the master regulator of autophagy and cell growth, MTORC1. Notably, in our study we demonstrate the relevance of the AMBRA1-mediated regulation of MYC in tumorigenesis, also identifying AMBRA1 as a tumor suppressor gene.
Environmental restrictions, such as nutrient shortage and cellular stressors, trigger a cell response consisting of autophagy induction and simultaneous inhibition of cell proliferation. The conservation among species of this metabolic reprogramming also suggests a reciprocal regulation of autophagy and cell proliferation, although our understanding of the molecular mechanism(s) linking these 2 pathways is far from conclusive. In this context, the mechanistic target of rapamycin complex 1 (MTORC1) is an exception, since it has been long known to regulate both autophagy and cell growth, although distinct MTORC1 substrates play independent roles in the 2 pathways. The depletion of some upstream regulators of autophagy, AMBRA1 among them, has been as well associated with increased proliferation.
AMBRA1 has emerged as a scaffold protein regulating autophagy in multiple ways. In “fed” conditions, AMBRA1 tethers the class III PtdIns3K complex at the cytoskeleton, negatively regulating autophagy; however, this inhibitory mechanism is lost upon starvation, when the ULK1 kinase phosphorylates AMBRA1. In a positive feedback control, AMBRA1 activates ULK1, an event that is prevented by the MTORC1-mediated phosphorylation of AMBRA1, thereby inhibiting autophagy. Finally, Antonioli and co-authors recently showed that AMBRA1 also establishes a feedback loop on the kinase MTORC1, thus enhancing autophagy (), and that AMBRA1 proteasomal degradation is responsible for autophagy termination after prolonged starvation.
Figure 1. AMBRA1-mediated regulation of MYC and its interplay with the autophagy pathway. We recently demonstrated that AMBRA1 enhances PPP2/PP2A phosphatase activity on phospho-Ser62 of MYC, resulting in the proteasomal degradation of the transcription factor and in preventing hyperproliferation and tumorigenesis. By contrast, the oncogene KIAA1524/CIP2A inhibits the activity of PPP2 in the same pathway. We also found that the AMBRA1-PPP2 role in MYC dephosphorylation is under MTORC1 control. Intriguingly, numerous crosstalk and feedback mechanisms between complexes regulating this pathway (MTORC1, AMBRA1-PPP2, KIAA1524/CIP2A-PPP2) and autophagy have been reported (see the figure for more details), supporting the evidence that autophagy and cell proliferation regulation are tightly coordinated.
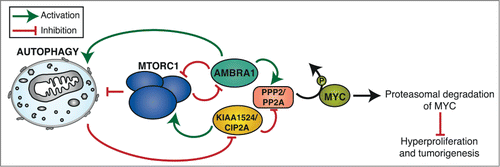
In our recent publication, we demonstrate that the scaffold protein AMBRA1 is more than that: a point of convergence of both autophagy and cell proliferation. AMBRA1 signals MTORC1 inhibition to the pro-proliferative factor and oncoprotein MYC, resulting in decreased cell proliferation and preventing tumorigenesis ().
First, we analyzed the levels of cell cycle regulators in murine embryonic fibroblasts (MEFs) defective for Ambra1, thus revealing an Ambra1 dose-dependent hyperphosphorylation of the RBL1/p107 (retinoblastoma-like 1), consistent with its inhibition and an increase in pro-proliferative molecules. Strikingly, the protein phosphatase for which RBL1 is a substrate, namely the serine/threonine phosphatase PPP2/PP2A, was identified as a new AMBRA1 interactor in 2 different high-throughput screenings. PPP2 has diverse cellular functions, as well as in cell proliferation and autophagy. Of note, MYC has been reported among the PPP2 substrates that are relevant to cell proliferation regulation. In detail, the phosphorylation of MYC on Ser62 (P-MYCS62), occurring upon growth-factor receptor activation, is removed by PPP2, triggering MYC proteasomal degradation and inhibition of proliferation. It is well known that direct interactors of PPP2 modulate the activity of the phosphatase toward specific substrates; indeed, we found that this is also the case for AMBRA1, whose deficiency results in MYC hyperphosphorylation at Ser62, MYC destabilization, and decreased cell proliferation, thus resembling PPP2 inhibition. In fact, PPP2 extracted from Ambra1-depleted cells, is inefficient in dephosphorylating P-MYCS62 in a phosphatase assay in vitro. This effect relies, at least in part, on the capability of AMBRA1 to recruit MYC at the phosphatase complex. These results strongly support a role for AMBRA1 as a positive regulator of PPP2 activity toward MYC.
In light of MTORC1's role in cell growth, autophagy, and AMBRA1 regulation, we wondered whether this key kinase could also play a role in the AMBRA1-mediated regulation of MYC. In fact, genetic or pharmacological inhibition of MTORC1 results in P-MYCS62 dephosphorylation and destabilization. Strikingly, AMBRA1 is needed in order to signal the MTORC1 inhibition to MYC, although it is not clear yet whether the MTORC1-mediated phosphorylation of AMBRA1 plays any role(s) in this context.
Along with the relevance of the AMBRA1-PPP2 complex in MYC regulation, there is the question of whether this complex is actually functional in the regulation of cell proliferation and tumorigenesis. To address this issue, we generated an AMBRA1 mutant defective for binding the catalytic subunit of PPP2 (AMBRA1PXP), which lacks 2 novel PPP2 binding sites. Functional assays performed with AMBRA1PXP allowed us to conclude that AMBRA1-PPP2 direct interaction is relevant to MYC regulation and cell proliferation modulation. By contrast, AMBRA1PXP seems to be fully competent for autophagy induction, even though we cannot rule out the existence of mechanism(s) of reciprocal regulation between AMBRA1 and PPP2 interplaying with autophagy.
Bearing in mind the role of MYC as an oncogene, we next examined the relevance of AMBRA1-mediated regulation of MYC in tumorigenesis. Significantly, we observed (i) an increased incidence of spontaneous tumors in mice heterozygous for the Ambra1 gene-trap mutation (Ambra1+/gt) and (ii) earlier and bigger Ambra1gt/gt masses, compared with wild-type ones, in a xenograft tumor assay. The lack of rescue after Ambra1gt/gt reconstitution with AMBRA1PXP suggests that, also in this context, the AMBRA1-PPP2 interaction is important.
Further, an inverse correlation is observed between AMBRA1 and P-MYCS62 in human cancer cell lines, with AMBRA1 overexpression being sufficient to affect not only MYC and cell proliferation, but also their pro-tumorigenic phenotype. Interestingly, AMBRA1 mutations are also associated with human cancer.
Overall, these findings reveal a role for AMBRA1 as a tumor suppressor gene, as has been demonstrated for other upstream regulators of autophagy, such as BECN1. Interestingly, we also recently showed that BECN1, as well as AMBRA1, affects MYC regulation, but probably through the indirect regulation of the MYC kinases MAPK1/ERK2 and MAPK3/ERK1, rather than the phosphatase PPP2.
The roles that we describe for AMBRA1 and BECN1 in this context are good examples of the intricate interplay between cell proliferation and autophagy, an issue we should bear in mind when manipulating autophagy in cancer. Along these lines, Jäättelä's group recently demonstrated that an endogenous inhibitor of PPP2, the oncogene KIAA1524/CIP2A, is a substrate of autophagy, as well as a modulator of MTORC1 signaling (). Interestingly, KIAA1524/CIP2A and AMBRA1 exert opposite regulatory effects on PPP2 and MTORC1, raising the question of whether these proteins could compete for the same binding sites and/or regulate one another in a feedback mechanism. Given the emerging relevance of the coordinated regulation of autophagy and cell proliferation in cancer research, AMBRA1-KIAA1524/CIP2A crosstalk, as well as all the regulation mechanisms at the boundary between autophagy and cell proliferation, are absolutely worth being studied.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from KBVU (R72-A4408), Lundbeck Foundation (R167–2013–16100), Novo Nordisk Foundation (7559), The Bjarne Saxhof Foundation, AIRC (IG2013-14170), and in part from FISM (2013), the Telethon Foundation (GGP14202), and the Italian Ministry of University and Research (FIRB Accordi di Programma 2011). VC is supported by the Lundbeck Foundation (R165–2013–15982). Also, FC lab in Copenhagen is part of the newly established Center of Excellence in Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation.